

Abstract
Purpose of review
Infantile hemangioma is a common vascular tumor with a unique lifecycle: rapid growth in infancy, followed by a period of involution, leading to complete regression. This review summarizes recent studies of molecular mechanisms of hemangioma formation and places new findings and hypotheses in the context of past accomplishments.
Recent findings
The new work identifies a novel signaling pathway for vascular growth factor and extracellular matrix regulation in vascular endothelial cells and provides a basis for novel therapeutic strategies. In hemangioma-derived endothelial cells defects in a vascular endothelial growth factor receptor/integrin complex reduce the expression of a vascular endothelial growth factor decoy receptor. As a consequence, hemangioma endothelial cells exhibit constitutive vascular endothelial growth factor signaling. Germ-line mutations in components of the growth factor receptor/integrin complex in some hemangioma patients, and somatic mutations in a phosphatase in sporadic hemangioma specimens, raise the possibility that hemangioma formation involves a combination of germline risk factor mutations and somatic mutations, similar to what recent studies have shown is the case for venous malformations.
Summary
Alterations in pathways that negatively control vascular endothelial growth factor signaling in vascular endothelial cells are responsible for the formation and rapid growth of infantile hemangiomas.
Keywords: Vascular anomalies, VEGF, NFAT, endothelium, hypoxia, cell trafficking
Introduction
Infantile hemangiomas (IH), belonging to a group of disorders called vascular anomalies, are benign tumors of vascular endothelial cells [1,2]. They occur as (mostly) sporadic, solitary cutaneous lesions in up to 10% of Caucasian newborns with a lower incidence in Asian and African populations. They are more frequent in females than in males (3:1), in premature babies, and in cases where gestation is associated with placental abnormalities [3,4]. Hemangioma lesions appear in the early postnatal period (1–2 weeks) and proliferate rapidly during the next 6–10 months. This proliferating phase is followed by a longer (up to 7 years) involuting phase, during which endothelial cell apoptosis dominates over proliferation. A final involuted phase results in complete regression and replacement of the vascular tumor with a fibrous, adipocyte-rich tissue [5].
In the present review, we will focus on emerging insights into the pathogenetic mechanisms of this common vascular tumor. We will discuss how recent studies of these mechanisms have led not only to elucidation of novel signaling pathways in vascular endothelial cells, but also to identification of therapeutic targets and strategies for treatment in cases where rapidly growing hemangiomas become clinically problematic.
Vascular malformations and tumors
The localized lesions of blood vessels classified as vascular anomalies (VAs) can affect any organ system but are most frequently diagnosed in the skin. While they may not be easily distinguishable to the non-specialist physician, VAs can be divided into malformations (capillary, venous, arterial, lymphatic and combined) and tumors (infantile hemangiomas, congenital hemangiomas and hemangioendotheliomas). This classification is based on distinct clinical, pathological and molecular differences between vascular tumors and malformations. However, there are also common features, particularly at the level of molecular pathogenetic mechanisms, making it important to consider signaling defects in hemangiomas in the broader context of defects causing other VAs.
Hemangiomas vary considerably in location, size, and aggressivity of growth, suggesting that they are unlikely to be due to a single molecular defect, but rather to a series of defects, along a common pathway of angiogenesis, that result in a rather uniform phenotype. This idea is supported by genetic evidence suggesting the existence of multiple hemangioma-associated gene loci in rare cases of familial hemangiomas [6]. Interestingly, studies of such families also indicate that different types of lesions – hemangiomas and malformations – segregate together, suggesting an overlap in the pathogenetic mechanisms of vascular tumors and malformations [7]. The report of a somatic hemangioma-associated missense mutation in VEGFR3 [8], a protein known to be involved in lymphatic malformations [9], supports this idea.
VEGF receptor 2 signaling is upregulated in hemangioma
We have recently reported that a major mechanism underlying hemangioma formation involves abnormalities in a novel pathway of regulation of VEGF-A (referred to as VEGF below) signaling in endothelial cells [10]. In studying the molecular characteristics and behavior of endothelial cells isolated from proliferating hemangioma lesions (hemECs) of several patients, we observed that VEGF receptor 1 (VEGFR1) transcripts and protein levels are abnormally low in hemECs compared to several different control endothelial cells, including foreskin-derived microvascular endothelial cells (HDMECs), umbilical vein endothelial cells (HUVECs), facial skin-derived microvascular endothelial cells and cord blood-derived endothelial progenitor cells (EPCs). These low levels of expression are associated with VEGF-dependent activation of VEGF receptor 2 (VEGFR2) and activation and/or increased expression of downstream signaling targets. Activities of multiple kinases in lysates of hemECs cultured without exogenous VEGF are similar to those of HDMECs treated with VEGF, and levels of HIF-1α, VEGF and glucose transporter-1 (GLUT-1) proteins are as high in unstimulated hemECs as in VEGF-stimulated HDMECs. This constitutive activation of VEGFR2 signaling is reduced to control levels in cultured hemECs induced to overexpress either full-length wild-type VEGFR1 or a tyrosine kinase-“dead” mutant VEGFR1. Therefore, constitutive activation of VEGF-dependent VEGFR2 signaling in hemangioma-derived endothelial cells is a consequence of reduced VEGF-binding VEGFR1 “decoy” function.
Abnormalities in an NFAT-controlling protein complex in hemangioma
One would expect the high levels of VEGFR2 signaling in hemangioma endothelial cells to result in increased levels of calcineurin activity and thus activation/nuclear translocation of NFAT transcription factors [11]. Paradoxically, hemECs in culture as well as hemangioma tissue in vivo exhibit reduced expression of known NFAT-regulated genes, such as genes encoding COX-2 [12], the calcineurin antagonist DSCR1 (Down Syndrome Critical Target Region 1) [13] and monocyte chemotactic protein (MCP) −1 [14]. Further analyses, including a demonstration that the VEGFR1 promoter contains a functional NFAT-binding site, indicate that the VEGFR1 gene is also an NFAT transcriptional target and that the low level expression of this VEGF receptor in hemECs is a consequence of repressed NFAT activation [10]. The repressed NFAT activation in hemECs has been traced back to abnormalities in a protein complex that includes VEGFR2, TEM8 (tumor endothelial marker 8 [15]; Anthrax toxin receptor 1 [16]) and β1 integrin (Figure 1).
Figure 1. Low level expression of VEGFR1 and constitutive VEGFR2 signaling in hemangioma endothelial cells.
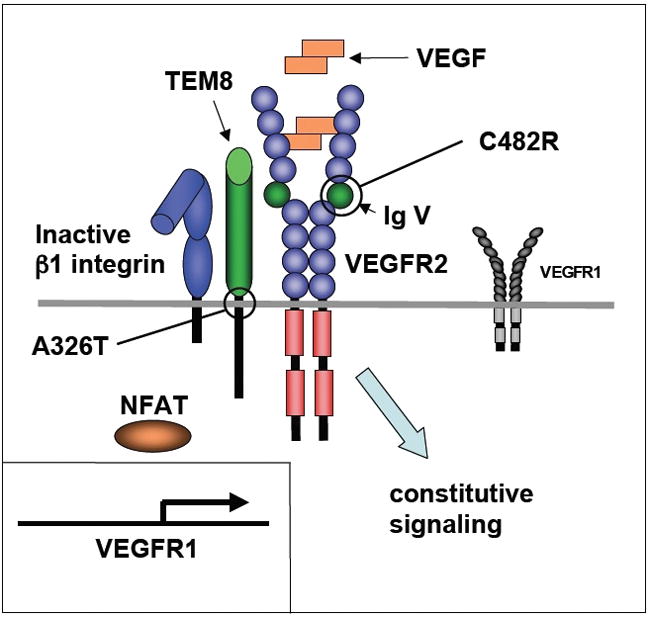
Defects in a complex of VEGFR2, TEM8 and β1 integrin in hemangioma endothelial cells keep β1 integrin in an inactive conformation and compromise the ability of the complex to stimulate activation/nuclear translocation of NFAT and transcription of the VEGFR1 gene. The consequences are low levels expression of VEGFR1 and VEGF-dependent constitutive activation of VEGFR2 signaling. In some hemangioma patients, the defects are caused by heterozygosity for a missense mutation (C482R) in immunoglobulin-like domain V of the VEGFR2 receptor or a missense mutation (A326T) in the transmembrane domain of TEM8 (also known as Anthrax toxin receptor 1).
Some hemangioma patients are heterozygous for germ-line missense mutations in the extracellular region of VEGFR2 or the transmembrane domain of TEM8, and the induced expression of mutant TEM8 in control endothelial cells (HDMEC) has been found to suppress the activity of β1 integrin and repress activation of NFAT [10]. Interestingly, suppressed activation of β1 integrin is a hallmark of all hemECs even in cases where mutations in VEGFR2 or TEM8 have not been found. This suppressed β1 integrin activation in all hemECs is associated with increased interactions between components of the VEGFR2/TEM8/β1 integrin complex since immune-complexes isolated with VEGFR2-specific antibodies contain substantially larger amounts of TEM8 and β1 integrin with lysates from hemECs than from control endothelial cells [10]. We believe, therefore, that the VEGFR2/TEM8/β1 integrin complex in endothelial cells must contain additional components, not yet identified, and that hemangioma-causing mutations in the genes of such components are likely to be found in future mutation screens.
The VEGFR2/TEM8/β1 integrin complex in microvascular endothelial cells controls transcription of VEGFR1
Treatment of control endothelial cells (HDMECs) with either β1 integrin stimulatory antibodies (or fibronectin) or VEGF results in stimulation of VEGFR1 transcription [10]. These effects are VEGFR2-, TEM8- and NFAT-dependent. In the case of VEGFR2, the studies [10] suggest that the VEGFR1-stimulatory function does not involve the cytoplasmic kinase domain, but requires the extracellular region. Furthermore, the effect of the hemangioma-associated missense mutation (C482R) in the extracellular region of VEGFR2 suggests a critical role for sequences in the immunoglobulin-like domain V outside the VEGF-binding region. The hemangioma-associated mutation helps define, therefore, an NFAT/VEGFR1-controlling functional domain in VEGFR2 that is distinct from the VEGF-binding and tyrosine kinase domains of the receptor (Figure 2).
Figure 2. Stimulation of VEGFR1 expression in normal microvascular endothelial cells is controlled by the VEGFR2/TEM8/β1 integrin complex.
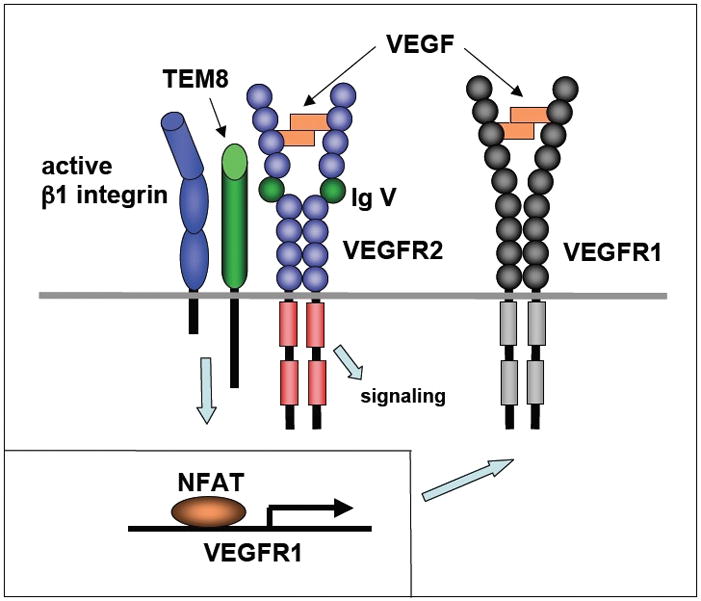
Stimulation of microvascular endothelial cells with VEGF or adhesion to extracellular matrix activates the VEGFR2/TEM8/β1 integrin complex. This results in stimulation of VEGFR1 transcription via activation of NFAT, high level expression of VEGFR1 protein and reduced VEGF-dependent signaling through VEGFR2.
The hemangioma-associated missense mutation in the transmembrane domain of TEM8 has the same dominant-negative effect on VEGFR1 expression as the soluble, extracellular domain of TEM8 when expressed in control microvascular endothelial cells. Interestingly, this soluble extracellular domain is encoded by a normal TEM8 splice variant, variant 3, which is co-expressed with two variants encoding transmembrane forms of the protein [16]. Production of a mixture of full-length TEM8 that can stimulate and a dominant-negative form that can repress VEGFR1 expression suggests the possibility that microvascular endothelial cells can regulate the VEGFR1 expression levels in response to VEGF and matrix stimulation, depending on circumstances. By reducing the amount of soluble TEM8 relative to the membrane-bound forms, cells could stimulate VEGFR1 expression and thus reduce VEGF-dependent activation of VEGFR2. In contrast, increasing the ratio of soluble TEM8 to the full-length forms would lead to reduced VEGFR1 levels and thus stimulation of VEGF-dependent VEGFR2 signaling. Given these possibilities, it will be of great interest to find out whether the ratio between TEM8 splice variants differs in highly proliferative and quiescent endothelial cells and whether changes in this ratio is associated with progression from proliferation to involution in hemangioma tumor tissues.
Endothelial progenitor cells in hemangioma
Several studies indicate that the endothelial cells within proliferating hemangioma lesions are arrested at an early developmental stage of differentiation. The evidence in support of this includes co-expression of the EPC marker CD133 and the lymphatic endothelial hyaluronan receptor-1 (LYVE-1) with the blood vascular marker CD34 [17,18]. Interestingly, expression levels of CD133 and LYVE-1 are greatly reduced in involuting hemangiomas. Dosanjh et al. [19] also found similarities between hemECs and fetal endothelial cells. Furthermore, proliferating phase hemECs exhibit an aberrant, increased migratory response to the angiogenesis inhibitor endostatin in the presence of VEGF [20], and Kahn et al. [21] demonstrated that this response to endostatin is intermediate between the responses of EPCs (stimulation) and mature HDMECs (inhibition). Undifferentiated EPCs therefore contribute to proliferating hemangiomas. As the tumors involute, the number of these undifferentiated endothelial cells are reduced.
Given previous findings that endothelial cells within proliferating hemangioma lesions exhibit X-chromosome inactivation patterns consistent with clonal expansion from a single cell or a very small number of cells [8,20], it is likely that the majority of EPCs within hemangiomas are generated locally as a result of mechanisms responsible for the initiation and rapid growth of the tumors. Less likely is the possibility that EPCs are continuously recruited into the growing tumors from the circulation. However, the clonality assays used in previous studies do not exclude the possibility that some small fraction of the progenitor cells identified in the lesions is derived from sources outside the tumors. In fact, Kleinman et al. [22] have reported that levels of mediators (VEGF and matrix metalloproteinase (MMP)-9) of bone marrow-derived EPC trafficking are increased in the blood from children with proliferating hemangiomas. These authors have also found that expression of such mediators, including stromal cell-derived factor (SDF)-1α, MMP-9, VEGF and HIF-1α, is increased in hemangioma tissue specimens.
Since the expression levels of VEGF, SDF-1α and MMP-9 are increased during tissue ischemia as a result of increased stabilization of HIF-1α [23–25], these data are consistent with previous discoveries that proliferating hemangioma endothelial cells express markers, including GLUT-1 and insulin-like growth factor (IGF)-2, that are typically found associated with hypoxia [26–28]. The demonstration by Jinnin et al. [10] of constitutive activation of VEGFR2 signaling in proliferating hemangioma-derived endothelial cells provides a molecular explanation for these findings. One of the downstream targets of this signaling pathway is HIF-1α, and both transcript and protein levels of this transcription factor are increased in hemECs as compared with control microvascular endothelial cells. The increased expression of VEGF, GLUT-1, IGF-2, MMP-9 and SDF-1α in hemangioma is therefore likely a direct consequence of the constitutive VEGFR2 signaling.
By what mechanisms are hemangioma lesions initiated?
As mentioned above, recent studies demonstrate that some hemangioma patients carry heterozygous missense mutations in VEGFR2 or TEM8 [10]. The mutations are germline mutations and they provide, therefore, no explanation for the finding that hemECs derived from their proliferating lesions exhibit X-chromosome inactivation patterns consistent with clonality, whereas non-endothelial cells from the same lesions do not [20]. Also, the mutations do not appear to cause systemic vascular abnormalities in affected individuals. This raises two important questions: First, how do the VEGFR2 and TEM8 mutations contribute to hemangioma formation, and second, what are the mechanisms responsible for the local initiation of the proliferating tumor?
The studies of Jinnin et al. [10] provide strong evidence for the conclusion that the VEGFR2 and TEM8 mutations represent risk factor mutations for hemangioma formation. The sequence changes in both VEGFR2 (C482R) and TEM8 (A326T) affect highly conserved amino acid residues and result in suppressed β1 integrin/NFAT activation (see Figure 1). The data suggest that the C482R mutation in VEGFR2 is a loss-of-function mutation in the context of the ability of VEGFR2 to control expression of VEGFR1 in an NFAT-dependent manner. The A326T mutation in TEM8 has a dominant-negative effect.
To the extent that the mutations do not cause systemic vascular abnormalities, they are similar to heterozygous germline mutations in other genes causing vascular malformations with localized lesions. For example, in the case of venous malformations recent data [29] strongly support the conclusion that a somatic mutational event is required for localized lesions to occur in individuals who are heterozygous for a germline mutation in the tyrosine kinase receptor TIE2 [30]. Since infantile hemangiomas predictably undergo a process of involution (except in rare cases of so-called Non-Involuting Congenital Hemangioma, NICH [31]) while venous malformations do not regress, it is possible that the lesion-triggering events in hemangioma may be reversible pathophysiological events rather than somatic mutations. Such events may involve processes (for example, perinatal hypoxia [32] or placental cell emboli [33]) that place stresses on the VEGFR2/TEM8/β1 integrin-dependent VEGFR1 control mechanism in microcapillary endothelial cells. In favor of hypoxia is the clinical observation of a blanched area of skin in the position of the future hemangioma, suggesting ischemia. However, somatic mutations cannot be ruled out. In fact, a somatic missense change in the kinase domain of VEGFR2 has already been reported in a case of sporadic hemangioma [8], and recent studies of a serine/threonine kinase, Snrk-1, and a dual specific phosphatase, Dusp-5, raise the intriguing possibility that somatic mutations in their genes may contribute to human vascular anomalies, including infantile hemangioma [34,35].
The genes for these two proteins have been shown to control angioblast populations during early vascular development, with Dusp-5 acting downstream and counteracting the effects of Snrk-1. Furthermore, treatment of human umbilical vein endothelial cells in vitro with Snrk-1 or Dusp-5 siRNAs results in defective migration and increased apoptosis, respectively, suggesting that the two proteins also function in differentiated endothelial cells [34,35]. Of substantial interest is the finding of Pramanik et al. [35] that a Ser-to-Pro substitution in Dusp-5 is present in cDNAs from several types of patient lesions: in 12 of 17 vascular and lymphatic malformations and 1 of 3 infantile hemangioma specimens. The authors present evidence that the sequence change is somatic rather than germ-line and they conclude that it likely results in instability of Dusp-5 protein. Whether the mutant transcripts are present in the endothelial cells of the patient tissues remains to be determined, but if the mutation occurs in an endothelial cell of an individual who is heterozygous for the germline mutations in VEGFR2 or TEM8, it could conceivably represent the triggering event in the formation of a localized hemangioma lesion. Dusp-5 preferentially dephosphorylates ERK in vitro [36] and knock-down of Dusp-5 in zebrafish embryos results in increased levels of phosphorylated ERK [35]. Thus, somatic reduction of Dusp-5 function in an endothelial cell with a defective VEGFR1 control pathway may be sufficient to push VEGFR2-mediated ERK activation to the hemangioma level [10].
What are the mechanisms responsible for hemangioma involution?
Loss of Dusp-5 function increases apoptosis in HUVECs in vitro [35]. In conjunction with other pro-apoptotic stimuli, somatic mutations in Dusp-5 may thus contribute to both the initiation and growth of hemangioma lesions as well as the subsequent process of involution. However, the involuting phase of the hemangioma life cycle is a complex process and likely involves interactions between endothelial and non-endothelial cells. Further studies are needed to fully define the critical involution-promoting cell types and interactions, but several studies provide some clues and the basis for hypotheses that can be experimentally tested.
Comparisons of apoptosis (as measured by TUNEL assays) between proliferating and involuting hemangiomas demonstrate that the rate of apoptosis is substantially increased as the tumors involute [37,38]. Understanding the mechanisms of this increase is therefore key to understanding the involution process. As discussed above, proliferating hemangiomas express high levels of HIF-1α protein and release factors, such as SDF-1α, that can induce recruitment of bone marrow-derived cells from the circulation into the tumors. Some of these cells are likely EPCs [22], but the majority may represent CD45-positive subpopulations of myeloid cells. Ritter et al. [32] found that proliferating hemangiomas contain large numbers of cells expressing markers (including CD45) for hematopoietic cells of the myeloid lineage, and lack of evidence for proliferation of such cells within the lesions led the authors to conclude that they most likely are recruited into the growing tumors from the circulation. Because of reduced numbers of such cells in involuted hemangioma specimens, the authors also suggested that the myeloid cells play a role in stimulating endothelial growth during the proliferating phase [32]. While these are reasonable conclusions, we believe the data are also consistent with the hypothesis that recruitment of myeloid cells into hemangiomas contributes more to involution than to proliferation of endothelial cells.
This hypothesis is based on a recent report [39] describing a similar HIF-1α/SDF-1α–dependent recruitment of bone marrow-derived myeloid cells, EPCs and pericyte progenitors into glioblastoma tumor tissue. Interestingly, the CD45-positive myeloid cells recruited into the tumor consist of several subpopulations, including cells expressing VEGFR1 and MMP-9 [39]. Within the glioblastoma environment, the expression of MMP-9 is sufficient to initiate angiogenesis by increasing the bioavailability of VEGF [39]. In contrast, a similar recruitment of VEGFR1 expressing myeloid cells into the hemangioma environment, with VEGF-dependent signaling at maximal and VEGFR1 expression at minimal levels, may result in increased VEGFR1 decoy function, reduced availability of VEGF and decreased activation of VEGFR2 signaling.
Conclusions
The recent work on molecular, genetic and cellular mechanisms in hemangioma and vascular malformations is providing insights not only into vascular disease mechanisms, but into normal endothelial control mechanisms as well. Coupled with a very large number of outstanding studies of fundamental mechanisms of angiogenesis in development and disease, not covered by this review, the work provides a strong basis for future studies. More work is clearly needed on the triggering mechanisms for hemangioma formation, the relative roles of germline and somatic mutations, and the cellular and molecular processes that result in hemangioma involution.
Finally, as these processes are better characterized, translational studies are needed to develop effective therapies for treatment of clinically problematic hemangiomas. About 80% of hemangiomas are located in the head and neck region and most are small, requiring no treatment. However, in about 20% of the cases their aggressive growth and/or their location relative to vital structures require therapeutic intervention. Current treatment options include the use of corticosteroids [5], recombinant interferon α [40], the immune response modifier Imiquimod [41], recombinant platelet-derived growth factor [42], and bleomycin [43]. In a recent study the beta-blocker propranolol was used to treat severe hemangiomas associated with cardiac complications [44]. The effects of these drugs on hemangioma growth are variable, side effects are of concern, and the drugs are not directed at targets known to be critical components of pathogenetic mechanisms in hemangioma formation and growth. The recent results of studies of hemangioma mechanisms will likely change this situation. Based on the results of Jinnin et al. [10], it would appear that locally administered antibodies to VEGF, inhibitors of VEGFR2, or agents that may stimulate components of the VEGFR2/TEM8/β1 integrin/NFAT pathway should be effective in treating rapidly growing and clinically problematic hemangiomas.
Acknowledgments
The authors wish to acknowledge the support from the National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health (P01 AR048564) and The John Butler Mulliken Foundation, Inc.
References and recommended reading
Papers of particular interest, published within the annual period of the review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Mulliken JB, Glowacki J. Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plast Reconstr Surg. 1982;69:412–422. doi: 10.1097/00006534-198203000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Vikkula M, Boon LM, Mulliken JB, Olsen BR. Molecular basis of vascular anomalies. Trends Cardiovasc Med. 1998;8:281–292. doi: 10.1016/s1050-1738(98)00024-3. [DOI] [PubMed] [Google Scholar]
- 3.Amir J, Metzker A, Krikler R, et al. Strawberry hemangioma in preterm infants. Pediatr Dermatol. 1986;3:331–332. doi: 10.1111/j.1525-1470.1986.tb00535.x. [DOI] [PubMed] [Google Scholar]
- 4.Haggstrom AN, Drolet BA, Baselga E, et al. Prospective study of infantile hemangiomas: demographic, prenatal, and perinatal characteristics. J Pediatr. 2007;150:291–294. doi: 10.1016/j.jpeds.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 5.Mulliken J, Young A. Vascular Birthmarks: Hemangiomas and Malformations. Philadelphia: WB Saunders Company; 1988. [Google Scholar]
- 6.Walter JW, Blei F, Anderson JL, et al. Genetic Mapping of a Novel Familial Form of Infantile Hemangioma. Am J Med Genet. 1999;82:77–83. doi: 10.1002/(sici)1096-8628(19990101)82:1<77::aid-ajmg15>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 7.Blei F, Walter J, Orlow SJ, et al. Familial segregation of hemangiomas and vascular malformations as an autosomal dominant trait. Arch Derm. 1998;134:718–722. doi: 10.1001/archderm.134.6.718. [DOI] [PubMed] [Google Scholar]
- 8.Walter JW, North PE, Waner M, et al. Somatic Mutation of Vascular Endothelial Growth Factor Receptors in Juvenile Hemangioma. Genes Chrom Cancer. 2002;33:295–303. doi: 10.1002/gcc.10028. [DOI] [PubMed] [Google Scholar]
- 9.Irrthum A, Karkkainen MJ, Devriendt K, et al. Congenital hereditary lymphedema caused by a mutation that inactivates VEGFR3 tyrosine kinase. Am J Hum Genet. 2000;67:295–301. doi: 10.1086/303019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10**.Jinnin M, Medici D, Park L, et al. Suppressed NFAT-dependent VEGFR1 expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat Med. 2008;14:1236–1246. doi: 10.1038/nm.1877. This study provides the first evidence that the VEGFR1 gene is a transcriptional target for NFAT and that a complex of proteins, including VEGFR2, TEM8, and β1 integrin, controls expression of VEGFR1 by an NFAT-dependent mechanism. It also provides genetic and biochemical evidence that sequences within the immunoglobulin-like domain V in the extracellular region of VEGFR2 are required for VEGFR2 function within the complex. The work further demonstrates that defects within the VEGFR2/TEM8/β1 integrin complex suppress activation of NFAT and transcription of NFAT target genes, including VEGFR1, in proliferating hemangioma endothelial cells in vitro as well as in hemangioma tissue in vivo. Using a candidate gene mutation screen, the authors show that the defects in some hemangioma patients are due to heterozygous missense mutations in VEGFR2 or TEM8. Finally, the study demonstrates that the loss of VEGFR1 decoy function in proliferating hemangioma endothelial cells results in VEGF-dependent constitutive VEGFR2 signaling and activation and/or increased transcript/protein levels of downstream targets, leading to in increased proliferation and migration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Armesilla AL, Lorenzo E, Gomez del Arco P, et al. Vascular endothelial growth factor activates nuclear factor of activated T cells in human endothelial cells: a role for tissue factor gene expression. Mol Cell Biol. 1999;19:2032–2043. doi: 10.1128/mcb.19.3.2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hernandez GL, Volpert OV, Iniquez MA, et al. Selective inhibition of vascular endothelial growth factor-mediated angiogenesis by cyclosporine A: roles of the nuclear factor of activated T cells and cyclooxygenase 2. J Exp Med. 2001;193:607–620. doi: 10.1084/jem.193.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hesser BA, Liang XH, Camenisch G, et al. Down syndrome critical region protein 1 (DSCR1), a novel VEGF target gene that regulates expression of inflammatory markers on activated endothelial cells. Blood. 2004;104:149–158. doi: 10.1182/blood-2004-01-0273. [DOI] [PubMed] [Google Scholar]
- 14.Satonaka H, Suzuki E, Nishimatsu H, et al. Calcineurin promotes the expression of monocyte chemoattractant protein-1 in vascular myocytes and mediates vascular inflammation. Circ Res. 2004;94:693–700. doi: 10.1161/01.RES.0000118250.67032.5E. [DOI] [PubMed] [Google Scholar]
- 15.St Croix B, Rago C, Velculescu V, et al. Genes expressed in human tumor endothelium. Science. 2000;289:1197–1202. doi: 10.1126/science.289.5482.1197. [DOI] [PubMed] [Google Scholar]
- 16.Bradley KA, Mogridge J, Mourez M, et al. Identifiction of the cellular receptor for anthrax toxin. Nature. 2001;414:225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 17.Yu Y, Flint AF, Mulliken JB, et al. Endothelial progenitor cells in infantile hemangioma. Blood. 2004;103:1373–1375. doi: 10.1182/blood-2003-08-2859. [DOI] [PubMed] [Google Scholar]
- 18.Dadras SS, North PE, Bertoncini J, et al. Infantile hemangiomas are arrested in an early developmental vascular differentiation state. Mod Pathol. 2004;17:1068–1079. doi: 10.1038/modpathol.3800153. [DOI] [PubMed] [Google Scholar]
- 19.Dosanjh A, Chang J, Bresnick S, et al. In vitro characteristics of neonatal hemangioma endothelial cells: similarities and differences between normal neonatal and fetal endothelial cells. J Cutan Pathol. 2000;27:441–450. doi: 10.1034/j.1600-0560.2000.027009441.x. [DOI] [PubMed] [Google Scholar]
- 20.Boye E, Yu Y, Paranya G, et al. Clonality and altered behavior of endothelial cells from hemangiomas. J Clin Invest. 2001;107:745–752. doi: 10.1172/JCI11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kahn ZA, Melero-Martin JM, Wu X, et al. Endothelial progenitor cells from infantile hemangioma and umbilical cord blood display unique cellular responses to endostatin. Blood. 2006;108:915–921. doi: 10.1182/blood-2006-03-006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleinman ME, Greives MR, Churgin SS, et al. Hypoxia-Induced Mediators of Stem/Progenitor Cell Trafficking Are Increased in Children With Hemangioma. Arterioscler Thromb Vasc Biol. 2007;27:2664–2670. doi: 10.1161/ATVBAHA.107.150284. [DOI] [PubMed] [Google Scholar]
- 23.Forsythe JA, Jiang BH, Iyer NV, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ceradini DJ, Kulkarni AR, Callaghan MJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 25.Himelstein BP, Koch CJ. Studies of type IV collagenase regulation by hypoxia. Cancer Lett. 1998;124:127–133. doi: 10.1016/s0304-3835(97)00463-1. [DOI] [PubMed] [Google Scholar]
- 26.North PE, Waner M, Mizeracki A, Mihm MC., Jr GLUT-1: a newly discovered immunohistochemical marker for juvenile hemangiomas. Hum Pathol. 2000;31:11–22. doi: 10.1016/s0046-8177(00)80192-6. [DOI] [PubMed] [Google Scholar]
- 27.Feldser D, Agani F, Iyer NV, et al. Reciprocal Positive Regulation of Hypoxia-inducible Factor 1a and Insulin-like Gowth Factor 2. Cancer Res. 1999;59:3915–3918. [PubMed] [Google Scholar]
- 28.Ritter MR, Dorrell MI, Edmonds J, et al. Insulin-like growth factor 2 and potential regulators of hemangioma growth and involution identified by large-scale expression analysis. Proc Natl Acad Sci U S A. 2002;99:7455–7460. doi: 10.1073/pnas.102185799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29**.Limaye N, Wouters V, Uebelhoer M, et al. Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nat Med. 2009;41:118–124. doi: 10.1038/ng.272. This study provides compelling evidence for a combination of germline and somatic mutations in the receptor tyrosine kinase TIE2 in familial venous malformations and for combinations of somatic mutations in the same gene in sporadic cases. The work also examines the effects of such TIE2 mutations on the expression, activation and cellular trafficking of the receptor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vikkula M, Boon LM, Carraway KL, 3rd, et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996;87:1181–1190. doi: 10.1016/s0092-8674(00)81814-0. [DOI] [PubMed] [Google Scholar]
- 31.Enjolras O, Mulliken JB, Boon LM, et al. Noninvoluting congenital hemangioma: a rare cutaneous vascular anomaly. Plast Reconstr Surg. 2001;107:1647–1654. doi: 10.1097/00006534-200106000-00002. [DOI] [PubMed] [Google Scholar]
- 32.Ritter MR, Reinisch J, Friedlander SF, et al. Myeloid cells in infantile hemangioma. Am J Pathol. 2006;168:621–628. doi: 10.2353/ajpath.2006.050618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.North PE, Waner M, Buckmiller L, et al. Vascular tumors of infancy and childhood: beyond capillary hemangioma. Cardiovasc Pathol. 2006;15:303–317. doi: 10.1016/j.carpath.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 34*.Chun CZ, Kaur S, Samant GV, et al. Snrk-1 is involved in multiple steps of angioblast development and acts via notch signalling pathway in artery-vein specification in vertebrates. Blood. 2008 Aug 22; doi: 10.1182/blood-2008-06-162156. [Epub ahead of print]. This study utilizes gain- and loss-of-function experiments in zebrafish to demonstrate that the serine/threonine kinase Snrk-1 is required for migration and maintenance of angioblasts and their differentiation to arterial and venous endothelial cells. The authors also report that knocking down Snrk-1 expression in human umbilical vein endothelial cells results in defective migration in vitro. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35**.Pramanik K, Chun CZ, Garnaas MK, et al. Dusp-5 ans Snrk-1 coordinately function during vascular development and disease. Blood. 2008 Oct 16; doi: 10.1182/blood-2008-06-162180. [Epub ahead of print]. This study provides evidence that the dual specific phosphatase Dusp-5, expressed in angioblasts and established vasculature, acts downstream of and counteracts the function of Snrk-1. The authors also report the intriguing finding that somatic mutations in Dusp-5 are present in a large proportion of tissue samples from patients with infantile hemangioma and vascular and lymphatic malformations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwak SP, Dixon JE. Multiple dual specificity protein tyrosine phosphatases are expressed and regulated differentially in liver cell lines. J Biol Chem. 1995;270:1156–1160. doi: 10.1074/jbc.270.3.1156. [DOI] [PubMed] [Google Scholar]
- 37.Razon MJ, Kraling BM, Mulliken JB, Bischoff J. Increased apoptosis coincides with onset of involution in infantile hemangioma. Microcirculation. 1998;5:189–195. [PubMed] [Google Scholar]
- 38.Frischer JS, Huang J, Serur A, et al. Biomolecular markers and involution of hemangiomas. J Pediatr Surg. 2004;39:400–404. doi: 10.1016/j.jpedsurg.2003.11.043. [DOI] [PubMed] [Google Scholar]
- 39*.Du R, Lu KV, Petritsch C, et al. HIF1α Induces the Recruitment of Bone Marrow-Derived Vascular Modulatory Cells to Regulate Tumor Angiogenesis and Invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. This study of hypoxia/HIF-1α-driven recruitment of bone marrow-derived myeloid cells into glioblastoma tumors provides an interesting parallel to the recruitment of such cells, by similar mechanisms, into infantile hemangiomas. The finding that the myeloid cells include VEGFR1-expressing populations, raises the possibility that such cells may help tip the balance from proliferation to involution in hemangioma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greinwald JH, Jr, Burke DK, Bonthius DJ, et al. An update on the treatment of hemangiomas in children with interferon α-2a. Arch Otolaryng–Head Neck Surg. 1999;125:21–27. doi: 10.1001/archotol.125.1.21. [DOI] [PubMed] [Google Scholar]
- 41.Hazen PG, Carney JF, Engstrom CW, et al. Proliferating hemangioma of infancy: successful treatment with topical 5% imiquimod cream. Pediatr Dermatol. 2005;22:254–256. doi: 10.1111/j.1525-1470.2005.22318.x. [DOI] [PubMed] [Google Scholar]
- 42.Metz BJ, Rubenstein MC, Levy ML, et al. Response of ulcerated perineal hemangiomas of infancy to becaplermin gel, a recombinant human platelet-derived growth factor. Arch Dermatol. 2004;140:867–870. doi: 10.1001/archderm.140.7.867. [DOI] [PubMed] [Google Scholar]
- 43.Muir T, Kirsten M, Fourie P, et al. Intralesional bleomycin injection (IBI) treatment or haemangiomas and congenital vascular malformations. Pediatr Surg Int. 2004;19:766–773. doi: 10.1007/s00383-003-1058-6. [DOI] [PubMed] [Google Scholar]
- 44.Leaute-Labreze C, Dumas de la Roque E, Hubiche T, et al. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008;358:2649–2651. doi: 10.1056/NEJMc0708819. [DOI] [PubMed] [Google Scholar]