

Abstract
Osmotically controlled implants yield precise zero-order drug delivery kinetics and are utilized in a number of applications. The implants deliver drugs for extended periods (weeks to years) and exhibit good in vivo/in vitro correlation. This paper reviews critical variables associated with these implants, with a focus on release rate testing. The extended-duration kinetics can be problematic when attempting to test for >70% cumulative delivery. An innovative scheme based on the scientific principles of operation of the system is described to ensure >70% delivery at the target rate and duration for the DUROS® Viadur® (leuprolide acetate) implant.
Key words: DUROS® system, leuprolide, osmosis, osmotic implants, Viadur® implant
INTRODUCTION
Zero-order delivery (i.e., constant delivery rate) is the desired target product performance in many extended-duration drug delivery applications. Zero-order delivery assures that plasma or tissue drug concentrations will not fall below minimum efficacious thresholds nor will the maximum concentrations exceed levels that are toxic or provoke uncomfortable side effects. Drug delivery systems based on the principles of osmosis have demonstrated the capacity to provide zero-order delivery in a number of human, animal, and research applications. These include systems administered orally to the gastrointestinal tract (oral osmotic systems), systems administered intraruminally to cattle, systems implanted in laboratory and food-producing animals, and systems implanted for human therapy (1–5). Implantable drug delivery systems offer a number of potential advantages including convenience and compliance for the patient, delivery of drugs that cannot be effectively delivered via the oral route, and the potential for more cost-effective health care delivery (e.g., reduced visits to the physician for administration of therapies).
The ALZET® osmotic pump is designed for implantation in laboratory animals. It has been implanted in multiple anatomical sites and in multiple animal species. It has been utilized in research described in over 10,000 scientific publications. The ALZET pump has allowed researchers to investigate novel therapies and therapeutic regimes with potential application to human therapy (6).
The DUROS® implantable osmotic system has been applied or investigated for a number of human therapies, with delivery durations ranging from several months to 1 year. The system is applicable to potent drugs and therapies that benefit from extended, precise delivery. Catheters can be included in the DUROS system design to enable site-specific delivery. DUROS systems include the commercially available Viadur® system for the delivery of leuprolide for prostate cancer. Other systems that are under development are the Chronogesic® system for the delivery of sufentanil for chronic pain, the Omega DUROS system for delivery of the protein omega interferon for treatment of hepatitis C viral infection, a catheterized DUROS system for intrathecal opioid delivery for chronic pain, and a catheterized DUROS system for local chemotherapy of brain tumors (7).
Osmotic systems are different in design from other extended-release drug delivery systems that rely on diffusion, dissolution, or erosion for their extended-release action. Osmotic systems tend to produce precise delivery kinetics and can provide highly predictable delivery characteristics, as described below. The regulatory and quality assurance scheme should derive from the scientific principles of operation of the drug delivery system, and, thus, the quality assurance scheme for an osmotic system would be expected to be somewhat different than the quality assurance scheme for dosage forms based on different scientific principles of action.
This paper describes the design and performance of osmotic implants and then discusses the critical variables associated with these implants, using the Viadur® (leuprolide acetate) implant as a case study example.
OSMOTIC IMPLANTS: DESIGN AND PRINCIPLES OF OPERATION
The diversity of delivery systems that can utilize osmotically driven drug release mechanisms is illustrated by the ALZET osmotic pump (veterinary medicine, research applications) and the DUROS implantable system (human medicine).
A schematic of the ALZET® osmotic pump for animal research is shown in Fig. 1. A solution of the drug or test agent is filled into the inner reservoir. Once implanted, body water is osmotically imbibed into the osmotic (salt) layer through a semipermeable membrane and compresses the reservoir at a rate determined by the composition of the membrane, resulting in steady delivery of the drug solution from the reservoir for a period of weeks to months (3,6).
Fig. 1.

Cross-sectional diagram of ALZET® osmotic pump. (Reprinted by permission of DURECT Corporation)
The DUROS® implantable osmotic system utilizes the same osmotic principles of operation. A schematic of the DUROS system is shown in Fig. 2. It resembles a miniature syringe and consists of an outer cylindrical reservoir (generally titanium alloy or a polymeric material) with a rate-controlling membrane at one end and an exit port at the other end. For site-directed therapy, a catheter can be attached to the exit port. Internal to the system are the osmotic engine (tablets containing primarily NaCl combined with other pharmaceutical excipients), the elastomeric piston, and the drug formulation in the drug reservoir. The rate-controlling membrane is composed of a specially designed semipermeable polyurethane polymer, chosen for a specific water permeability and the ability not to foul during in vivo operation. For subcutaneous implantation, the dimensions can range from 10 mm outside diameter (OD) ×45 mm length (L) to 4 mm OD × 45 mm L or smaller.
Fig. 2.

Cross-sectional diagram of the DUROS® osmotic implant. (Reprinted by permission from (5))
In operation, body water is drawn from the surrounding tissue through the semipermeable membrane of the DUROS system and into the osmotic engine by osmosis. The rate of water uptake is controlled by the permeability of the membrane and the osmotic pressure of the osmotic engine (i.e., the osmotic pressure of NaCl). The resulting expansion of the osmotic engine results in steady displacement of the piston and pumping of the drug formulation at a controlled rate.
Mathematically, the drug delivery rate (dM/dt) from the DUROS system is described by the following equation:
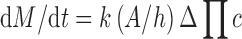 |
1 |
where:
- k
Membrane permeability
- A
Membrane area
- h
Membrane thickness
- ΔΠ
Osmotic pressure difference (osmotic engine vs. tissue)
- c
Drug concentration
In order to achieve a steady, zero-order delivery rate (dM/dt) over the time course of action of the osmotic implant, all of the variables (k, A, h, ΔΠ, and c) must remain constant over time.
Regarding the membrane permeability, k, osmotic drug delivery systems can be implanted in a number of sites in the body for either systemic or localized delivery. A preferred implantation location is the subcutaneous space. Implantation provokes the foreign body response, which can vary in its severity in relation to the biocompatibility of the implanted system. The foreign body response is not a static phenomenon; as is well known, it progresses from initial protein absorption and acute inflammation to chronic inflammation and the formation of a fibrotic capsule. A variety of different cell types are recruited to the implant surface in a time-defined progression. The thickness of the fibrotic capsule can depend on the biocompatibility of the implanted drug delivery system.
DUROS osmotic systems have been engineered to be highly biocompatible. While it is theoretically possible for the foreign body response to impact overall system performance in a time-dependent manner (e.g., membrane “fouling”), DUROS systems tend to have minimal encapsulation (8) and, as shown below, deliver at a constant rate in vivo, indicating that the membrane permeability, k, in the above equation, remains constant over time in vivo and is unaffected by the foreign body response to the DUROS system.
The membrane area, A, and the membrane thickness, h, are held constant by the features of the design. The membrane is produced by injection molding. From a chemistry, manufacturing, and controls (CMC) perspective, it should be noted that variations in the molding process or variations in the polyurethane feed raw material could lead to variations in membrane area, thickness, or permeability.
ΔΠ is the osmotic pressure difference between the tissue at the outer edge of the membrane and osmotic pressure in the osmotic engine. The osmotic engine contains mainly NaCl, present as a solid. Once a small amount of water is imbibed into the engine, a saturated aqueous solution of sodium chloride is present, and the osmotic pressure in the osmotic engine compartment is that of a saturated solution of NaCl. If sufficient NaCl is initially present in the osmotic engine, solid NaCl will be present throughout the travel stroke of the piston, a saturated NaCl solution will always be present in the osmotic engine compartment, and the osmotic pressure will always be that of a saturated solution of NaCl. Physiologic processes hold the tissue osmotic pressure essentially constant, especially when considering the relatively small tissue osmotic pressure compared with that of a saturated solution of NaCl. Thus, ΔΠ will be constant for the delivery lifetime of the system.
c is the concentration of drug in the drug formulation pumped from the system. DUROS formulations can be aqueous solutions, nonaqueous solutions, or suspensions. For solution formulations, c is constant provided that the drug is stable in the solution at body temperature. For suspensions, the drug formulation must be both physically and chemically stable, i.e., the drug must be chemically stable in the formulation and the suspension must not settle or separate.
PERFORMANCE OF OSMOTIC IMPLANTS
The Viadur® (leuprolide acetate implant) system contains the gonadotropin-releasing hormone (GnRH) analog leuprolide acetate dissolved in dimethyl sulfoxide. Subassemblies without the drug formulation are sterilized by gamma irradiation; then, the drug formulation is sterile-filtered and aseptically filled into the final implant. The Viadur® system has dimensions of 4 mm OD × 45 mm L and a drug reservoir volume of 150 μl. (For additional details on the Viadur implant, see (9).)
The in vitro release rate of leuprolide from the Viadur® system is shown in Fig. 3. Steady delivery of leuprolide is observed for 1 year. These data were obtained from systems placed into individual test tubes containing 8 ml of 37°C unstirred phosphate-buffered saline, with changeover to fresh test tubes at regular intervals (9).
Fig. 3.

In vitro release of leuprolide from the Viadur® implant in 37°C phosphate-buffered saline, n = 12. (Reprinted from (9) with permission from Elsevier)
Similar zero-order in vitro delivery profiles have been observed for the delivery of sufentanil from the DUROS Chronogesic system (5) and delivery of interferon from the OMEGA DUROS system (10).
The primary therapeutic option for the palliative treatment of advanced prostate cancer is lowering of circulating androgen levels. This lowering can be achieved by the continuous administration of GnRH analogs, such as leuprolide. In a clinical study, subcutaneous implantation of a single Viadur® system into the upper arm of prostate cancer patients maintained constant serum concentrations of leuprolide for a full year (Fig. 4). Additionally, leuprolide concentrations were maintained when the implants were removed and new Viadur systems implanted. In response to the steady delivery of leuprolide, testosterone levels were observed to decrease below the castrate threshold and be maintained at desired levels (9,11,12).
Fig. 4.

Serum leuprolide concentrations for patients treated with the Viadur® implant. (Reprinted from (9) with permission from Elsevier)
When DUROS systems are explanted, the system can be assayed for the remaining drug content. This assay can then be compared with the original drug content for the lot of systems under investigation, and the amount of drug delivered in vivo was determined. This assessment was undertaken for Viadur® systems implanted into rats, dogs, and humans. In rats, three lots of Viadur systems were tested, and the systems were explanted at 3, 6, 9, and 12 months. These systems were compared to systems tested for release rate in vitro. The results are shown in Fig. 5, where it is seen that there is good agreement between cumulative delivery in vivo and in vitro for each lot and that the cumulative delivery increases linearly with time. The average ratio of cumulative amount delivered in vitro to cumulative amount delivered in vivo at 12 months was 1.04 ± 0.03. In dogs at 12 months, the ratio was 1.05 ± 0.06, and in humans, the ratio was 1.03 ± 0.08. Greater than 99% drug stability (by RP–HPLC assay) was observed both in vivo (rat, dog, and human) and in vitro (9).
Fig. 5.

Comparison of cumulative leuprolide delivery in vivo (rats) and in vitro for three lots of Viadur® implants. (Data normalized with respect to average amount delivered in vivo for all three lots at 12 months; reprinted from (9) with permission from Elsevier)
TESTING FOR CRITICAL CMC VARIABLES
The testing scheme for a controlled-release system should assure that the system will deliver therapy to the patient as demonstrated in the clinical trials. The testing scheme should also ensure conformance to product labeling.
Key attributes for osmotic implants include the following:
Biocompatibility
Drug content and purity
Drug stability/degradants (shelf life and while implanted)
Sterility
Pyrogenicity/endotoxins
Drug release rate
Biocompatibility
Biocompatibility derives from the system design and the materials of construction specified in the design. The raw materials must be controlled to ensure that the materials are clean and consistent and that the raw material manufacturer does not change their formulation or process in a manner that adversely affects biocompatibility. Generally, design (and subsequent manufacturing) should avoid sharp corners or edges, and the surface roughness of parts should be monitored. The manufacturing processes for the implant must be controlled for cleanliness via process design and process monitoring.
Drug Content
Drug content should meet compendial specifications. Specification on initial drug purity should also consider the long-duration nature of the system. In system and process design, it is important to consider manufacturing variations that will occur batch to batch in drug concentration and batch to batch in drug reservoir volume. These variations must be monitored and controlled.
Drug Stability/Degradants
Drug stability should be extensively studied in the system development phase. Specifications for impurities and degradants should be based on regulatory guidelines and information on the properties (toxicity, biological activity) of the respective impurity or degradant. While it is desirable that all degradants and impurities be present in the systems tested in the nonclinical program, it must be recognized that this does not always occur. DUROS systems (and other nonerodible implants) are somewhat unique in that the systems can be explanted intact at the end of the delivery duration, and it is possible to assess drug stability after in vivo exposure. This assay can provide additional assurance regarding the levels of impurities and degradants administered to the patient; however, it is the opinion of the author that it is performance during nonclinical testing and the test results (i.e., initial assay) for the systems in the nonclinical studies that should have the preponderance of weight in setting the drug purity specifications for lot release.
Sterility
As parenteral products, osmotic implants must be sterile. The appropriate tests and in process controls depend on the method of sterilization chosen for commercial use. Of particular concern are aseptic processes, where the process must be validated according to appropriate guidelines and where ongoing monitoring is essential to ensuring product quality.
Pyrogenicity/Endotoxins
Osmotic implants should be nonpyrogenic. Endotoxin testing methods should be developed for each specific implant design and validated, with manufacturing lots tested for endotoxin levels versus appropriate specifications.
Drug Release Rate
Verification of the drug release rate of extended- or controlled-release systems is an essential test for manufactured lots. “Dose dumping” (i.e., “burst” or initial release of a significant quantity of the system drug contents) is a significant concern due either to the toxicity (therapeutic index) of the drug being delivered or concerns about the drug loading being depleted prior to the labeled duration of the system. Conceptually, the release profile can be divided into three main intervals:
Initial release
Steady state
Late time (>70% release)
Specifications on initial release should be based on the toxicity/therapeutic index of the drug. If an initial burst can be tolerated by the patient without adverse effects, then wider specifications on the initial interval may be appropriate.
The in vivo/in vitro correlation observed for the Viadur® implant would seem to form a solid basis for in vitro QC release rate testing. However, the challenge was the impracticality of performing a 1-year release rate test for lot clearance.
Acceleration of the release test via temperature is a possibility, but temperature can only be increased up to the point where polymeric components begin to lose physical integrity. Sufficient acceleration was not observed at temperatures where the system components maintain their physical integrity. Additionally, drug stability at elevated temperature can also be a concern.
How, then, could it be assured that each implant lot would deliver an efficacious amount of leuprolide for 12 months? The solution proposed during the development and regulatory approval process was to reference the basic scientific principles underlying the system operation, as shown by the osmotic system theory predictions for release rate as given in Eq. 1.
Because the Viadur® implant is a constant, zero-order delivery rate system, the delivery rate observed at early time accurately predicts the overall average delivery rate for the system over 365 days. Thus, the amount of drug delivered at 365 days (the equivalent of testing the system to full labeled duration) can be correlated to release during early time intervals:
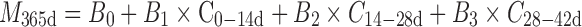 |
2 |
C0–14d, C14–28d, and C28–42d are the release during 0–14, 14–28, and 28–42 days; B0, B1, B2, and B3 are correlation constants; and M365d is the amount of drug delivered at 365 days. C0–14d represents a start-up interval; it includes thermal effects (day 1), membrane hydration, and other start-up phenomena (9). By day 14, the system is delivering at steady state: C14–28d and C28–42d represent steady-state delivery intervals. (The inclusion of two steady-state intervals improves the overall correlation.) The results of this correlation are shown in Fig. 6, where it is seen that there is excellent agreement between the predicted values of M365d and the actual experimental values of M365d.
Fig. 6.

Ratio of M 365 predicted from Eq. 2 to actual M 365 for eight lots of Viadur® implants
The proposed control scheme was as follows:
Conduct an abbreviated in vitro release rate test (37°C, PBS, unstirred) and set specifications on C0–14d, C14–28d, and C28–42d, where the specification on C0–14d represents a specification on initial burst/start-up and the specifications on C14–28d and C28–42d represent specifications on both the steady-state release rate and the cumulative delivery at 365 days (M365d) from the correlation.
Assay the NaCl content of the tablet to assure that sufficient NaCl is present in each batch of tablets such that a saturated solution is always present in the osmotic engine (i.e., implies constant driving potential, ΔΠ).
Assay the drug content. This assay implies the presence of target amount and the correct concentration.
Assay the drug purity. Based on the formulation development work, this ensures drug stability for the labeled lifetime of 365 days.
Referencing Eq. 1, it can be seen that this control scheme and the supporting development work ensures that all variables in the equation (k, A, h, ΔΠ, and c) are held constant over the 365-day delivery duration of the implant.
The US FDA’s response to this proposed control scheme was as follows: “The proposed Viadur™ cumulative leuprolide acetate release rate method and specifications are acceptable on an interim basis.” However, the agency continued that “…an accelerated in vitro release rate procedure is recommended as a Phase 4 commitment…” (http://www.fda.gov/cder/foi/nda/2000/02-1088_VIADUR_admincorres_P2.pdf, FDA website, accessed May 2007).
CONCLUSIONS
Understanding the scientific principles of a controlled- or extended-release system is essential to achieving target product performance and quality. The quality scheme for a product must consider standard pharmaceutical attributes and attributes that are unique to a novel or innovative dosage form. The quality scheme should derive from the scientific principles of the novel dosage form (as elucidated by a development program based on Quality by Design principles). Attempting to utilize testing schemes developed for different dosage forms with different principles of operation increases product development complexity and is not reflective of good regulatory science and can ultimately deny the public access to novel and efficacious new products.
Osmotic implantable systems provide excellent zero-order drug delivery profiles both in vitro and in vivo. Due to the long duration of drug delivery, there can be practical and technical limits to testing to >70% release within a reasonable amount of time. However, by exploiting the scientific principles underlying the operation of osmotic dosage forms, it is possible to develop a scientifically based scheme for assuring the long-term delivery performance of the systems without having to test for the entire delivery duration in real time. These alternative test methods have been shown to be discriminating, insuring that the marketed product provides the targeted characteristics for in vivo drug release.
Osmotic implantable systems represent a relatively new and innovative class of products that deliver with drugs precise zero-order kinetics. These implants tend to be less sensitive to external variables (such as boundary limitations and stirring effects) than other systems and, due to their osmotic principle of operation, therefore can yield a high degree of correlation between in vitro release and in vivo product performance. As a consequence of their long duration of in vivo activity, these systems have the potential to improve therapy, patient compliance, and patient quality of life while offering an opportunity for novel, scientifically based quality and regulatory paradigms.
References
- 1.Wong PSL, Gupta SK, Stewart BE. Osmotically controlled tablets. In: Rathbone MJ, Hadgraft J, Roberts MS, editors. Modified-release drug delivery technology. 1. New York: Marcel Dekker; 2003. pp. 101–14. [Google Scholar]
- 2.Zingerman JR, Cardinal JR, Chern RT, Holste J, Williams JB, Eckenhoff B, et al. The in vitro and in vivo performance of an osmotically controlled delivery system-IVOMEC SR® bolus. J Control Rel. 1997;47:1–11. doi: 10.1016/S0168-3659(96)01610-0. [DOI] [Google Scholar]
- 3.Theeuwes F, Yum SI. Principles of the design and operation of generic osmotic pumps for the delivery of semisolid or liquid drug formulations. Ann Biomed Eng. 1976;4:343–53. doi: 10.1007/BF02584524. [DOI] [PubMed] [Google Scholar]
- 4.Magruder J. Pumps/osmotic: VITS veterinary implant. In: Mathiowitz E, editor. Encyclopedia of controlled drug delivery. New York: Wiley; 1999. pp. 906–9. [Google Scholar]
- 5.Wright JC, Johnson RM, Yum SI. DUROS® osmotic pharmaceutical systems for parenteral and site-directed therapy. Drug Deliv Technol. 2003;3:3–11. [Google Scholar]
- 6.ALZET® Osmotic pumps. http://www.ALZET.com. Accessed 3 May 2009.
- 7.Wright JC, Culwell J. Long-term controlled delivery of therapeutic agents by the osmotically driven DUROS® implant. In: Rathbone MJ, Hadgraft J, Roberts MS, Lane ME, editors. Modified-release drug delivery technology. 2. New York: Informa Healthcare; 2008. pp. 143–9. [Google Scholar]
- 8.Cukierski MJ, Johnson PA, Beck JC. Chronic (60-week) toxicity study of DUROS implants in dogs. Int J Toxicol. 2001;20:369–81. doi: 10.1080/109158101753333659. [DOI] [PubMed] [Google Scholar]
- 9.Wright JC, Leonard ST, Stevenson CL, Beck JC, Chen G, Jao RM, et al. An in vivo/in vitro comparison with a leuprolide osmotic implant for the treatment of prostate cancer. J Control Rel. 2001;75:1–10. doi: 10.1016/S0168-3659(01)00358-3. [DOI] [PubMed] [Google Scholar]
- 10.Yang B, Rohloff C, Mercer R, Horwege K, Negulescu C, Lautenbach S, et al. Continuous Delivery of Stabilized Proteins and Peptides at Consistent Rates for at Least 3 Months from the DUROS® Device. Am Assoc Pharm Sci Annual Meeting. 2008; T3150.
- 11.Fowler JE, Flanagan M, Gleason DM, Klimberg IW, Gottesman JE, Sharifi R, et al. Evaluation of an implant that delivers leuprolide for 1 year for the palliative treatment of prostate cancer. Urology. 2000;55:639–42. doi: 10.1016/S0090-4295(00)00479-9. [DOI] [PubMed] [Google Scholar]
- 12.Fowler JE, Gottesman JE, Reid CF, Andriole GL, Soloway MS, et al. Safety and efficacy of an implantable leuprolide delivery system in patients with advanced prostate cancer. J Urol. 2000;164:730–4. doi: 10.1016/S0022-5347(05)67291-6. [DOI] [PubMed] [Google Scholar]