

Abstract
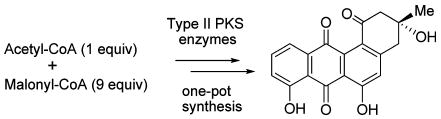
A one-pot enzymatic total synthesis of angucycline antibiotic rabelomycin was accomplished, starting from acetyl-CoA and malonyl-CoA, using a mixture of polyketide synthase (PKS) enzymes of the gilvocarcin, ravidomycin, and jadomycin biosynthetic pathways. The in vitro results were compared to in vivo catalysis using analogous sets of enzymes.
The benz[a]anthraquinone antibiotics, commonly named angucyclines,1,2 are well-known for their broad range of biological activities including antibiotic, antitumor, antiviral, antifungal, platelet aggregation, and enzyme inhibitory effects. The angucyclinone rabelomycin (5) is a commonly found, simple member of this family. It was first isolated from the culture broth of Streptomyces olivaceus ATCC21549 in 1970 and was shown to have antibacterial activities comparable to those of tetrangomycin against Gram-positive microorganisms.3 It is also a proposed intermediate of the biosyntheses of various other members of the angucycline family, such as urdamycin,4 oviedomycin,5 and landomycin E.6
Many of the biologically active angucycline group antibiotics, including rabelomycin (5), have attracted synthetic chemists, and 5 along with other angucyclinones became model compounds for the development of efficient, synthetic routes toward this class of compounds.2,7–10 Most of these procedures require multistep chemical transformations, which are often relatively tedious and time-consuming and suffer from overall low production yield.
Recent progress in elucidating the biosynthetic pathways of various secondary metabolites has made it possible to genetically engineer molecules of interest in a tailor-made fashion, including polyketides and sugars. Following up on this, in vitro enzymatic syntheses emerged as a powerful tool to efficiently produce structurally complex biomolecules and also to provide an alternative to conventional chemical synthesis.11–15 The highly regio- and stereospecific nature of enzymatic catalysis facilitates a multistep enzymatic synthesis in a single pot, which is usually impossible in conventional chemical syntheses. Such an enzymatic approach also avoids the unwanted degradation of unstable intermediates, in addition to the minimal use of hazardous chemicals and minimal production of waste.16 In this communication, we report an efficient one-pot enzymatic synthesis of rabelomycin (5), as a typical representative of the angucycline family.
Previous in vivo expressions demonstrated that jadomycin PKS enzymes comprising ketosynthases KSα and KSβ (JadAB, KSβ is also known as chain-length factor, CLF), acyl carrier protein (ACP) JadC, ketoreductase (KR) JadE, and the two cyclases (CYC) JadD and JadI are sufficient to generate angucyclinones UWM6 (4) and rabelomycin (5).17 Through inactivation and cross complementation of early post-PKS oxygenase encoding genes of the gilvocarcin and jadomycin pathways, we have demonstrated that both natural products share a common biosynthetic route. Rabelomycin is a shunt product of both pathways.18 Therefore, gilvocarcin PKS enzymes were initially chosen for the reconstitution of the rabelomycin pathway in vitro. KS GilA and CLF GilB were coexpressed as a heterodimer in Streptomyces lividans TK64 and isolated as dimer-complex. The recombinant malonyl-CoA:ACP transacylase (MCAT) GilP and PKS-associated KR GilF were produced in Escherichia coli. However, the expression of soluble ACP GilC, CYC GilK, and CYC GilG in E. coli failed and resulted exclusively in the formation of inclusion bodies. Therefore, these enzymes were replaced by the homologous enzymes RavC (ACP), JadD (CYC), and RavG (CYC) from the recently cloned ravidomycin pathway and the jadomycin pathway, respectively (see Supporting Information, Figure S1).19,20 RavC appeared as a mixture of apo- and holo-form (∼ 3:4). A malonyl-CoA synthetase (MatB) from Rhizobium trifolii was used to generate malonyl-CoA in situ through the co-incubation with sodium malonate, ATP, and CoA.21
Having all of the proposed necessary biosynthetic enzymes for the production of rabelomycin (5) in hand, we started the in vitro experiments. Incubation of a mixture of acetyl-CoA, malonyl-CoA, and NADPH with various combinations of overexpressed and isolated enzymes (GilAB and RavC; GilABF and RavC; GilABFP and RavC; GilABFP, JadD and RavC; GilABFP, RavG, and RavC) failed to produce any metabolite. However, when CYC JadD was added to the previous most complex mixture, i.e., using now a mixture of acetyl-CoA, malonyl-CoA, enzymes GilAB, RavC, GilP, GilF, RavG, and JadD (relative enzyme stoichiometry = 1:114:13:29:53:15 μmol, pH 7.6), a gradual color change from colorless to light yellow and on to red-brown was observed over a 2 h time period. The ACP RavC could be replaced by its homologues JadC or RavC1 with the same results. HPLC analyses of the mixture revealed the formation of one major compound showing retention time, mass, and UV spectra identical to the data of rabelomycin (5, Figure 1). Purification of this compound through HPLC afforded 1.1 mg of rabelomycin (∼ 80% yield). The structure of the product was further confirmed by comparison of its 1H and 13C NMR data with those reported for rabelomycin (see Supporting Information).3,7 The production of rabelomycin was completely abolished when either acetyl-CoA or malonyl-CoA was removed from the enzyme mixture. These in vitro results were in contrast to earlier in vivo experiments, where the combined heterologous expression of the jadomycin minimal PKS (encoded by jadABC), the PKS-associated KR (encoded by jadE), and one cyclase (encoded by jadD) in Streptomyces lividans TK64 still produced partly cyclized compounds. SEK43 (8) and UWM4 (9) were found in the absence of the second cyclase (encoded by jadI, a homologous gene of ravG), while RM20b (6) and RM20c (7) were reported to be produced through the coexpression of the tetracenomycin minimal PKS and the PKS-associated KR of the jadomycin pathway (encoded by jadE, Scheme 1, Figure 2).17,22 Because we failed to produce the partly cyclized shunt metabolites 6–9 in vitro, we reasoned that the minimal PKS (GilAB, RavC) might require a complete set of cyclases (RavG and JadD) in addition to GilF and GilP to establish a functional multienzyme complex in vitro. In the case of the in vivo experiments, host enzymes might be assisting minimal PKS enzymes to direct the formation of SEK43 (8) and UWM4 (9). The lack of production of rabelomycin (5) in the absence of either acetyl-CoA or malonyl-CoA clearly indicated that both building blocks are crucial to generate the angucyclinone rabelomycin. In contrast, it was previously reported for the enterocin PKS that it is capable of synthesizing acetate-primed nonaketides from malonyl-CoA alone.12
Figure 1.
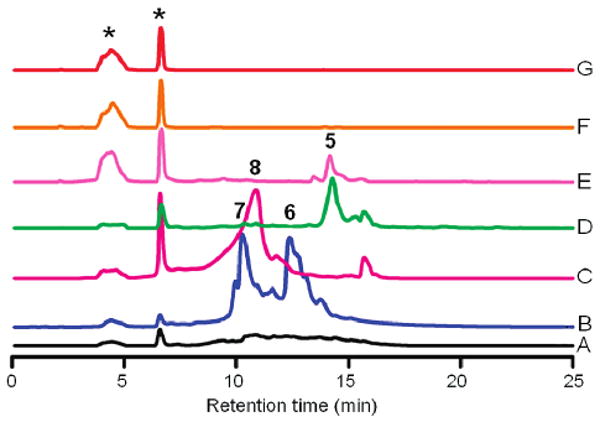
HPLC analyses of the compounds generated from in vivo (traces A–D, 96 h) and in vitro (traces E–G, 2 h) experiments: (A) crude extract of S. lividans TK64/pUL201PW (in vivo control); (B) S. lividans TK64/pRab2; (C) S. lividans TK64/pRab3; (D) S. lividans TK64/pRab4; (E) enzymes GilABFP, RavCG and JadD with acetyl-CoA and malonyl-CoA; (F) GilABFP, RavC and JadD with acetyl-CoA and malonyl-CoA; (G) enzymes GilFP, RavCG and JadD with acetyl-CoA and malonyl-CoA (in vitro control). Compounds that were produced in the controls are marked by asterisks.
Scheme 1.
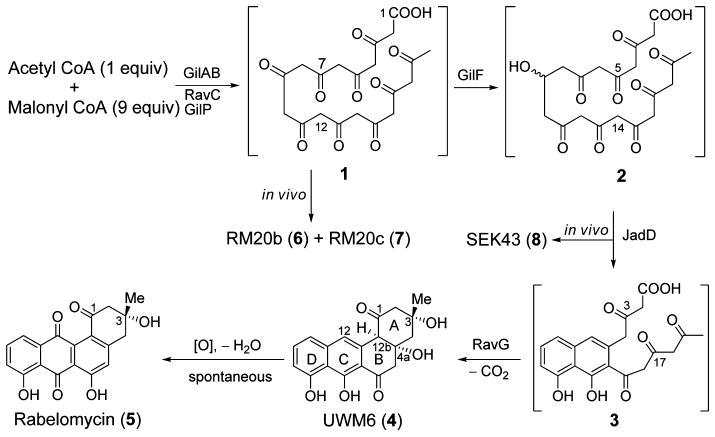
Proposed Biosynthesis of Rabelomycin (5) Involving Enzymes from Various Pathways
Figure 2.
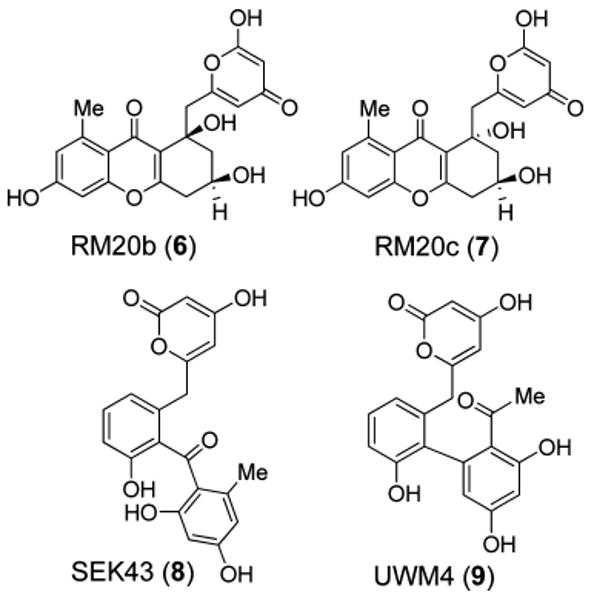
Structures of metabolites produced in in vivo experiments: see text and Figure 3.
To further validate the above stated hypothesis, namely, that unidentified host strain enzymes may have supported the formation of partly cyclized PKS-shunt products such as 8 and 9, the genes corresponding to the above listed enzymes of the in vitro experiment were cloned into the pUWL201PW vector23 to generate in vivo expression constructs in the heterologous host S. lividans TK64 (Figure 3, see Supporting Information for details). To minimize cloning steps in the in vivo experiments, gilC was used to express the ACP (instead of ravC, which would have matched exactly the in vitro experiments). Confirming Hutchinson's earlier in vivo results, expression of pRab1 (containing genes encoding the minimal PKS and PKS-associated KR) and pRab2 (like pRab1 plus MCAT gene gilP) in S. lividans TK64 accumulated the spontaneously cyclized decaketides RM20b (6) and RM20c (7).
Figure 3.
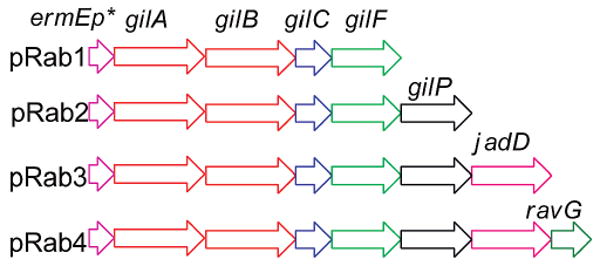
In vivo expression constructs.
The over 50-fold higher production of 6 and 7 in the case of pRab2 compared to pRab1 is probably due to the excess supply of MCAT GilP, which serves as a catalytic subunit of the PKS complex, which in the case of pRab1 needed to be recruited from the fatty acid synthase of the host strain. Again, confirming Hutchinson et al., expression of pRab3 (= pRab2 plus CYC gene jadD) in S. lividans led to the production of SEK43 (8), for which the JadD-mediated regiospecific C7–C12 cyclization of the nascent polyketide is required (Figure 2). The completely cyclized angucyclinones rabelomycin (5) and UWM6 (4) were only observed in vivo through the expression of pRab4, in which cyclase gene ravG was added. Compounds 4 and 5 were observed initially in a 1:1 ratio (after 48 h), but within 4 days only 5 could be observed (Figure 1, trace D).
The lack of UWM6 (4) production in the in vitro experiment could be due to the spontaneous oxidation of 4 during the course of the experiment. Slightly alkaline conditions (pH 7.6) may have facilitated C4a–C12b dehydration of 4, leading to an aromatization of ring B, which then allows spontaneous oxidation at the C-12 position to yield 5 (Scheme 1). Such a spontaneous oxidation of 4 to 5 has been reported earlier.17
In summary, we accomplished a highly efficient one-pot enzymatic synthesis of rabelomycin (5). To the best of our knowledge, it is the first report of a total enzymatic synthesis of an angucycline group antibiotic. The enzymatic total synthesis of tetracenomycins by Hutchinson et al.11,24 and of enterocin and wailupemycins by Moore et al.12 are the only other examples of metabolites to be synthesized in vitro using type II PKS enzymes thus far. The in vitro reconstitution of the rabelomycin biosynthetic pathway reported here lays the groundwork for further enzymatic syntheses of other angucycline or angucycline-derived natural products and to further interrogate biosynthetic steps of angucyclinones and angucyclinone-derived metabolites.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health (CA 102102) to J.R. The mass spectrometry facility of the University of Kentucky is acknowledged for their service, and we are grateful to Prof. Yi Tang, UCLA, for providing us with the MatB-expression construct and to Professor Mark Zabriskie, Orgeon State University, for providing us with plasmid pXY200.
Footnotes
Supporting Information Available: Experimental details, full characterization of compounds, and copies of 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Rohr J, Thiericke R. Nat Prod Rep. 1992;9:103–137. doi: 10.1039/np9920900103. [DOI] [PubMed] [Google Scholar]
- 2.Krohn K, Rohr J. Top Curr Chem. 1997;188:127–195. [Google Scholar]
- 3.Liu WC, Parker WL, Slusarchyk DS, Greenwood DL, Graham SF, Meyers E. J Antibiot. 1970;23:437–441. doi: 10.7164/antibiotics.23.437. [DOI] [PubMed] [Google Scholar]
- 4.Faust B, Hoffmeister D, Weitnauer G, Westrich L, Haag S, Schneider P, Decker H, Künzel E, Rohr J, Bechthold A. Microbiology. 2000;146:147–154. doi: 10.1099/00221287-146-1-147. [DOI] [PubMed] [Google Scholar]
- 5.Lombó F, Abdelfattah MS, Brana AF, Salas JA, Rohr J, Mendez C. ChemBioChem. 2009;10:296–303. doi: 10.1002/cbic.200800425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu L, Ostash B, Rix U, Nur-e-Alam M, Mayers A, Luzhetskyy A, Méndez C, Salas JA, Bechthold A, Fedorenko V, Rohr J. J Org Chem. 2005;70:631–638. doi: 10.1021/jo0483623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krohn K, Khanbabaee K. Angew Chem, Int Ed Engl. 1994;33:99–100. [Google Scholar]
- 8.Krohn K, Boeker N, Floerke U, Freund C. J Org Chem. 1997;62:2350–2356. doi: 10.1021/jo9622587. [DOI] [PubMed] [Google Scholar]
- 9.Carreno MC, Urbano A. Synlett. 2005:1–25. [Google Scholar]
- 10.Krohn K, Agocs A, Bäuerlein C. J Carbohydr Chem. 2003;22:579–583. [Google Scholar]
- 11.Shen B, Hutchinson CR. Science. 1993;262:1535–1540. doi: 10.1126/science.8248801. [DOI] [PubMed] [Google Scholar]
- 12.Cheng Q, Xiang L, Izumikawa M, Meluzzi D, Moore BS. Nat Chem Biol. 2007;3:557–558. doi: 10.1038/nchembio.2007.22. [DOI] [PubMed] [Google Scholar]
- 13.Thibodeaux CJ, Melancon CE, 3rd, Liu HW. Angew Chem, Int Ed. 2008;47:9814–9859. doi: 10.1002/anie.200801204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He XM, Liu HW. Annu Rev Biochem. 2002;71:701–754. doi: 10.1146/annurev.biochem.71.110601.135339. [DOI] [PubMed] [Google Scholar]
- 15.Roessner CA, Scott AI. Chem Biol. 1996;3:325–330. doi: 10.1016/s1074-5521(96)90114-3. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi H, Liu YN, Liu HW. J Am Chem Soc. 2006;128:1432–1433. doi: 10.1021/ja0562144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kulowski K, Wendt-Pienkowski E, Han L, Yang KQ, Vining LC, Hutchinson CR. J Am Chem Soc. 1999;121:1786–1794. [Google Scholar]
- 18.Kharel MK, Zhu L, Liu T, Rohr J. J Am Chem Soc. 2007;129:3780–3781. doi: 10.1021/ja0680515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han L, Yang K, Ramalingam E, Mosher RH, Vining LC. Microbiology. 1994;140:3379–3389. doi: 10.1099/13500872-140-12-3379. [DOI] [PubMed] [Google Scholar]
- 20.Kharel MK, Nybo ES, Shepherd MD, Rohr J. ChemBioChem. 2010;11:523–532. doi: 10.1002/cbic.200900673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.An JH, Kim YS. Eur J Biochem. 1998;257:395–402. doi: 10.1046/j.1432-1327.1998.2570395.x. [DOI] [PubMed] [Google Scholar]
- 22.Meurer G, Gerlitz M, Wendt-Pienkowski E, Vining LC, Rohr J, Hutchinson CR. Chem Biol. 1997;4:433–443. doi: 10.1016/s1074-5521(97)90195-2. [DOI] [PubMed] [Google Scholar]
- 23.Doumith M, Weingarten P, Wehmeier UF, Salah-Bey K, Benhamou B, Capdevila C, Michel JM, Piepersberg W, Raynal MC. Mol Gen Genet. 2000;264:477–85. doi: 10.1007/s004380000329. [DOI] [PubMed] [Google Scholar]
- 24.Bao W, Wendt-Pienkowski E, Hutchinson CR. Biochemistry. 1998;37:8132–8138. doi: 10.1021/bi980466i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.