

Abstract
Dried blood spots are increasingly being used in drug development. This commentary considers the pharmacokinetic issues that arise and compares these with those attached to plasma, the mainstay matrix. A common implicit use of these matrices is as a surrogate for plasma water, and to this extent, the critical assumption made is constancy in fraction unbound for plasma and, additionally for blood, constancy of hematocrit and blood cell affinity of compound. Often, these assumptions are reasonable and either matrix suffices, but not always. Then the value of one over the other matrix depends on the magnitude of the blood-to-plasma concentration ratio of drug, its clearance, and the cause of the deviation from constancy. Additional considerations are the kinetics of distribution within blood and those arising when the objective is assessment or comparison of bioavailability. Most of these issues can be explored and addressed in vitro prior to the main drug development program.
Key words: dried blood spots, drug development, pharmacokinetics
Plasma has been the mainstay matrix for measurement of systemic concentration of compounds used in the assessment and evaluation of pharmacokinetics (PK) and pharmacodynamics (PD), both efficacy and safety, in drug discovery and development. An alternative, dried blood spots (DBS), has recently gained increasing popularity, with some distinct practical advantages but also technical issues (1–5), although the method was employed in pediatrics as early as 1963 (6). Solutions to many of these analytical and methodological issues have been addressed (7–14). However, there does not appear to have been any associated commentary on the PK and related issues that DBS raise. This communication is intended to help those thinking of using DBS as an approach to measure systemic drug concentration. Most of the comments equally apply to metabolites.
The issues raised by DBS are, in principle, the same as those raised by the use of whole blood in that estimates of concentration from DBS should be the same as those in the original blood samples from which they are prepared. Historically, the main arguments in favor of plasma over blood have been the greater ease of storage and chemical analysis and the homogeneity of plasma compared to clotted blood. With respect to pharmacokinetics, the main argument in favor of whole blood has principally been based on physiological considerations. Namely, unlike plasma clearance, there is an upper bound to organ blood clearance; it is organ blood flow (with the extraction ratio approaching the upper limit of 1). Organ blood clearance then becomes perfusion rate-limited and more sensitive to changes in blood flow than to other processes, such as enzymatic activity and plasma protein binding (15). Also, in the situation that blood clearance exceeds hepatic blood flow, it provides a clue as to the significant involvement of additional organs or tissues in the elimination of a compound. In addition, knowing the hepatic extraction ratio (given by the ratio of hepatic blood clearance to hepatic blood flow) allows an estimate of the maximum oral bioavailability of a compound to be made (16). However, because plasma rather than blood is commonly measured, to make the above conclusions, it becomes necessary to convert plasma clearance to blood clearance using the blood-to-plasma concentration ratio. In recent years, it has become recognized that the blood-to-plasma ratio, coupled with plasma protein binding, also helps in the prediction of tissue distribution of moderately strong bases (17).
Essentially all events occurring within the body, whether related to PK or PD, are driven by unbound concentration. Hence, it may be argued that we should be directly measuring unbound rather than total concentration. However, there are many technical issues surrounding the determination of unbound concentration that have severely limited this measurement in practice. Microdialysis offers a potential way forward, although this technique has its own technical issues that are likely to limit its wide or routine use. So, total measurement is here to stay, at least for the foreseeable future. The fundamental question is: Under what circumstances is total drug measurement in whole blood (read DBS) or plasma an adequate surrogate for unbound compound? The corollary question is: When blood or plasma ceases to be an appropriate surrogate, what steps need to be taken to make the necessary correction?
To explore these issues, it is helpful to understand the relationships between unbound, total plasma, and total whole blood concentrations, displayed diagrammatically in Fig. 1. Here, it is seen that binding can be to constituents in both plasma and on or in blood cells, while the blood cell membrane can act as a barrier either slowing or totally impeding movement of compound into blood cells. At equilibrium, assuming no degradation in blood, the relationships are:
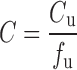 |
1 |
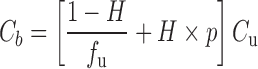 |
2 |
where Cu, C, and Cb are the unbound, total plasma, and total blood concentration, respectively, fu is the fraction unbound in plasma, H is the hematocrit, and ρ is the blood cell-to-unbound plasma concentration ratio, a measure of the affinity that the blood cells have for the compound. In pharmacokinetics, the ratio Cu/Cb is commonly denoted by fub, although clearly it is not the fraction of unbound drug in blood as no assumption is made in Eq. 2 as to whether drug enters blood cells to distribute into the intracellular aqueous space. From these relationships, it is seen that plasma concentration is proportional to unbound concentration when fu is constant and for blood concentration to be proportional to unbound concentration, in addition to fu, H and ρ need to be constant. When, as often is the case, these conditions hold reasonably well, then either plasma or blood concentration can equally be used to reflect unbound concentration. The problem arises when one or more of these conditions are not met.
Fig. 1.

Schematic of events occurring within blood. The extent and kinetics of distribution into cells depends upon the permeability of the cell membrane and the affinity of compound for constituents within plasma and blood cell
Consider first plasma. Some drugs, such as caffeine and aminoglycoside antibiotics, do not bind to plasma proteins (fu = 1); for such drugs, C = Cu, and arguably, plasma is the matrix to measure (although as argued subsequently, blood is as useful as plasma for these drugs). However, the majority of drugs do bind to plasma proteins. Then possible changes in fu need to be kept in mind. The value of fu can increase due to saturation of the available binding sites (when the molar concentration of the compound, or a displacer, in plasma approaches the molar concentration of the binding sites). It may also vary appreciably with age, disease, such as renal and hepatic disease, and in a variety of other conditions, such as pregnancy and excessive burns (18). Under these circumstances, to provide the correct interpretation of the underlying events one should measure either unbound drug concentration directly or determine fu (or a ratio of fu values, control and altered condition). In addition, binding often differs between animal species and man, and here again, when undertaking safety assessment based on relative unbound exposure across species, correction for such differences in binding should be made.
Now consider blood where the additional parameters H and ρ are potential issues. Normally, hematocrit is relatively constant and not of concern, although it is lower in some conditions such as anemia, where the hematocrit can fall from its usual value of 0.45 to as low as 0.2, which needs to be taken into account in the interpretation of blood measurements. Some compounds, particularly hydrophilic ones, such as many antibiotics, are too polar and large to enter blood cells, and for these compounds, blood cells act simply to dilute plasma, with Cb given by
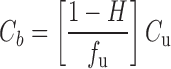 |
3 |
from which it is seen that the situation is essentially the same as for plasma. Frequently, such compounds, e.g., aminoglycosides, also do not bind to plasma proteins (fu = 1), or do so poorly, so that Cb approximates to (1 − H)⋅Cu, in which case blood provides a reasonable measure of unbound drug. Some other drugs, such as caffeine and ethanol, freely enter blood cells but do not bind to cell constituents or plasma proteins. For these drugs, Cb is approximately equal to Cu, and again, there is no danger of misinterpreting blood values. However, these are exceptions. Most drugs not only enter blood cells but also bind there such that ρ can be a critical issue.
The major blood cells of interest are erythrocytes; drugs may also have a high affinity for white cells and platelets, but the fractional volumes of these are so low as in general not to materially affect the global value of ρ. Drugs vary in their affinity to constituents on or in erythrocytes. Acidic drugs tend to have little affinity for erythrocytes, whereas moderately strong bases (pKa > 7), predominantly ionized at physiological pH, bind reasonably avidly to acidic phospholipids there through ion pairing (19). Apart from hemoglobin, one of the most abundant proteins in erythrocytes is carbonic anhydrase to which some sulfonamide-based inhibitors, such as acetozolamide and chlorthalidone, avidly bind. Cyclosporine and tacrolimus have a very high affinity for intracellular cyclophillin. In these examples, cellular binding is much greater than plasma protein binding (ρ >> 1/fu), so that Eq. 2 reduces to
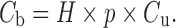 |
4 |
Now blood concentration is directly proportional to unbound concentration and insensitive to changes in plasma protein binding, and to this extent, blood is potentially a superior matrix to plasma. However, blood concentration is sensitive to ρ, which can and does change, for example, due to saturation of binding over the range of therapeutic concentrations, at least for cyclosporine (20). Under these circumstances, caution should be exercised when interpreting blood data. These drugs also exhibit high blood-to-plasma concentration ratios. This is evident by dividing Eq. 2 by Eq. 1, yielding
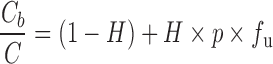 |
5 |
and setting ρ >> 1/fu. For example, for tacrolimus, Cb/C = 10–20, and fu = 0.07 (21,22) from which it is seen that when H = 0.45, ρ = 300–617. One issue with attempting to use plasma as the analytical matrix for PK and PD studies with such drugs is that any hemolysis introduces significant errors in the measurement. Continuing with tacrolimus, calculation shows that if a tolerance of 10% is the allowed limit in the increase in plasma concentration due to hemolysis, then this is met when only 0.5% hemolysis occurs. This is one of the reasons why blood, rather than plasma, has been used during drug development, for example, of tacrolimus and cyclosporine and in their subsequent therapeutic drug monitoring (23,24). Another reason for using blood is that erythrocyte binding of such compounds is temperature-dependent (18) so that if one wished to obtain an estimate of the plasma concentration that existed in vivo at the time of collection, the blood sample would need to be kept at 37°C during the separation of plasma, which creates technical problems in routine clinical practice.
The blood-to-plasma concentration ratio offers a useful diagnostic as to whether plasma protein binding or blood cell affinity is likely to be the more important when considering DBS. Thus, examination of Eq. 5 shows that the latter predominates when Cb/C > 1.5, since then, for normal hematocrit (0.45), fu × ρ > 2, or ρ > 2/fu, from which, on substitution into Eq. 2, it is seen that H × ρ is the more influential term in the relationship between Cb and Cu (fub). In this example, it implies that fu can vary twice as much as ρ to have the same impact on Cb for a given Cu. Generally, fu varies by not more than a factor of 3, and often less, for drugs bound to albumin, but it can vary more for those bound predominantly to α1-acid glycoprotein (18). Unfortunately, at present, virtually nothing is known about the magnitude of inter- and intra-subject variability in ρ for drugs. Factors to consider are disease, age, genetics, co-medication, and gender, in addition to drug concentration and temperature. Some drugs may be substrates or inhibitors of one of the many transporters that reside in the erythrocytic membrane. Clearly, given that fub approximates to 1/(H × ρ) when Cb/C > 2, it is important to define and understand sources of variability in ρ for such compounds when applying DBS in drug development. One may be tempted in preliminary investigations to use the blood-to-plasma ratio as the measure of variability in ρ. However, for such compounds, the ratio approximates to H × fu × ρ, which tends to inflate the estimate of variability in ρ but could also disguise it, if for any reason fu and ρ vary inversely.
An additional consideration arises from the fact that often times, with the availability of highly sensitive assays, which allow for the need of only minute sample volumes, blood is collected from heel or finger pricks. However, such samples are initially contaminated with interstitial fluid that not only dilutes blood but also may have concentrations of compound that differ from that in plasma. This problem is overcome by discarding the initial fluid collected. Even so, capillary blood tends to reflect more arterial blood perfusing the lancing site than venous blood draining it, particularly when the heel or finger is immersed in warm water to stimulate blood flow. Furthermore, these sites are at locations different than those of the usual sites of venous sampling, with the potential for differences in drug concentration, given that the concentration in venous blood reflects events occurring on passage of blood through the associated tissue bed. Such differences are expected to occur particularly in the early moments following drug administration, before distribution equilibrium has been achieved. Even so, such differences are likely to be generally small and certainly should be essentially nonexistent once distribution equilibrium of drug has occurred.
As mentioned previously, distribution of drug between plasma and blood cells takes time. Usually, this occurs very rapidly and is of little practical concern, but occasionally it may be so. The higher the blood cell affinity and plasma protein binding and the lower cell membrane permeability, the longer it takes for equilibrium to be achieved. Certainly, it takes several minutes for distribution equilibrium to occur for cyclosporine and related compounds (25), whereas it takes much longer for chlorthalidone, with the blood cell concentration peaking several hours after plasma following drug administration (26). Accordingly, when whole blood is the measurement matrix, such temporal delays may have an impact on the interpretation of PK and PD for the common situation in which response is better related to unbound drug in plasma than drug in blood cells. Much depends on which process, PK or PD, is the rate-limiting step over the period of interest. In the special case of carbonic anhydrase inhibitors, such as chlorthalidone, where the erythrocyte is one of the primary target sites, PD is expected to correlate better with blood than plasma measurements, especially during the period that distribution equilibrium between these sites has yet to be achieved.
Choice of matrix can influence the accuracy of estimation of bioavailability. Commonly, the estimate is taken to be the ratio of AUCs following extravascular and iv administrations (or after two extravascular administrations when the interest is in relative bioavailability), normalizing for any differences in dose. An implicit assumption in this method is constancy of clearance. When blood cell binding is saturated following either or both routes of administration, blood clearance may no longer be constant; much depends on the rate-limiting step in clearance. When blood clearance is high and perfusion rate-limited, saturation has little to no impact on clearance, and the ratio of blood AUCs provides an accurate measure of bioavailability. However, when clearance is low, plasma clearance is proportional to fu and blood clearance to fub, which is seen from Eq. 2 to depend on ρ. Accordingly, as ρ varies with concentration under saturating conditions, so will blood clearance. Under these circumstances, the ratio of blood AUCs no longer provides an accurate estimate of bioavailability. Whether plasma provides a more accurate estimate depends on whether or not plasma protein binding is also saturated. With cyclosporine, ρ but not fu exhibits concentration-dependent binding, and in this case, plasma is the better choice of matrix for assessment (27). How much error in estimation is incurred by using blood depends on the characteristic of the drug, which can be explored through simulation and set against the practicalities involved. A qualifier is needed here. One method of overcoming the problem of nonlinearity is to make the concentration–time profiles between treatments be as close to each other as possible, as then the nonlinearity in blood cell binding applies to both treatments equally at all times, and the AUC ratio method now applies. In practice, this is not easy to achieve as it implies prior knowledge of both the shape of the profile and bioavailability, so that the input profile of say an iv administration is appropriate, although the use of an iv radiolabeled tracer, which mixes with but does not disturb the pool, would suffice. A related issue is bioequivalence assessment. In such cases, as the test is for similarity of profiles, blood is arguably as suitable as plasma (27).
CONCLUSION
The preceding analysis should be put into perspective. For drugs showing little variability in fu and ρ under conditions of study, there is little concern in the use of DBS as an alternative to plasma. However, caution should be exercised when variability in either fu or ρ is large. Then it is important to identify, quantify, and, where possible, correct for, or accommodate, the variability, especially when interpreting PK data or linking PK to PD. For drugs with a blood-to-plasma ratio approaching the lower limit of 0.55, indicating minimal blood cell uptake, general concern is with variability in fu (about which much is known) when using DBS measurement as a surrogate for unbound drug in plasma. For drugs with larger values of the blood-to-plasma ratio, especially >2, variability in blood cell affinity becomes the critical factor. Fortunately, the magnitude and determinants of much of the variability can be evaluated in vitro using blood obtained from the study population of interest, which should be undertaken prior to implementing the use of DBS in a drug development program. So too should the study of the influence of drug concentration, temperature, and kinetics of blood cell distribution.
References
- 1.ter Heine R, Rosing H, van Gorp ECM, Mulder JW, Beijnen JH, Huitema ADR. Quantification of etravirine (TMC125) in plasma, dried blood spots and peripheral blood mononuclear cell lysate by liquid chromatography tandem mass spectrometry. J Pharm Biomed Anal. 2009;49:393–400. doi: 10.1016/j.jpba.2008.10.040. [DOI] [PubMed] [Google Scholar]
- 2.Barfield M, Spooner N, Lad R, Parry S, Fowles S. Application of dried blood spots combined with HPLC-MS/MS for the quantification of acetaminophen in toxicokinetic studies. J Chromatograph B. 2008;870:32–7. doi: 10.1016/j.jchromb.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 3.Li W, Tse FLS. Dried blood spot sampling in combination with LC-MS/MS for quantitative analysis of small molecules. Biomed Chromatog. 2010;24:49–65. doi: 10.1002/bmc.1367. [DOI] [PubMed] [Google Scholar]
- 4.van der Heijden J, de Beer Y, Hoogtanders K, Christiaans M, de Jong GJ, Neef C, et al. Therapeutic drug monitoring of everolimus using the dried blood spot methd in combination with liquid chromatography–mass spectrometry. J Pharm Biomed Anal. 2009;50:664–70. doi: 10.1016/j.jpba.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 5.Liang X, Li Y, Barfield M, Ji QC. Study of dried blood spots techniques for the determination of dextromethorphan and its metabolite dextrorphan in human whole blood by LC-MS/MS. J Chromatograph B. 2009;877:799–806. doi: 10.1016/j.jchromb.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 6.Guthrue R, Suzi A. A simple phenylalanine method for detecting phenylketouria in large populations of newborn infants. Pediatrics. 1963;23:338–43. [PubMed] [Google Scholar]
- 7.Spooner N, Lad R, Barfield M. Dried blood spots as a sample collection technique for the determination of pharmacokinetics in clinical studies: considerations for the validation of a quantitative bioanalytical method. Anal Chem. 2009;81:1557–63. doi: 10.1021/ac8022839. [DOI] [PubMed] [Google Scholar]
- 8.Beaudette P, Bateman KP. Discovery stage pharmacokinetics using dried blood spots. J Chromatograph B. 2004;809:153–8. doi: 10.1016/j.jchromb.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 9.Edelbroeck PM, van der Heijden J, Stolk LML. Dried blood spot methods in therapeutic drug monitoring: methods, assays and pitfalls. Ther Drug Monit. 2009;31:327–36. doi: 10.1097/FTD.0b013e31819e91ce. [DOI] [PubMed] [Google Scholar]
- 10.Holub M, Tuschl K, Ratschmann R, Strnadová KA, Muhl A, Heinze G, et al. Influence of hematocrit and localization of punch in dried blood spots on levels of amino acids and acylcarnitines measured by tandem mass spectrometry. Clin Chim Acta. 2006;373:27–31. doi: 10.1016/j.cca.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 11.Mei JV, Alexander JR, Adam BW, Hannon WH. Use of filter paper for the collection and analysis of human whole blood specimens. J Nutrition. 2001;131:1631S–6. doi: 10.1093/jn/131.5.1631S. [DOI] [PubMed] [Google Scholar]
- 12.Damen CWN, Rosing H, Schellens JHM, Biejnen JH. Application of dried blood spots combined with high-performance liquid chromatography coupled with electrospray ionisation tandem mass spectrometry for simultaneous quantification of vincristine and actinomycin-D. Anal Bioanal Chem. 2009;394:1171–82. doi: 10.1007/s00216-009-2775-z. [DOI] [PubMed] [Google Scholar]
- 13.McDade TW, Williams S, Snodgrass JJ. What a drop can do: dried blood spots as a minimally invasive method for integrating biomarkers into population-based research. Demography. 2007;44:899–925. doi: 10.1353/dem.2007.0038. [DOI] [PubMed] [Google Scholar]
- 14.Déglon J, Thomas A, Cataldo A, Mangin P, Staub C. On-line desorption of dried blood spot: a novel approach for the direct LC/MS analysis of µ-whole blood samples. J Pharm Biomed Anal. 2009;49:1034–9. doi: 10.1016/j.jpba.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Rowland M, Tozer TN. Clinical pharmacokinetics: concepts and applications. 4. Baltimore: Lippincott, Williams & Wilkins; 2010. pp. 157–68. [Google Scholar]
- 16.Rowland M. Influence of route of administration on drug availability. J Pharm Sci. 1972;61:70–4. doi: 10.1002/jps.2600610111. [DOI] [PubMed] [Google Scholar]
- 17.Rodgers T, Rowland M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm Res. 2007;24:918–33. doi: 10.1007/s11095-006-9210-3. [DOI] [PubMed] [Google Scholar]
- 18.Tozer TN. Implications of altered plasma protein binding in disease states. In: Benet LZ, Massoud N, Gambertoglio JG, editors. Pharmacokinetic basis of drug treatment. New York: Raven Press; 1983. pp. 173–93. [Google Scholar]
- 19.Rodgers T, Leahy D, Rowland M. Physiologically-based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J Pharm Sci. 2005;94:1259–76. doi: 10.1002/jps.20322. [DOI] [PubMed] [Google Scholar]
- 20.Kawai R, Matthew D, Tanaka C, Rowland M. Physiologically-based pharmacokinetics of cyclosporine A: extension to tissue distribution kinetics in rats and scale-up to human. J Pharmacol Expt Therap. 1998;287:457–68. [PubMed] [Google Scholar]
- 21.Piekoszewski W, Jusko J. Plasma protein binding of tacrolimus in humans. J Pharm Sci. 1992;82:340–1. doi: 10.1002/jps.2600820325. [DOI] [PubMed] [Google Scholar]
- 22.Akbas SH, Ozdem S, Caglar S, Tuncer M, Gurkan A, Yucetin L, et al. Effects of some hematological parameters on whole blood tacrolimus concentration measured by two immunoassay-based analytical methods. Clin Biochem. 2005;38:552–7. doi: 10.1016/j.clinbiochem.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 23.Hoogtanders K, van der Heijden J, Christiaans M, Edelbroeck P, van Hoof JP, Stolk LML. Therapeutic drug monitoring of tacrolimus with the dried blood spot method. J Pharm Biomed Anal. 2007;44:658–64. doi: 10.1016/j.jpba.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 24.Mee AV, Wong PY, Sun C, Oei L, Elliott S, Naik N, et al. Monitoring of cyclosporine concentrations by using dry blood-spot samples. J Clin Lab Analysis. 1991;5:74–7. doi: 10.1002/jcla.1860050114. [DOI] [PubMed] [Google Scholar]
- 25.Kawai R, Lemaire M, Steimer J-L, Bruelisauer A, Niederberger W, Rowland M. Physiologically-based pharmacokinetic study on a cyclosporin derivative, SDZ IMM 125. J Pharmacokin Pharmacodyn. 1994;22:327–65. doi: 10.1007/BF02353860. [DOI] [PubMed] [Google Scholar]
- 26.Fleuren HLJ, van Rossum M. Nonlinear relationship between plasma and red blood cell pharmacokinetics of chlorthalidone in man. J Pharmacokin Pharmacodyn. 1977;5:359–75. doi: 10.1007/BF01061696. [DOI] [PubMed] [Google Scholar]
- 27.Legg B, Rowland M. Saturable binding of cyclosporin A to erythrocytes: estimation of binding parameters in renal transplant patients and implications for bioavailability assessment. Pharm Res. 1988;5:80–5. doi: 10.1023/A:1015932032609. [DOI] [PubMed] [Google Scholar]