

Abstract
γ-Hydroxybutyric acid (GHB), a drug of abuse, exhibits saturable renal clearance and capacity-limited metabolism. The objectives of this study were to construct a mechanistic toxicokinetic (TK) model describing saturable renal reabsorption and capacity-limited metabolism of GHB and to predict the effects of inhibition of renal reabsorption on GHB TK in the plasma and urine. GHB was administered by iv bolus (200–1,000 mg/kg) to male Sprague-Dawley rats and plasma and urine samples were collected for up to 6 h post-dose. GHB concentrations were determined by LC/MS/MS. GHB plasma concentration and urinary excretion were well-described by a TK model incorporating plasma and kidney compartments, along with two tissue and two ultrafiltrate compartments. The estimate of the Michaelis-Menten constant for renal reabsorption (Km,R) was 0.46 mg/ml which is consistent with in vitro estimates of monocarboxylate transporter (MCT)-mediated uptake of GHB (0.48 mg/ml). Simulation studies assessing inhibition of renal reabsorption of GHB demonstrated increased time-averaged renal clearance and GHB plasma AUC, independent of the inhibition mechanism assessed. Co-administration of GHB (600 mg/kg iv) and l-lactate (330 mg/kg iv bolus plus 121 mg/kg/h iv infusion), a known inhibitor of MCTs, resulted in a significant decrease in GHB plasma AUC and an increase in time-averaged renal clearance, consistent with the model simulations. These results suggest that inhibition of renal reabsorption of GHB is a viable therapeutic strategy for the treatment of GHB overdoses. Furthermore, the mechanistic TK model provides a useful in silico tool for the evaluation of potential therapeutic strategies.
Key words: gamma-hydroxybutyrate, kidney reabsorption, pharmacokinetic model, renal clearance, toxicokinetics
INTRODUCTION
γ-Hydroxybutyrate (GHB) is an endogenous four-carbon chain carboxylic acid, which is produced primarily from γ-aminobutyric acid (GABA) in mammalian brain (1). GHB can also be formed from γ-butyrolactone and 1,4-butanediol by hydrolysis or oxidation catalyzed by lactonases or alcohol and aldehyde dehydrogenases in the body (2). GHB is a proposed neurotransmitter with its own specific receptor identified in the brain (3). In addition, GHB interacts with the GABAB receptor via direct and/or indirect interactions (4,5). GHB's physiological and toxicological effects include sedation, euphoria, growth hormone stimulation, anticoagulation, inhibition of intestinal mobility, and respiratory depression (6–8). GHB may also influence brain metabolism by reducing energy substrate consumption and exert tissue-protective effects under anoxic conditions (9).
GHB has been used to treat alcohol and heroin dependence in Europe (10), sleep disorders in Europe (11), and is approved in the USA as Xyrem® for the treatment of patients with narcolepsy who experience episodes of cataplexy. However, GHB is best known for its illicit use and abuse. GHB is abused as a popular steroid alternative by body builders, as a recreational drug at nightclubs and rave parties, and as a means of drug-facilitated sexual assaults (6,12). In the USA, over 7,100 GHB overdose cases and 65 GHB related deaths from 1990–2000 have been reported (13). The availability of GHB has been highly restricted since its classification as a schedule I controlled drug in 2000; however, it is easily synthesized, with instructions for home production and kits containing the necessary material available through internet Web sites. Additionally, the GHB precursors, γ-butyrolactone and 1,4-butanediol are available as health supplements and industrial solvents and are used as a GHB substitute or in combination with GHB by abusers (2,14). Currently, GHB overdoses are treated mainly with supportive care, such as intubation and mechanical ventilation for respiratory depression, with no specific therapies available for clinical use (14,15).
The nonlinear toxicokinetics (TK) of GHB has been demonstrated in rats (16) and humans (17,18). The mechanisms underlying the nonlinear TK of GHB include capacity-limited metabolism (16,17), capacity-limited oral absorption (17,19), and saturable renal reabsorption (20). GHB is extensively reabsorbed in the proximal tubules of the kidney mediated by pH-dependent and sodium-dependent monocarboxylate transporters (MCTs and SMCTs; 20,21). Renal clearance of GHB increases with increasing dose due to saturation of active renal reabsorption. Following GHB overdoses, the metabolic clearance of GHB decreases due to saturation of enzymatic pathways; however, renal clearance can be further increased by inhibiting the renal reabsorption of GHB thereby increasing overall GHB elimination (20). l-lactate is an endogenous energy source, which is transported by MCTs across biological barriers (22). Our previous studies have demonstrated that l-lactate could significantly inhibit the uptake of GHB in membrane vesicles from rat kidney cortex in vitro (21) and that l-lactate also could reduce the renal reabsorption of GHB significantly in vivo (20). Inhibition of active renal reabsorption represents a novel therapeutic strategy for the treatment of GHB overdose. However, a mechanistic model describing the saturable renal reabsorption of GHB and its interaction with MCT inhibitors has not been developed. Such a model would provide insight into the influence of inhibition of renal reabsorption on plasma GHB concentrations and urinary excretion profiles and allow for the evaluation of multiple therapeutic strategies prior to their in vivo assessment.
The objectives of this study were (1) to construct and validate a mechanistic model for GHB toxicokinetics describing saturable renal reabsorption and capacity-limited metabolism, and (2) to predict the consequences of inhibition of transporter-mediated renal reabsorption on GHB toxicokinetics in plasma and urine.
METHODS
Chemicals and Reagents
Sodium GHB, l-lactate, and formic acid were purchased from Sigma-Aldrich (St. Louis, Missouri). Deuterated GHB (GHB-d6) was purchased from Cerrilliant (Round Rock, Texas). Ketamine and xylazine were obtained from Henry Schein (Melville, New York). Acetic acid and High Performance Liquid Chromtography (HPLC)-grade methanol, acetonitrile, and water were purchased from Honeywell Burdick and Jackson (Morristown, New Jersey).
Animals and Surgery
Male Sprague-Dawley rats (Harlan, Indianapolis, Indiana) weighing 280–320 g were used throughout the study. The animal housing room had controlled environmental conditions with temperature and relative humidity of approximately 20 ± 2°C and 40–70% and artificial lighting that alternated on a 12-h light/dark cycle. All animal care and experimental protocols were approved by the Institutional Animal Care and Use Committee at the University at Buffalo. The rats had cannulas implanted in the right jugular vein and the left femoral vein (interaction study only), as previously described (20), and were kept in individual cages for 2 to 3 days after surgery prior to the start of the experiments.
Experimental Design
GHB TK
GHB was dissolved in sterile water (200 mg/ml) followed by filtration with a 0.2-µm filter for sterility. Rats were randomly assigned to dose groups, and GHB (200, 400, 600 or 1,000 mg/kg) was administered by iv bolus injection into the jugular vein cannula (N = 7–10 per dose group). Rats were placed in metabolic cages for the duration of the study. Blood samples (200 µl) were collected at 0, 5, 10, 20, 30, 60, 90, 120, 180, 240, 300, and 360 min and transferred to heparinized 0.6-ml centrifuge tubes. Plasma was separated by centrifugation at 1,000g for 5 min at 4°C. Urine samples were collected between 0–60, 60–120, 120–240, and 240–360 min. All samples were stored at −80°C until analysis.
GHB and l-lactate interaction study
Rats were randomly assigned to receive GHB alone (600 mg/kg iv) or GHB plus lactate (600 mg/kg iv GHB; 330 mg/kg iv bolus plus 121 mg/kg/h iv infusion of l-lactate; N = 3 and 5, respectively). GHB was administered via the jugular vein cannula and l-lactate was administered via the femoral vein 5 min after GHB was given. Rats were housed in metabolic cages for the duration of the study. Blood samples (200 μl) were taken from the jugular vein cannula at 0, 3 (lactate group only), 5 (GHB alone), 10, 20, 30, 60, 90, 120, 180, 210, 240, and 300 min and were transferred to heparinized 0.6-ml tubes. Urine samples were collected as detailed in the toxicokinetics section. All samples were stored at −80°C until analysis. The time of onset and offset of sedative/hypnotic effect [loss and return to righting reflex (LRR and RRR)] was determined in all animals. The duration of sedative/hypnotic effect (sleep time) was determined as the difference between RRR and LRR.
Plasma and Urine Sample Preparation and LC/MS/MS Analysis
GHB was extracted from plasma using anion exchange solid phase extraction as previously described (23). Briefly, 5 µl of GHB-d6 (1 mg/ml) and 5 µl GHB stock solution (or double-distilled water for samples) were added to 50 µl plasma. Acetonitrile (0.4 ml) was added to precipitate plasma proteins followed by centrifugation at 10,000g for 20 min. An aliquot of 0.2 ml supernatant was aspirated and added to 0.8 ml double-distilled water. Bond Elut SAX cartridges (100 mg resin, 1 ml volume, Varian, Palo Alto, California) were preconditioned, washed, and samples and standards eluted as previously described (24). The eluent was evaporated to dryness under a stream of N2 gas and reconstituted with 1.25 ml of 0.1% formic acid in double-distilled water and 5% acetonitrile.
Urine samples were prepared using a previously described method with minor modifications (25). Briefly, 10 µl of GHB-d6 (200 µg/ml) and 10 µl GHB stock solution (standards) or double-distilled water (samples) were added to 50 µl urine. Double-distilled water (930 µl) was added followed by 1 ml of acetonitrile to precipitate proteins. Samples were centrifuged for 20 min at 10,000g, and the supernatant was collected for LC/MS/MS analysis.
GHB in plasma and urine was measured using a previously developed assay (24) with slight modifications. LC/MS/MS analysis was conducted on an Agilent 1100 series HPLC with an online degasser, binary pump, and autosampler (Agilent Technologies, Palo Alto, California) connected to a PE Sciex API 3000 triple–quadrupole tandem mass spectrometer with a turbo ion spray (Applied Biosystems, Foster City, California). Seven microliters of plasma or urine sample was injected onto an Xterra MS C18 column (250 × 2.1 mm id, 5-µm particle size; Waters Co., Milford, Massachussets). GHB was separated using gradient elution with a flow rate of 0.2 ml/min, 100 to 90% A over 5 min, 90 to 10% A from 5 to 7.5 min, and 10% to 100% A from 7.5 to 112 min. Mobile phase A was comprised of 0.1% formic acid in HPLC-grade water with 5% acetonitrile and mobile phase B contained 0.1% formic acid in acetonitrile with 5% HPLC-grade water. Compound specific mass spectrometer parameters and linear calibration ranges are detailed in Felmlee et al. (23).
Population TK Modeling
Nonlinear mixed effects modeling in NONMEM VI (level 1.1, NONMEM Project Group, Icon Development Solutions, Ellicott City, Maryland, USA) was used for population model development. All modeling was conducted using the first-order conditional estimation method and the ADVAN9 differential equation solver. The NONMEM objective function, standard diagnostic plots, individual fits, and visual predictive checks (VPCs) were used for model development. VPCs were generated by simulating plasma and urinary excretion profiles from 0 to 360 min for each dose for 1,000 rats based on the population TK model. The mean and 10th and 90th percentiles for each dose were determined for each time point in SAS version 4.1 (SAS Institute, Cary, North Carolina) and were plotted with the observed data to evaluate if the model well-described the data.
Structural, Parameter Variability, and Observational Models
A mechanistic TK model was developed to describe the saturable renal clearance of GHB. A schematic representation of the model is depicted in Fig. 1, and parameter descriptions are listed in Table I. The differential equations for the model in Fig. 1 are listed below (Eqs. 1–8) with the initial conditions of each compartment set to zero. Initial Michaelis-Menten parameter estimates for metabolic clearance and renal reabsorption were obtained from previous GHB infusion studies (20) and in vitro studies (21). Plasma concentration and urinary excretion data from all doses were modeled simultaneously.
Fig. 1.

Final structural model for population toxicokinetic analysis. Refer to Table I for parameter descriptions
Table I.
Pharmacokinetic Parameters Obtained from Fitting Data to a Mechanistic Toxicokinetic Model with Nonlinear Metabolism and Nonlinear Renal Elimination
| Parameter | Units | Description | Final estimate (CV% for between-subject variability) |
|---|---|---|---|
| V plasma | ml | Plasma volume | 10.5a |
| V tissue1 | ml | Volume of the first tissue compartment | 75.9 (16%) |
| V tissue2 | ml | Volume of the second tissue compartment | 26.8 |
| V kidney | ml | Kidney volume | 4.0a |
| V ulf1 | ml | Volume of the first ultrafiltrate compartment | 3.0a |
| V ulf2 | ml | Volume of the second ultrafiltrate compartment | 1.0a |
| CL D | ml/min | Distributional clearance (tissue 1) | 26.9 |
| CL D2 | ml/min | Distributional clearance (tissue 2) | 3.07 |
| Q R | ml/min | Renal blood flow | 12.5a |
| V max,m | mg/min | Maximum metabolic elimination rate | 0.581 |
| K m,m | mg/ml | Metabolic Michaelis-Menten constant | 0.054 |
| GFR | mg/min/kg | Glomerular filtration rate | 10 |
| V max,R | mg/min | Maximum renal reabsorption rate | 2.34 |
| K m,R | mg/ml | Renal reabsorption Michaelis-Menten constant | 0.46 |
| UF | ml/min | Urine flow | 0.1a (114%) |
| ε plasma | Proportional error for plasma | 2.5% | |
| ε urine | Proportional error for urine | 46% | |
| OBJ | Objective function | −578.4 |
aParameter was fixed to physiological value
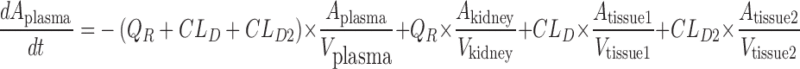 |
1 |
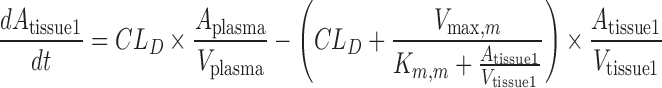 |
2 |
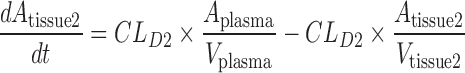 |
3 |
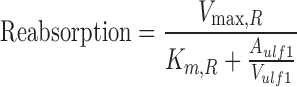 |
4 |
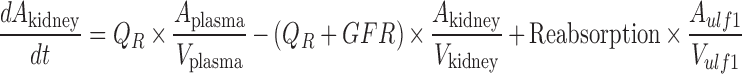 |
5 |
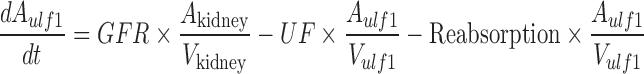 |
6 |
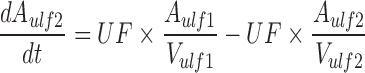 |
7 |
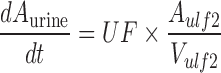 |
8 |
The parameter variability model that was applied was previously described for population analysis of GHB pharmacokinetics in rats (26). Briefly, an exponential model was used to describe between-subject variability, Pi = θ exp(ηi), where Pi and ηi are the individual parameter and the random effect describing between-subject variability for the ith rat, and θ is the population mean. The variance of ηi was reported as described in Fung et al. (26) and is presented in Table I.
The residual variability for both plasma concentrations and urinary excretion was described by a proportional log error model:
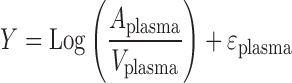 |
9 |
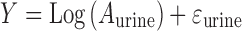 |
10 |
where Aplasma/Vplasma and Aurine are individual predictions with no random error and Y is the individual prediction incorporating proportional error.
Model Simulations
Simulations were conducted in NONMEM VI using the population TK model parameters to assess the impact of competitive, noncompetitive, and uncompetitive inhibition of renal reabsorption on plasma and urinary excretion profiles. GHB was administered by iv bolus (200, 400, 600, or 1,000 mg/kg) in the presence and absence of transport inhibitor. Equation 4 was modified to Eq. 11 (competitive), Eq. 12 (noncompetitive), and Eq. 13 (uncompetitive) to account for the presence of inhibitor, where R equals I/Ki and the inhibitor is administered at steady-state.
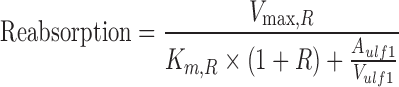 |
11 |
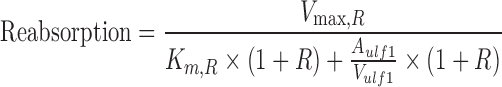 |
12 |
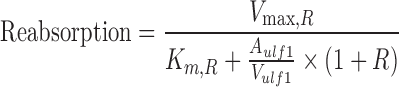 |
13 |
Statistical and Non-Compartmental Analyses
Plasma AUCs were calculated in WinNonLin version 5.2 (Pharsight Corp., Mountain View, California). Time-averaged renal clearance was calculated based on Eq. 14. Statistical analyses were conducted in GraphPad Prism version 4.0 (GraphPad Inc., San Diego, California). Significant differences between means were determined by a Student's t test, and P < 0.05 was considered statistically significant.
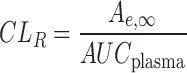 |
14 |
RESULTS
GHB Toxicokinetics
The plasma GHB concentrations and urinary GHB excretion following iv administration of GHB were well captured with the mechanistic model incorporating saturable renal reabsorption and capacity-limited metabolic clearance. The model is a hybrid physiological model with some parameters, such as plasma volume and urine flow, fixed to physiological values (Table I) (27–29). We found that a model incorporating two ultrafiltrate compartments was required to adequately capture the urinary excretion of GHB with active renal reabsorption from only the first ultrafiltrate compartment consistent with reabsorption from the proximal tubule. Two tissue distribution compartments (fast and slow distribution) were necessary to describe plasma GHB concentrations which is consistent with previous compartmental models used to describe GHB plasma concentrations (26). Protein binding was not incorporated into the model as GHB has negligible plasma protein binding.
Figure 2 illustrates the experimentally observed GHB plasma concentrations and urinary GHB excretion following administration of 200 (Fig 2a, b), 400 (Fig 2c, d), 600 (Fig 2e, f), or 1,000 mg/kg (Fig 2g, h) GHB doses. The solid lines represent the population fitting for the final structural model depicted in Fig. 1 with plasma and urine data from all doses fitted simultaneously. The 10th and 90th percentiles are depicted in Fig. 2 and were generated by simulating model output. Final TK parameter estimates, between-subject (residual) variability and random variability values are listed in Table I. The final estimate for the renal reabsorption Michaelis-Menten constant (0.46 mg/ml; Km,R) is in good agreement with experimentally determined Km (0.48 mg/ml) obtained from uptake studies in MCT1 expressing MDA-MB231 cells (21).
Fig. 2.

VPC plots following iv administration of GHB for the final population model detailed in Fig. 1 and Table I. Solid squares represent individual data points (N = 7–10 rats per dose). The plotted lines are based on 1,000 simulated plasma concentration-time (panels a, c, e, and g) and urinary excretion (b, d, f, and h) profiles for each dose. The mean (solid line) and 10th (dotted line) and 90th (dashed line) percentiles were calculated from the predicted concentrations. GHB doses: a, b 200 mg/kg; c, d 400 mg/kg; e, f 600 mg/kg; g, h 1,000 mg/kg
Simulation of Inhibition of Renal Reabsorption
We simulated the plasma concentration and urinary excretion profiles of GHB at various doses in the presence of a hypothesized transport inhibitor which inhibits the active renal reabsorption of GHB competitively (Fig. 3 and Table II), noncompetitively, or uncompetitively (data not shown). GHB was administered by iv bolus, and interactions were examined using a constant inhibitor concentration in the ultrafiltrate. We assumed that the hypothetical inhibitor would alter active renal reabsorption and not influence tissue distribution or metabolism of GHB. The plasma concentration of GHB decreased markedly at all the doses tested in the presence of a competitive inhibitor, at R values equal to 1, 10, or 100 (Fig. 3). The total amount of GHB excreted in the urine and the renal clearance of GHB increased greatly in the presence of inhibitor (Table II), and this effect was more evident at mid-range doses of GHB (400 and 600 mg/kg) than at high doses of GHB (1,000 mg/kg). The AUC decreased with increasing R, while the dose-averaged renal clearance increased, independent of the inhibition mechanism employed. This decrease in GHB plasma concentration and AUC correlates with the pharmacological effects of GHB, as demonstrated previously (23,25,30).
Fig. 3.

Simulations of GHB-transporter inhibitor interactions in rats. GHB was administered IV in the absence (solid line) or presence of a fixed concentration of inhibitor where R (I/K i) is equal to 1 (dashed line), 10 (dotted line), and 100 (dot/dash line). Simulations of plasma concentration-time and urinary excretion profiles were conducted based on competitive inhibition of renal reabsorption. GHB doses: a, b 200 mg/kg; c, d 400 mg/kg; e, f 600 mg/kg; g, h 1,000 mg/kg
Table II.
Changes in Plasma AUC (min mg/ml) and Time-averaged Renal Clearance (milliliter per minute) Based on Simulations of Competitive Inhibition of Renal Reabsorption Using the Mechanistic Toxicokinetic Model
| GHB+inhibitor | ||||||||
|---|---|---|---|---|---|---|---|---|
| GHB alone | R = 1 | R = 10 | R = 100 | |||||
| GHB dose (mg/kg) | AUC | CL R | AUC | CL R | AUC | CL R | AUC | CL R |
| 200 | 29.3 | 0.197 | 27.4 | 0.319 | 21.0 | 0.921 | 14.4 | 2.394 |
| 400 | 83.0 | 0.331 | 76.2 | 0.447 | 56.6 | 0.968 | 37.5 | 2.159 |
| 600 | 133.3 | 0.540 | 124.6 | 0.636 | 96.1 | 1.072 | 63.6 | 2.100 |
| 1,000 | 208.2 | 0.823 | 198.8 | 0.902 | 165.2 | 1.244 | 117.1 | 2.060 |
GHB Toxicokinetics in the Presence of l-lactate
To confirm the results of the simulation study, a competitive inhibitor of MCT-mediated GHB uptake, l-lactate, was co-administered with GHB (600 mg/kg iv bolus), and GHB plasma concentrations and urinary excretion were measured. l-lactate was administered 5 min post-GHB by iv infusion (121 mg/kg/h) with an iv bolus loading dose (330 mg/kg) to achieve steady-state. The TK of GHB was significantly altered by the co-administration of l-lactate. A significant decrease in plasma AUC (twofold) and increase in renal clearance (twofold) was observed (Table III). The results obtained with l-lactate are similar to the results obtained when R equals 10 (Table II); however, the plasma AUC value is lower than expected with changes in renal clearance alone suggesting alterations in tissue distribution. These TK alterations resulted in a significant decrease in the sedative/hypnotic effect of GHB as measured by sleep time (Table III).
Table III.
Changes in Plasma AUC and Time-averaged Renal Clearance (CL R) in Rats Following Administration of GHB Alone (600 mg/kg) or in Combination with l-lactate (330 mg/kg iv Bolus Followed by 121 mg/kg/h iv Infusion)
| AUC (mg*min/ml) | CLR (ml/min) | Sleep Time (min) | |
|---|---|---|---|
| GHB alone (N = 3) | 131 ± 18.8 | 0.415 ± 0.06 | 126 ± 15.1 |
| GHB + l-lactate (N = 5) | 74.8 ± 9.4* | 0.910 ± 0.10* | 87.0 ± 11.1* |
Results are presented as mean ± SD
AUC is the area under the plasma concentration versus time curve; CL R is renal clearance; sleep time is defined as the time difference between loss of righting reflex and return of righting reflex (RRR-LRR)
*P < 0.05 when compared using a Student's t test
DISCUSSION
Illicit use of GHB is prevalent due to its sedative/hypnotic and euphoric effects, and there is currently no available therapeutic strategy for the treatment of GHB overdose. GHB exhibits complex TK with capacity-limited metabolism (16,17) and intestinal absorption (17,19) and saturable renal reabsorption (20). In the proximal tubules GHB transport is mediated by MCTs and SMCTs that become saturated with increasing GHB doses leading to increased urinary excretion (20). Previous studies have demonstrated the relationship between GHB concentrations in plasma and brain and sedative/hypnotic effect (23), suggesting that an understanding of GHB TK will aid in the development of an effective therapeutic strategy to reduce GHB concentrations in brain and plasma by increasing GHB clearance in overdose situations. The present study represents the first mechanistic TK model for GHB that incorporates saturable renal reabsorption and capacity-limited metabolic clearance through the simultaneous fitting of plasma concentration and urinary excretion data. The final mechanistic model was used to evaluate the inhibition of active renal reabsorption by a hypothetical inhibitor via multiple mechanisms as a novel therapeutic strategy for the treatment of GHB overdose. We demonstrated in simulation studies that time-averaged renal clearance increases and plasma AUC decreases when active renal reabsorption is inhibited, independent of the mechanism of inhibition. In rats, co-administration of GHB and l-lactate, a competitive MCT inhibitor, resulted in decreased GHB plasma AUC and increased time-averaged renal clearance consistent with the simulation results.
Previous models describing renal reabsorption have focused on passive reabsorption and solvent drag (31–33) with only three models, to the best of our knowledge, considering active renal reabsorption (34–36). A model incorporating reabsorption via passive diffusion is not appropriate for GHB as it is fully ionized in urine (pKa ∼4) and therefore cannot diffuse passively through the lipoidal membrane of the proximal renal tubule. GHB may be reabsorbed via solvent drag through aqueous channels in its ionized form; however, this was not included in the present model as it is not an identifiable parameter in the present model. Jusko and Levy (1970; 34) developed a pharmacokinetic model for the active renal reabsorption of riboflavin and simultaneously fit riboflavin plasma concentrations and urinary excretion. The riboflavin model, with minor modifications to account for nonlinear metabolism, was unable to describe GHB plasma concentrations and urinary excretion likely due to the substantially higher extent of active renal reabsorption of GHB. Based on an ascorbic acid renal reabsorption model (35), it appears that additional renal compartments are required to describe the urinary excretion of compounds that are extensively reabsorbed in the proximal tubule. The two ultrafiltrate compartments in the model represent the proximal and distal tubules of the kidney with active reabsorption occurring in only the proximal compartment. The inclusion of a compartment representing the distal tubules accounts for the time delay between filtration at the glomerulus and appearance of drug in the urine due to urine flow. We extended the ascorbic acid model to incorporate volumes in the ultrafiltrate compartments and connected these compartments by urine flow to improve the physiological relevance of the model. We evaluated the inclusion of multiple compartments to adequately describe GHB urinary excretion. Due to the complexity of GHB TK and the need to estimate multiple nonlinear parameters, we evaluated hybrid physiological models which allowed us to fix a number of parameters to physiological values.
In the current study, a population TK modeling approach was used to develop a mechanistic hybrid physiological model to simultaneously fit GHB plasma concentration and urinary excretion. The population TK model was able to identify saturable renal reabsorption and capacity-limited metabolic clearance consistent with previous studies conducted at steady-state concentrations of GHB (20,25,30). The present study is the first time both sets of nonlinear parameters (renal reabsorption and metabolism) have been estimated simultaneously. VPCs showed that the population model well-described the central tendencies and variability of the observed plasma concentrations over the entire dose range (Fig. 2). Cumulative GHB urinary excretion was well-described (central tendency and variability) for most doses; however, a slight mis-prediction of the central tendency was observed at the 1,000 mg/kg dose. This may be due to the presence of a low affinity transporter not accounted for in the population model or the method of urine collection (metabolic cage versus bladder cannula).
Previous studies investigating GHB TK have employed one- or two-compartment models and typically did not use a population PK modeling approach. Fung et al. (26) used a population PK approach to described GHB PK in rats and found that a two-compartment model with capacity-limited metabolic clearance best described GHB plasma concentrations. We found that two tissue compartments fit the plasma GHB concentrations better than a single compartment likely due to the inclusion of sampling points in the initial rapid distribution phase. Our estimate for the metabolic Michaelis-Menten constant (0.054 mg/ml; Km,m) is within the range of estimates reported in the literature (0.0095–0.338 mg/ml; 16,20,26,37) and similar to the Km,m value (0.063 mg/ml) obtained from steady-state infusion studies (20). The Km,m value in the present study is lower than those estimated previously from iv bolus data (0.075 and 0.338 mg/ml; 16,37); however, the previous models only included a single capacity-limited elimination term which may not provide a true estimation of the metabolic Michaelis-Menten constant. Both iv bolus studies used GHB doses similar to those administered in the present study and exhibited nonlinear renal clearance resulting from saturation of renal reabsorption. The metabolic intrinsic clearance (Vmax,m/Km,m) was estimated to be 0.011 L/min which is consistent with previous literature reports (0.012–0.046 L/min) (16,26,37). Although our estimate is slightly lower than literature reports, this may be due to the simultaneous estimation of capacity-limited metabolism and saturable renal reabsorption parameters over a wider dose range.
Active renal reabsorption of GHB is mediated by multiple MCT isoforms in the proximal tubule of the kidney in rats and humans (20,38). In vitro studies conducted in rat kidney membrane vesicles (21), HK-2 (human kidney) cells (38,39), and rat MCT1-transfected MDA-MB231 cells (21,40) have demonstrated the involvement of MCT1, MCT2, and MCT4 in the renal reabsorption of GHB. In addition, GHB reabsorption is mediated by SMCTs in the renal proximal tubule (41). Based on the population TK model, the affinity of GHB for renal reabsorption (Km,R) was estimated at 0.46 mg/ml which is close to the range of in vitro affinities determined for MCT isoforms (0.48–1.9 mg/ml; 21,42). In vitro studies assessing SMCT-mediated GHB transport have not been conducted in renal cells lines; however, results in transfected oocytes (41) and rat FRTL cells (43) show that SMCT1 has a greater affinity for GHB (0.17 and 0.07 mg/ml, respectively) than MCT isoforms. It may not be possible to distinguish between these two separate transport processes in vivo as the affinities are within fivefold of one another. To differentiate between MCT isoforms and SMCTs would require specific and selective inhibitors for individual transporters which are currently unavailable.
Inhibition of the MCT-mediated renal reabsorption represents a novel therapeutic strategy for the treatment of GHB overdoses. Previous studies have demonstrated the relationship between GHB exposure and plasma concentrations and the sedative/hypnotic effect of GHB (23,30) indicating that therapeutic strategies designed to reduce plasma GHB concentrations by increasing clearance would lead to reduced sedative/hypnotic effect. Simulations carried out using the developed population TK model demonstrated that inhibition of active renal reabsorption by multiple inhibition mechanisms markedly altered GHB TK with increased time-averaged renal clearance and decreased plasma AUC. We further demonstrated that the co-administration of GHB and l-lactate resulted in similar parameter changes to those obtained in the simulation studies for competitive inhibition and is consistent with previous studies conducted assessing the impact of l-lactate on GHB TK (20,25). The simulations conducted using the population TK model assume that co-administration of a hypothetical MCT inhibitor does not alter the distribution of GHB which may influence the predicted alterations in plasma GHB concentrations and urinary excretion. Further studies should be conducted to assess the impact of MCT inhibitors on the tissue distribution of GHB specifically focusing on alterations of GHB brain distribution. Additionally, studies investigating the impact and timing of l-lactate administration following oral dosing of GHB should be conducted to more accurately mimic the clinical situation.
The use of GABAB receptor antagonists has been proposed as a therapeutic strategy for treating GHB overdose (44). Administration of GABAB receptor antagonists SCH 50911 and CGP 46381 prior to GHB reduced the sedative/hypnotic effect of GHB (45); however, studies did not evaluate whether the antagonists were effective when administered after GHB, thereby replicating the therapeutic situation. The use of l-lactate to inhibit MCT-mediated GHB reabsorption would likely be easier to translate to the clinic as l-lactate preparations are already approved for use in humans. In addition, l-lactate and a GABAB receptor antagonist could be used in combination as they work through different targets, and their interaction may be synergistic. The potential for side effects may also be greater with the use of GABAB antagonists as they may alter endogenous GABA function. All potential strategies should be evaluated to find the optimal detoxification regime(s).
The mechanistic nature of the developed TK model will allow for the evaluation of multiple therapeutic strategies, including altered metabolism, and the impact on overall GHB time-course in plasma and urine. Currently, the model is limited to predictions of alterations in GHB TK, specifically plasma GHB concentrations and urinary excretion. While plasma GHB concentrations correlate with the sedative/hypnotic effect of GHB, the brain represents the site of action. GHB concentrations in the brain extracellular fluid, as measured by microdialysis, correlate with its sedative/hypnotic effect (23, 24), and the TK model presented here will be expanded to incorporate the time-course of GHB in the brain. Additional information regarding the tissue distribution of GHB could be utilized to develop a whole-body physiological model incorporating the mechanistic model of renal reabsorption developed in the current study which would allow for the impact of MCT inhibitors on tissue distribution to be assessed. Furthermore, the current model could be expanded to incorporate toxicodynamic endpoints such as respiratory depression which would permit the simultaneous prediction of TK/TD consequences during the evaluation of multiple therapeutic strategies.
CONCLUSIONS
The toxicokinetics of GHB were well-described by a mechanistic model incorporating active renal reabsorption and capacity-limited metabolic clearance. The affinity of GHB for renal reabsorption (0.46 mg/ml) was similar to in vitro estimates for MCT-mediated uptake in cell culture studies (0.48–1.9 mg/ml; 21, 42). Inhibition of renal reabsorption leads to decreased plasma GHB AUC and increased time-averaged renal clearance independent of inhibition mechanism. These results support the development of a therapeutic strategy aimed at inhibiting the renal reabsorption of GHB to decrease plasma GHB concentrations, thereby reducing the sedative/hypnotic effect of GHB. The mechanistic model of GHB TK will provide a useful tool for the in silico evaluation of novel therapeutic strategies.
Acknowledgements
This work was supported by the National Institute of Health, National Institute of Drug Abuse [Grant DA 023223]. MAF received a Graduate Fellowship from Pfizer Global Research and Development.
Abbreviations
- GABA
γ-aminobutyric acid
- GFR
glomerular filtration rate
- GHB
γ-hydroxybutyrate
- LC/MS/MS
liquid chromatography-tandem mass spectrometry
- MCT
monocarboxylate transporter
- SMCT
sodium-dependent monocarboxylate transporter
- TK
toxicokinetics
References
- 1.Roth RH, Giarman NJ. Natural occurrence of gamma-hydroxybutyrate in mammalian brain. Biochem Pharmacol. 1970;19:1087–1093. doi: 10.1016/0006-2952(70)90370-9. [DOI] [Google Scholar]
- 2.Snead OC, 3rd, Gibson KM. Gamma-hydroxybutyric acid. N Engl J Med. 2005;352(26):2721–2732. doi: 10.1056/NEJMra044047. [DOI] [PubMed] [Google Scholar]
- 3.Maitre M. The gamma-hydroxybutyrate signalling system in brain: organization and functional implications. Prog Neurobiol. 1997;51(3):337–361. doi: 10.1016/S0301-0082(96)00064-0. [DOI] [PubMed] [Google Scholar]
- 4.Hechler V, Ratomponirina C, Maitre M. Gamma-hydroxybutyrate conversion into GABA induces displacement of GABAB binding that is blocked by valproate and ethosuximide. J Pharmacol Exp Ther. 1997;281(2):753–760. [PubMed] [Google Scholar]
- 5.Mathivet P, Bernasconi R, De Barry J, Marescaux C, Bittiger H. Binding characteristics of gamma-hydroxybutyric acid as a weak but selective GABAB receptor agonist. Eur J Pharmacol. 1997;321(1):67–75. doi: 10.1016/S0014-2999(96)00916-8. [DOI] [PubMed] [Google Scholar]
- 6.Okun MS, Boothby LA, Bartfield RB, Doering PL. GHB: an important pharmacologic and clinical update. J Pharm Pharm Sci. 2001;4(2):167–175. [PubMed] [Google Scholar]
- 7.Carai MA, Agabio R, Lobina C, Reali R, Vacca G, Colombo G, et al. GABA(B)-receptor mediation of the inhibitory effect of gamma-hydroxybutyric acid on intestinal motility in mice. Life Sci. 2002;70(25):3059–3067. doi: 10.1016/S0024-3205(02)01553-9. [DOI] [PubMed] [Google Scholar]
- 8.Franconi F, Miceli M, Alberti L, Boatto G, Coinu R, De Montis MG, et al. Effect of gamma-hydroxybutyric acid on human platelet aggregation in vitro. Thromb Res. 2001;102(3):255–260. doi: 10.1016/S0049-3848(01)00249-3. [DOI] [PubMed] [Google Scholar]
- 9.Mamelak M. Gammahydroxybutyrate: an endogenous regulator of energy metabolism. Neurosci Biobehav Rev. 1989;13(4):187–198. doi: 10.1016/S0149-7634(89)80053-3. [DOI] [PubMed] [Google Scholar]
- 10.Gallimberti L, Spella MR, Soncini CA, Gessa GL. Gamma-hydroxybutyric acid in the treatment of alcohol and heroin dependence. Alcohol. 2000;20(3):257–262. doi: 10.1016/S0741-8329(99)00089-0. [DOI] [PubMed] [Google Scholar]
- 11.Mamelak M, Scharf MB, Woods M. Treatment of narcolepsy with gamma-hydroxybutyrate. A review of clinical and sleep laboratory findings. Sleep. 1986;9(1 Pt 2):285–289. doi: 10.1093/sleep/9.1.285. [DOI] [PubMed] [Google Scholar]
- 12.Schwartz RH, Milteer R, LeBeau MA. Drug-facilitated sexual assault (“date rape”) South Med J. 2000;93(6):558–561. [PubMed] [Google Scholar]
- 13.Shannon M, Quang LS. Gamma-hydroxybutyrate, gamma-butyrolactone, and 1,4-butanediol: a case report and review of the literature. Pediatr Emerg Care. 2000;16(6):435–440. doi: 10.1097/00006565-200012000-00017. [DOI] [PubMed] [Google Scholar]
- 14.Dietze PM, Cvetkovski S, Barratt MJ, Clemens S. Patterns and incidence of gamma-hydroxybutyrate (GHB)-related ambulance attendances in Melbourne, Victoria. Med J Aust. 2008;188(12):709–711. doi: 10.5694/j.1326-5377.2008.tb01851.x. [DOI] [PubMed] [Google Scholar]
- 15.Mason PE, Kerns WP., 2nd Gamma hydroxybutyric acid (GHB) intoxication. Acad Emerg Med. 2002;9(7):730–739. doi: 10.1111/j.1553-2712.2002.tb02154.x. [DOI] [PubMed] [Google Scholar]
- 16.Lettieri JT, Fung HL. Dose-dependent pharmacokinetics and hypnotic effects of sodium gamma-hydroxybutyrate in the rat. J Pharmacol Exp Ther. 1979;208(1):7–11. [PubMed] [Google Scholar]
- 17.Ferrara SD, Zotti S, Tedeschi L, Frison G, Castagna F, Gallimberti L, et al. Pharmacokinetics of gamma-hydroxybutyric acid in alcohol dependent patients after single and repeated oral doses. Br J Clin Pharmacol. 1992;34(3):231–235. doi: 10.1111/j.1365-2125.1992.tb04129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palatini P, Tedeschi L, Frison G, Padrini R, Zordan R, Orlando R, et al. Dose-dependent absorption and elimination of gamma-hydroxybutyric acid in healthy volunteers. Eur J Clin Pharmacol. 1993;45(4):353–356. doi: 10.1007/BF00265954. [DOI] [PubMed] [Google Scholar]
- 19.Arena C, Fung HL. Absorption of sodium gamma-hydroxybutyrate and its prodrug gamma-butyrolactone: relationship between in vitro transport and in vivo absorption. J Pharm Sci. 1980;69(3):356–358. doi: 10.1002/jps.2600690331. [DOI] [PubMed] [Google Scholar]
- 20.Morris ME, Hu K, Wang Q. Renal clearance of gamma-hydroxybutyric acid in rats: increasing renal elimination as a detoxification strategy. J Pharmacol Exp Ther. 2005;313(3):1194–1202. doi: 10.1124/jpet.105.083253. [DOI] [PubMed] [Google Scholar]
- 21.Wang Q, Darling IM, Morris ME. Transport of gamma-hydroxybutyrate in rat kidney membrane vesicles: role of monocarboxylate transporters. J Pharmacol Exp Ther. 2006;318(2):751–761. doi: 10.1124/jpet.106.105965. [DOI] [PubMed] [Google Scholar]
- 22.Halestrap AP, Price NT. The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem J. 1999;343(Pt 2):281–299. doi: 10.1042/0264-6021:3430281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Felmlee MA, Roiko SA, Morse BL, Morris ME (2010) Concentration-effect relationships for the drug of abuse γ-hydroxybutryic acid. J Pharmacol Exp Ther. doi: 10.1124/jpet.109.165381 [DOI] [PMC free article] [PubMed]
- 24.Raybon JJ, Boje KM. Pharmacokinetics and pharmacodynamics of gamma-hydroxybutyric acid during tolerance in rats: effects on extracellular dopamine. J Pharmacol Exp Ther. 2007;320(3):1252–1260. doi: 10.1124/jpet.106.113886. [DOI] [PubMed] [Google Scholar]
- 25.Wang Q, Wang X, Morris ME. Effects of l-lactate and d-mannitol on gamma-hydroxybutyrate toxicokinetics and toxicodynamics in rats. Drug Metab Dispos. 2008;36(11):2244–2251. doi: 10.1124/dmd.108.022996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fung HL, Tsou PS, Bulitta JB, Tran DC, Page NA, Soda D, et al. Pharmacokinetics of 1, 4-butanediol in rats: bioactivation to gamma-hydroxybutyric acid, interaction with ethanol, and oral bioavailability. AAPS J. 2008;10(1):56–69. doi: 10.1208/s12248-007-9006-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang SS, Fears R, Noirot S, Mbanya JN, Yudkin J. Changes in metabolism of rat kidney and liver caused by experimental diabetes and by dietary sucrose. Diabetologia. 1982;22(4):285–288. doi: 10.1007/BF00281307. [DOI] [PubMed] [Google Scholar]
- 28.Welch WJ, Deng X, Snellen H, Wilcox CS. Validation of miniature ultrasonic transit-time flow probes for measurement of renal blood flow in rats. Am J Physiol. 1995;268(1 Pt 2):F175–178. doi: 10.1152/ajprenal.1995.268.1.F175. [DOI] [PubMed] [Google Scholar]
- 29.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10(7):1093–1095. doi: 10.1023/A:1018943613122. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Wang Q, Morris ME. Pharmacokinetic interaction between the flavonoid luteolin and gamma-hydroxybutyrate in rats: potential involvement of monocarboxylate transporters. AAPS J. 2008;10(1):47–55. doi: 10.1208/s12248-007-9001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall S, Rowland M. Relationship between renal clearance, protein binding and urine flow for digitoxin, a compound of low clearance in the isolated perfused rat kidney. J Pharmacol Exp Ther. 1984;228(1):174–179. [PubMed] [Google Scholar]
- 32.Katayama K, Ohtani H, Kawabe T, Mizuno H, Endoh M, Kakemi M, et al. Kinetic studies on drug disposition in rabbits. I. Renal excretion of iodopyracet and sulfamethizole. J Pharmacobiodyn. 1990;13(2):97–107. doi: 10.1248/bpb1978.13.97. [DOI] [PubMed] [Google Scholar]
- 33.Komiya I. Urine flow dependence of renal clearance and interrelation of renal reabsorption and physicochemical properties of drugs. Drug Metab Dispos. 1986;14(2):239–245. [PubMed] [Google Scholar]
- 34.Jusko WJ, Levy G. Pharmacokinetic evidence for saturable renal tubular reabsorption of riboflavin. J Pharm Sci. 1970;59(6):765–772. doi: 10.1002/jps.2600590608. [DOI] [PubMed] [Google Scholar]
- 35.Graumlich JF, Ludden TM, Conry-Cantilena C, Cantilena LR, Jr, Wang Y, Levine M. Pharmacokinetic model of ascorbic acid in healthy male volunteers during depletion and repletion. Pharm Res. 1997;14(9):1133–1139. doi: 10.1023/A:1012186203165. [DOI] [PubMed] [Google Scholar]
- 36.Granero L, Gimeno MJ, Torres-Molina F, Chesa-Jimenez J, Peris JE. Studies on the renal excretion mechanisms of cefadroxil. Drug Metab Dispos. 1994;22(3):447–450. [PubMed] [Google Scholar]
- 37.Van Sassenbroeck DK, De Paepe P, Belpaire FM, Buylaert WA. Characterization of the pharmacokinetic and pharmacodynamic interaction between gamma-hydroxybutyrate and ethanol in the rat. Toxicol Sci. 2003;73(2):270–278. doi: 10.1093/toxsci/kfg079. [DOI] [PubMed] [Google Scholar]
- 38.Wang Q, Lu Y, Morris ME. Monocarboxylate transporter (MCT) mediates the transport of gamma-hydroxybutyrate in human kidney HK-2 cells. Pharm Res. 2007;24(6):1067–1078. doi: 10.1007/s11095-006-9228-6. [DOI] [PubMed] [Google Scholar]
- 39.Wang Q, Lu Y, Yuan M, Darling IM, Repasky EA, Morris ME. Characterization of monocarboxylate transport in human kidney HK-2 cells. Mol Pharm. 2006;3(6):675–685. doi: 10.1021/mp060037b. [DOI] [PubMed] [Google Scholar]
- 40.Wang Q, Morris ME. Flavonoids modulate monocarboxylate transporter-1-mediated transport of gamma-hydroxybutyrate in vitro and in vivo. Drug Metab Dispos. 2007;35(2):201–208. doi: 10.1124/dmd.106.012369. [DOI] [PubMed] [Google Scholar]
- 41.Ganapathy V, Thangaraju M, Gopal E, Martin PM, Itagaki S, Miyauchi S, et al. Sodium-coupled monocarboxylate transporters in normal tissues and in cancer. AAPS J. 2008;10(1):193–199. doi: 10.1208/s12248-008-9022-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Q, Morris ME. The role of monocarboxylate transporter 2 and 4 in the transport of gamma-hydroxybutyric acid in mammalian cells. Drug Metab Dispos. 2007;35(8):1393–1399. doi: 10.1124/dmd.107.014852. [DOI] [PubMed] [Google Scholar]
- 43.Cui D, Morris ME. The drug of abuse gamma-hydroxybutyrate is a substrate for sodium-coupled monocarboxylate transporter (SMCT) 1 (SLC5A8): characterization of SMCT-mediated uptake and inhibition. Drug Metab Dispos. 2009;37(7):1404–1410. doi: 10.1124/dmd.109.027169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jensen K, Mody I. GHB depresses fast excitatory and inhibitory synaptic transmission via GABA(B) receptors in mouse neocortical neurons. Cereb Cortex. 2001;11(5):424–429. doi: 10.1093/cercor/11.5.424. [DOI] [PubMed] [Google Scholar]
- 45.Carai MA, Colombo G, Brunetti G, Melis S, Serra S, Vacca G, et al. Role of GABA(B) receptors in the sedative/hypnotic effect of gamma-hydroxybutyric acid. Eur J Pharmacol. 2001;428(3):315–321. doi: 10.1016/S0014-2999(01)01334-6. [DOI] [PubMed] [Google Scholar]