

Abstract
Multiple N-methylation is a novel technology to improve bioavailability of peptides and increase receptor subtype selectivity. This technique has been applied here to the superpotent but non-selective cyclic peptide MT-II. A library of all possible 31 backbone N-methylated derivatives has been synthesized and tested for binding and activation at melanocortin receptor subtypes 1, 3, 4 and 5. It turned out that selectivity is improved with every introduced N-methyl group, resulting in several N-methylated selective and potent agonists for the hMC1R. The most potent of these derivatives is N-methylated on four out of five amide bonds in the cyclic structure. Its solution structure indicates a strongly preferred backbone conformation which resembles other a-MSH analogs but possesses much less flexibility and in addition distinct differences in the spatial arrangement of individual amino acid side chains.
Introduction
N-Methylation of peptide bonds is long known1–3 and has often been used to modify biological properties of bioactive peptides.4 However, it has become evident only recently that multiple N-methylation is a novel technology to improve the pharmacological properties of peptides5 and in extreme cases even achieve oral bioavailability6 such as found for the antibiotic Cyclosporin.7,8 Here we present a complete N-methylation library of the cyclic analogue of a-MSH, Ac-Nle-c[Asp-His-DPhe-Arg-Trp-Lys]-NH2 (MT-II), a biologically important and potent but non-selective agonist for the melanocortin receptor subtype family.9–10 The goal of our present study was to obtain new peptidic compounds with melanocortin receptor activity and improved selectivity, possessing pharmacological properties superior to those of MT-II. Therefore, we exchanged in a systematic manner His, DPhe, Arg, Trp and Lys by their Nα-methylated analogs and studied all of the 31 (25-1=31) possible MT-II-derivatives with one or more methylated backbone nitrogens (Figure 1).
Figure 1.

MT-II (Ac-Nle-cyclo(5β→10ε)(Asp5-His6-DPhe7-Arg8-Trp9-Lys10)-NH2) with sites of N-methylation indicated by arrows.
The melanocortin receptors are members of the 7 transmembrane (TM) spanning G-protein coupled receptor (GPCR) superfamily. Five melanocortin receptor subtypes (MCR1–5) have been discovered thus far.8,11–17 They all intracellularly mediate their effects by activating pathways that are cyclic adenosine monophosphate (cAMP) dependent. The diverse MCRs are distributed widely in mammalian tissues, and regulate numerous functions in the body including skin and hair coloration,18–20 inflammation21 and immunomodulation,22 steroid production and release,23,24 cardiovascular functions,25,26 energy homeostasis,27 feeding behaviour,28–30 penile erection and sexual behaviour31–33 and many other functions. These very diverse biological activities generally are associated with only 1 or 2 of the melanocortin receptors that are expressed in different parts of the body. Thus the search for highly selective and potent agonist and antagonist analogues is critical.
The natural endogenous agonistic ligands a-melanocyte stimulating hormone (a-MSH, Ac-Ser1-Tyr2-Ser3-Met4-Glu5-His6-Phe7-Arg8-Trp9-Gly10-Lys11-Pro12-Val13-NH2), β-MSH, γ-MSH and adrenocorticotropin (ACTH) all are derived from the same precursor protein, pro-opiomelanocortin (POMC). Structure-function examinations of a-MSH and of a-MSH fragments have lead to the identification of His-Phe-Arg-Trp as the critical pharmacophore for pigmentary activity and other functions of the melanotropin peptides.19,20,34–41 Truncation of the α-MSH sequence, keeping amino acids 4–10, exchange of Met4 with Nle, Glu5 with Asp, Phe7 with DPhe and Gly10 with Lys, acetylation of the Nle4-α-nitrogen, amidation of the C-terminal carboxyl function, and lactam cyclization resulted in the lactam bridged compound MT-II, a super potent but non-selective agonist at 4 melanocortin receptors MC1R, MC3R, MC4R, MC5R9,10,41 (not at the MC2R which has ACTH as its ligand, and where a-MSH and its analogues are not active).
A major focus in synthesising new melanotropins is gaining selectivity and improved pharmacological properties that are suitable for medical applications of these compounds. When creating peptidic ligands one possible way to obtain these properties is N-methylation of the amide bonds,4–6,42–44 since substitution of amide protons with methyl groups can result in receptor subtype selectivity,45–48 but also other pharmacological properties can be improved, such as metabolic stability6,49,51 lipophilicity,49–51 enhanced potency,52–55 enhanced bioavailability6,56,57 and conformational rigidity.48,51,58 It may also turn an agonist into an antagonist.57
Application of the N-alkyl scan concept59 to the highly active and selective cyclic pentapeptide cyclo(-RGDfV-)60 has led to one of the most active (0.5 nM) and selective inhibitors for the αvβ3 integrin,55,61 the analog cyclo(-Arg-Gly-Asp-DPhe-(Me)Val-], Cilengitide,55,61 which is now in clinical phase III for treatment of glioblastoma. N-Alkylation is also present in natural occurring peptides from plants, marine sources and various microorganisms. Several of these compounds exhibit biological activity like antibiotic,62–64 antitumor65–67 and immunosuppressor activity.68
Peptide Design
Ac-Nle4-c[Asp5, DPhe7, Lys10]α-MSH(4–10)-NH2 (MT-II) was chosen as a template for the design of a combinatorial library to search for more selective melanotropin peptides. Some N-methyl amino acids are commercially available, but most are expensive. However, several methods for synthesizing N-methylated amino acids in solution and on solid supports have been developed.69 We decided to do the N-methylations on a solid support, which gives flexibility in library design and facilitated coupling, because no coupling of N-methylated amino acids to an N-methylated peptide is necessary.6 We used the procedure originally described by Miller and Scanlan70 which has been optimized by Biron et al. and which is compatible with all commonly used amino acids.71
A major advantage of the o-NBS protecting group coupled to the free amine prior to N-methylation is that deprotection with mercaptoethanol is selective for N-methylated derivatives and does not occur when the protected amine is not alkylated.71 Hence it provides an inherent purification step during synthesis.
Couplings on Nα-methylamino acids are known to be more challenging than normal couplings.72 Hence, these couplings were performed with HATU and HOAt instead of TBTU and HOBt using DIEA as base in NMP yielding in complete couplings after 3h. Nevertheless, some peptides (3, 13, 25) could not be purified as sharp peaks by HPLC and were observed as broad peaks or mixtures of two peaks with identical mass, even after repeated synthesis. When the peaks were separated and reinjected, similar chromatograms were observed. This indicated the presence of conformational isomers rather than diastereomers. To study the influence of N-methylation on the conformation of the pharmacophore of MT-II, a library of MT-II analogues with single and multiple N-methylation in the core sequence of MT-II have been designed and synthesized (Figure 1 and Table 1).
Table 1.
Binding Assay of N-Methylated MT-II Analogues at hMCRs
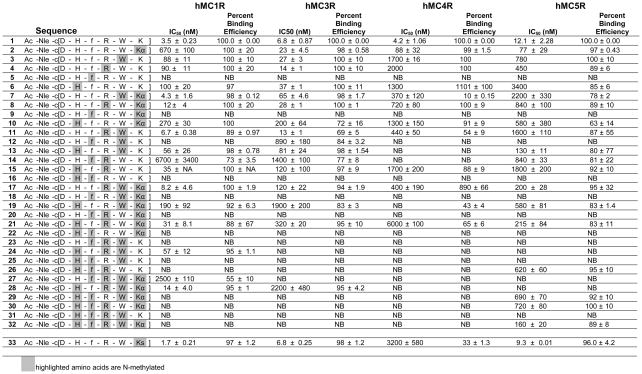 |
IC50 = concentration of peptide at 50% specific binding (N=4). NB = 0% of 125I-NDP-α-MSH displacement observed at 10 μM. Percent Binding Efficiency = maximal % of 125I-NDP-α-MSH displacement observed at 10 μM.
Results and Discussion
Biological data
Competitive binding assays with [125I]-[Nle4,DPhe7]-α-MSH (NDP-α-MSH), and adenylate cyclase assays were performed on HEK293 cells stable expressing the hMC1R, hMC3R, hMC4R, and hMC5R (Table 1 and Table 2, respectively).
Table 2.
cAMP Assay of N-Methylated MT-II Analogues at hMCRs
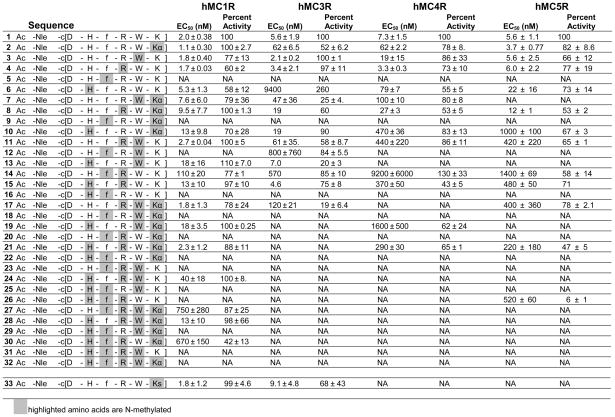 |
EC50 = Effective concentration of peptide that was able to generate 50% maximal intracellular cAMP accumulation (N=4). Percent Activity = % of cAMP produced at 10 μM ligand concentration, in relation to MT-II. NA = 0% cAMP accumulation observed at 10 μM. The peptides were tested at a range of concentration from 10−10 to 10−5 M.
In the first group of peptides with the single N-methylation screen (peptides 2–6), it is revealed that NMe-DPhe caused a total loss of binding as well as adenylate cyclase activities (Tables 1 and 2) at the hMC1R, hMC3R, hMC4R and hMC5R for peptide 5. For the rest of the core sequence single N-methylation does not cause this drastic loss of binding activities (Table 1). The cAMP assay data (Table 2) show that the N-methylated peptides 2, 3 and 6 still retain full agonist or partial agonist activity toward all subtypes of melanocortin receptors. This phenomenon parallels earlier studies that have shown that DPhe is the most critical amino acid for the binding and cAMP activities to hMCRs.43,73,75 The NMe-DPhe7 substitution apparently led to a conformation which prevents interaction of the aromatic ring with the 3rd and the 6th transmembrane binding domains aromatic groups.76 The NMe-His6 analog reveals increased binding selectivity for the hMC1R and hMC3R. This result parallels our earlier observation that constraining the His residue can lead to potent antagonists especially for the hMC3R.77
The second group of peptides are dimethylated derivatives in the core sequence of MT-II (peptides 7–16). In this group, some exciting results are observed. It is first demonstrated that any site of N-methylation combined with NMe-DPhe7 will lose the binding as well as cAMP activities at all hMCRs (peptides 9, 12, 14, 16) confirming the results from mono-methylation. N-Methylation combined with NMe-Lys reduces binding at the hMC4R and hMC5R (peptide 7, 8, 10). The cAMP functional assays show that except 7 at the hMC5R these peptides retain partial agonist activities. N-Methylation combined with NMe-Trp increases the selective agonist activity at the hMC1R (peptides 11, 13), and selective antagonist activity at hMC3R (peptide 13) and lead to large decreases in binding and functional activity at the hMC4R and hMC5R resulting in increased selectivity of peptide 12 for the hMC3R. The combination of NMe-His and NMe-Arg (peptide 15) reduces the binding at the hMC4R and the hMC5R, therefore increase the agonist selectivity for the hMC1R and hMC3R.
More interesting results are found in the 3rd group of peptides (peptides 17–26), tri-N-methylated derivatives in the core sequence of MT-II caused a loss in binding affinities (peptides 18, 20, 22, 23, 25, 26), especially, with any of the combinations with NMe-DPhe. In this group, as long as DPhe is not N-methylated, most ligands will retain binding activity at hMCRs (peptides 17, 19, 21, 24), but they all have reduced cAMP activity (Table 2) except at the hMC1R (partial agonists). Tri-N-methylations generally increase the agonist selectivity for the hMC1R (peptides 17, 19, 21, 24). It can be seen from Table 1 that 24 has totally lost activity for all receptors but the hMC1R, which still exhibits good binding affinity and >1000 fold selectivity vs the hMC3, hMC4 and hMC5 receptors. The compound has agonist activity as well (Table 2).
Selectivity, but also loss of activity, are further shown for the five tetra-N-methylated MT-II derivatives (peptides 28–32). Tetra-N-methylation caused a loss in binding affinities and cAMP activities at all subtypes of hMCRs except for peptide 28, which has potent binding affinity (14 nmol) at the hMC1R and has full agonist activity. Thus, a highly potent and selective agonist of hMC1R has been obtained. Peptides 29, 30 and especially 32 are weak, but selective antagonists for the hMC5R.
In addition to 31 Nα-methylated analogs, one ε-N-methylated lysine analogue (peptide 33) was investigated. It is interesting to note that in the case of peptide 33, there was a loss in binding to the hMC4R, while selective antagonist activity was seen for the hMC5R in the nanomolar range (peptide 33 is still a full agonist at the hMC1R and the hMC3R). Thus far there are few cases of selective peptide antagonists at the hMC5R.
It has been demonstrated that the MC1R, which is activated selectively by the fourfold N-methylated peptide 28 is involved in pain, in immune response, and in melanoma cancer.78 Thus, highly selective MC1R ligands have potential for treatment of pigmentary disorders,18–20,79,80 treatment of pain,78 cancer therapy,81,82 and inflammatory disorders of the skin. The hMC1R is just one subtype of hMCRs in the melanocortin system. It is mainly distributed in the peripheral system in mammals, and the other subtypes of hMCRs are distributed in both of the peripheral and the central nervous system.18 Designing highly constrained, lipophilic hMC1R selective N-methylated MT-II derivatives will have widest application for specific targeting of disease.
NMR Conformational Studies
The conformation of the highly potent and selective compound 28 (Figure 2) was investigated by NMR spectroscopy, restrained distance geometry calculations (DG), restrained 150 ps MD simulation in explicit water (rMD) and by unrestrained 30 ns MD simulation in explicit water (MD). Based on NMR assignments (Table 3) and other NMR data (ROEs, homo- and heteronuclear scalar coupling constants, HN temperature gradients) and on restrained and unrestrained molecular dynamics a distinct and preferred structure could be derived for the peptide backbone. The amide bridged side chains of the residues Asp5 and NMe-Lys10 that are also part of the cyclic core were found to be flexible. The resulting good agreement between measured and calculated distances clearly indicates a preferred backbone conformation of the structure obtained from the restrained MD simulation (Table 4).83 In a very extended (30 ns) unrestrained MD simulation in explicit water this structure proved to be stable except for slight changes of four backbone dihedral angles in the range of 20° to 40° (Table 5). Indicators for a reliable structure can be seen in the predominantly parallel orientation of CO(i) to CαHα (i+1) bond vectors84 and in an overall high dispersion of backbone chemical shifts (Hα: 4.231 to 6.021 ppm (Figure 3), HMe: 1.559 to 3.115 ppm). In our hands a comparative attempt to discuss the spectral data of non-N-methylated MT-II by one single preferred conformation failed. There was considerable backbone dynamics as indicated by a high heterogeneity of the best 30 out of 50 MT-II conformers obtained from DG calculations (RMSD of the backbone carbon and nitrogen atoms of 1.26 Å). Moreover, a low dispersion of chemical shifts (Hα: 4.198 to 4.644 ppm (Figure 3), HN: 7.871 to 8.541 ppm), a lack of strong differentiation of all seven backbone HN temperature gradients (−8.61 to −5.24 ppb/K) and of amide proton exchange rates as estimated by ROESY exchange peaks, as well as a smaller preference of distinct sidechain conformations was observed. Altogether, these indicators suggest a more flexible peptide backbone of MT-II as compared to peptide 28.
Figure 2.

Molecular structure of compound 28
Table 3.
Resonance Assignment of Peptide 28 (Sodium acetate-d4 buffer (50 mM), pH 4.5, 298 K). Chemical Shifts are Referenced on Sodium 3-(trimethylsilyl)propionate-2,2,3,3-d4 (1H at 0.000 ppm)
| HN (HNMe) | Hα | Hβ | Hγ | Hδ | Hε | Hζ | Hη | others | |
|---|---|---|---|---|---|---|---|---|---|
| Nle2 | 8.166 | 4.231 | 1.653 1.701 |
1.281 | 1.320 | 0.893 | - | - | HAcetyl: 2.062 |
| Asp3 | 8.328 | 5.183 | proR: 2.271 proS: 2.521 |
- | - | - | - | - | - |
| His4 | (3.115) | 5.392 | proR: 3.272 proS: 3.063 |
- | 2: 7.084 | 1: 8.563 | - | - | - |
| D-Phe5 | 7.657 | 4.668 | proR: 2.901 proS: 2.661 |
- | 7.152 | 7.327 | 7.320 | - | - |
| Arg6 | (1.559) | 5.164 | proR: 1.245 proS: 1.518 |
1.168 1.255 |
3.084 | 7.144 | - | - | - |
| Trp7 | (2.695) | 6.021 | proR: 3.287 proS: 3.216 |
- | 1: 7.249 | 1: 10.180 3: 7.639 |
2: 7. 535 3: 7.150 |
2: 7.321 | - |
| Lys8 | (2.774) | 5.238 | 1.914 1.914 |
1.146 1.291 |
1.470 1.594 |
3.046 3.530 |
8.174 | - | HAmide: 7.1457.575 |
Table 4.
Comparison between the Experimentally Derived Distance Restraints (dlow), (dupp) and Calculated (drMD) Interproton Distances of Compound 28 as obtained from Restrained MD Calculation. Violations of Upper Bounds (positive sign) and of Lower Bounds (negative sign) are Given in the Last Column (dviol)
| interproton distance | dlow [Å] | dupp [Å] | dMD [Å] | dviol [Å] | |
|---|---|---|---|---|---|
| Asp5 HβproR | Asp5 Hα | 2.32 | 2.84 | 2.629 | |
| Asp5 HβproS | Asp5 Hα | 2.58 | 3.15 | 3.050 | |
| Asp5 Hα | NMe-His6 HMe | 2.08 | 3.19 | 2.758 | |
| Asp5 HβproR | NMe-His6 Hα | 4.13 | 5.05 | 4.635 | |
| Asp5 HβproR | NMe-His6 HMe | 2.52 | 3.68 | 3.286 | |
| Asp5 HβproS | NMe-His6 Hα | 4.73 | 5.78 | 5.248 | |
| Asp5 Hα | NMe-His6 Hα | 4.15 | 5.07 | 4.608 | |
| Asp5 HβproR | DPhe7 HN | 3.15 | 3.85 | 3.520 | |
| Asp5 HβproS | DPhe7 HN | 3.43 | 4.19 | 4.100 | |
| Asp5 HβproR | NMe-Trp9 HMe | 2.62 | 3.78 | 2.716 | |
| Asp5 Hα | NMe-Lys10 Hζ | 3.06 | 3.74 | 4.014 | +0.27 |
| Asp5 HβproR | NMe-Lys10 Hζ | 2.77 | 3.38 | 3.054 | |
| NMe-His6 HMe | NMe-His6 Hα | 3.21 | 4.43 | 3.913 | |
| NMe-His6 Hα | DPhe7 HN | 2.05 | 2.51 | 2.390 | |
| NMe-His6 Hβ | DPhe7 HN | 3.71 | 5.23 | 4.326 | |
| NMe-His6 HMe | DPhe7 HN | 3.4 | 4.64 | 4.334 | |
| NMe-His6 Hα | DPhe7 Hα | 3.78 | 4.62 | 4.500 | |
| NMe-His6 HMe | NMe-Trp9 HMe | 3.24 | 4.97 | 3.689 | |
| NMe-His6 HMe | NMe-Trp9 Hδ1 | 3.4 | 4.64 | 3.660 | |
| DPhe7 HN | DPhe7 Hα | 2.53 | 3.09 | 3.053 | |
| DPhe7 Hβ | DPhe7 HN | 2.32 | 3.53 | 3.187 | |
| DPhe7 HN | NMe-Arg8 Hα | 3.77 | 4.61 | 4.748 | +0.14 |
| DPhe7 Hα | NMe-Arg8 HMe | 2.11 | 3.22 | 2.679 | |
| DPhe7 Hβ | NMe-Arg8 Hβ | 3.08 | 5.16 | 4.752 | |
| DPhe7 Hβ | NMe-Arg8 HMe | 2.65 | 4.52 | 4.450 | |
| DPhe7 Hα | NMe-Trp9 Hε1 | 4.37 | 5.34 | 4.472 | |
| NMe-Arg8 HMe | NMe-Arg8 Hα | 3.17 | 4.39 | 3.872 | |
| NMe-Arg8 Hα | NMe-Trp9 HMe | 2.08 | 3.18 | 2.779 | |
| NMe-Arg8 Hβ | NMe-Trp9 Hα | 4.23 | 5.87 | 5.526 | |
| NMe-Arg8 HMe | NMe-Trp9 Hα | 3.98 | 5.29 | 5.034 | |
| NMe-Arg8 HMe | NMe-Trp9 HMe | 3.34 | 5.06 | 4.816 | |
| NMe-Arg8 Hα | NMe-Trp9 Hα | 3.75 | 4.59 | 4.579 | |
| NMe-Arg8 HMe | NMe-Trp9 Hδ1 | 3.3 | 4.53 | 4.760 | +0.23 |
| NMe-Arg8 HMe | NMe-Trp9 Hε1 | 3.27 | 4.5 | 4.184 | |
| NMe-Arg8 HMe | NMe-Trp9 Hε3 | 3.63 | 4.9 | 4.359 | |
| NMe-Arg8 HMe | NMe-Trp9 Hη2 | 3.6 | 4.86 | 4.123 | |
| NMe-Arg8 HMe | NMe-Trp9 Hζ2 | 3.45 | 4.7 | 3.806 | |
| NMe-Arg8 HMe | NMe-Trp9 Hζ3 | 2.89 | 4.08 | 4.327 | +0.25 |
| NMe-Arg8 Hα | NMe-Lys10 HMe | 3.32 | 4.56 | 4.472 | |
| NMe-Arg8 Hβ | NMe-Lys10 HMe | 3.26 | 5.19 | 5.333 | +0.14 |
| NMe-Trp9 HMe | NMe-Trp9 Hα | 3.15 | 4.37 | 3.905 | |
| NMe-Trp9 Hα | NMe-Lys10 HMe | 2.23 | 3.35 | 2.996 | |
| NMe-Trp9 HMe | NMe-Lys10 Hα | 3.56 | 4.83 | 4.889 | +0.06 |
| NMe-Trp9 HMe | NMe-Lys10 Hζ | 3.34 | 4.58 | 4.504 | |
| NMe-Trp9 HMe | NMe-Lys10 Hδ | 3.63 | 5.59 | 5.450 | |
| NMe-Trp9 HMe | NMe-Lys10 Hγ1 | 2.85 | 4.04 | 3.964 | |
| NMe-Trp9 HMe | NMe-Lys10 Hγ2 | 2.85 | 4.04 | 3.057 | |
| NMe-Trp9 Hα | NMe-Lys10 Hα | 3.78 | 4.62 | 4.620 | |
| NMe-Trp9 Hα | Amide11 HN | 3.95 | 5.52 | 5.457 | |
| NMe-Lys10 Hβ | NMe-Lys10 HMe | 2.31 | 4.14 | 3.617 | |
| NMe-Lys10 Hδ | NMe-Lys10 Hα | 2.57 | 3.84 | 4.258 | +0.42 |
| NMe-Lys10 Hδ | NMe-Lys10 Hζ | 2.83 | 4.16 | 3.447 | |
| NMe-Lys10 Hδ | NMe-Lys10 Hβ | 2.42 | 4.36 | 2.532 | |
| NMe-Lys10 Hδ1 | NMe-Lys10 HMe | 4.33 | 5.66 | 5.053 | |
| NMe-Lys10 Hδ2 | NMe-Lys10 HMe | 4.33 | 5.66 | 5.731 | +0.07 |
| NMe-Lys10 Hε | NMe-Lys10 Hγ | 2.99 | 5.06 | 3.218 | |
| NMe-Lys10 Hγ | NMe-Lys10 Hα | 2.57 | 3.85 | 2.597 | |
| NMe-Lys10 Hγ | NMe-Lys10 Hζ | 2.88 | 4.22 | 3.798 | |
| NMe-Lys10 Hγ | NMe-Lys10 HMe | 2.96 | 4.87 | 4.133 | |
| NMe-Lys10 HMe | NMe-Lys10 Hα | 3.22 | 4.44 | 3.897 | |
| NMe-Lys10 Hα | Amide11 HN | 2.87 | 4.21 | 3.349 | |
| NMe-Lys10 Hβ | Amide11 HN | 2.94 | 4.99 | 3.889 | |
| NMe-Lys10 HMe | Amide11 HN | 3.43 | 5.38 | 4.116 | |
Table 5.
Φ and Ψ Dihedral Angles of the Average Structure from the Restrained MD (rMD) and from the Trajectory of the Unrestrained MD (MD)
| Amino acid residue | ΦrMD [°] | ΦMD [°] | ΨrMD [°] | ΨMD [°] |
|---|---|---|---|---|
| Nle4 | −101 | −75.5 +/− 39.6 | 109 | 29.4 +/− 82.5 |
| Asp5 | 71 | −89.7 +/− 32.7 | 144 | 112.7 +/− 16.9 |
| NMe-His6 | −98 | −102.4 +/− 15.7 | 78 | 117.4 +/− 19.2 |
| DPhe7 | 96 | 75.0 +/− 18.8 | −126 | −116.0 +/− 10.1 |
| NMe-Arg8 | −135 | −122.9 +/− 8.0 | 80 | 83.2 +/− 9.9 |
| NMe-Trp9 | −120 | −136.6 +/− 14.0 | 63 | 96.0 +/− 12.7 |
| NMe-Lys10 | −114 | −120.7 +/− 9.9 | 0 | 83.0 +/− 63.4 |
Figure 3.

Hα regions of 1H NMR spectra of peptide 28 (A and B) and MT-II (C and D). Spectra A and C were detected in 50 mM sodium acetate-d4 D2O buffer (pH 4.5), B and D were detected in DMSO-d6. Numbers refer to Hα atoms of the respective residues in the a-MSH sequence.
Structure of the peptide backbone of 28
The conformation of compound 28 is shown in Figure 4. The ROE pattern demonstrates that all peptide bonds are trans configurated. As most of the amide bonds are N-methylated, turn-structures are not only defined by intramolecular hydrogen bonds.85 We think that steric effects and dipole orientation such as the above mentioned parallel orientation of CO(i) to CαHα (i+1) bond vectors84 contribute most strongly to the conformation of smaller N-methylated cyclic peptides.
Figure 4.
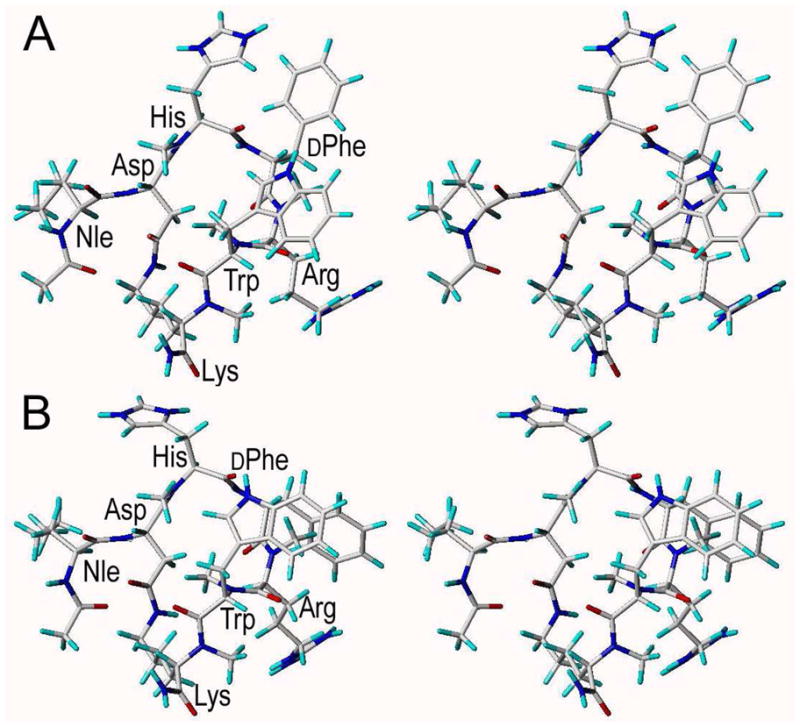
Stereoviews of the solution structure of peptide 28 as determined by NMR spectroscopy and MD calculations. The conformers shown in A and B possess the same cyclic core structure. The different side chain conformations of NMe-His6, DPhe7, and NMe-Arg8 in A and B, respectively, reflect dynamics in χ dihedral angles observed within the MD.
According to the NMe-His6 backbone dihedral angles (Φ=−98°, Ψ=78°) the structure obtained from restrained MD simulation possesses an inverted γ-turn85 centered at NMe-His6. A distance of 3.4 Å between the Asp5 carbonyl oxygen and the DPhe7 amide nitrogen, a hydrogen bond angle of 141° and a moderately negative temperature gradient of DPhe7 HN (−5.92 ppb/K in the aqueous buffer, −5.55 ppb/K in DMSO) indicate that the hydrogen bond within this inverted γ-turn is rather weak and protection form the solvent is incomplete. According to the minimal requirement for β-turns,86 which consists in a distance of less than 7 Å between Cαi and Cαi+3, the inverted γ-turn is located within a β-turn ranging from Asp5 to NMe-Arg8 (Cα-Cα distance: 6.4 Å). A distance of 7.6 Å between the α-carbon atoms of NMe-His6 and NMe-Trp9 almost fulfills the criterion for a second overlapping β-turn, which is close to type II’ β-turn geometry, as DPhe7 Φ, DPhe7 Ψ, NMe-Arg8 Φ, and NMe-Arg8 Ψ possess dihedral angles of 96°, −126°, −135° and 80°, respectively. The overlapping turns result in a virtually complete helical twist (α-turn) that extends from residues Asp5 to NMe-Trp9. Hydrophobic clustering of the NMe-Trp9 N-methyl group with the Asp5 Hβ, NMe-Arg8 Hα, NMe-Lys10 Hγ atoms and the NMe-His6 N-methyl group (indicated by the presence of ROESY cross peaks between the NMe-Trp9 methyl protons and the Asp5 Hβ, NMe-Arg8 Hα, NMe-Lys10 Hγ, NMe-His6 N-methyl protons) seems to stabilize this helical twist. Within the unrestrained 30 ns MD simulation starting from the structure of the restrained MD, slight changes occurred in a few backbone dihedral angles as compared to the average structure from the restrained MD simulation. Asp5 Ψ, NMe-His6 Ψ, DPhe7 Φ and NMe-Trp9 Ψ were most affected and changed from 144° to 113°, 78° to 117°, 96° to 75° and 63° to 96°, respectively (Table 5). The RMSD between the atoms of the peptide backbone from Asp5 to NMe-Lys10 of structures obtained from the unrestrained and restrained MD simulation is 1.0 Å. According to the NMe-His6 backbone dihedral angles (Φ=−102°, Ψ=117°) the inverted γ-turn is less pronounced in the structure obtained from unrestrained MD simulation. Upper bounds of some distance restraints within the cyclic core structure (Asp5Hβ - DPhe7HN; Asp5Hβ - NMe-His6HMe; DPhe7HN - NMe-Arg8Hα; NMe-Arg8Hα - NMe-Lys10HMe; NMe-Trp9HMe - NMe-Lys10Hα) were violated during the unrestrained MD simulation. This can be traced back directly to the aforementioned changes in backbone dihedral angles. As illustrated in more detail in the sections describing the side chain dynamics and structure calculations, we think that the changes in the backbone dihedral angles were caused by artificial strains in the amide linked Asp5 and NMe-Lys10 sidechains. These were introduced within the DG calculation as our structure calculation protocol did not take conformational averaging explicitly into account. Accordingly, the conformer obtained from restrained MD seems to be the best structural model for the peptide backbone and we focused the analysis of side chain conformation on this structure.
In consideration of the high binding affinity to hMC1R (IC50=14 nm) and the strong restriction that cyclization and fourfold N-methylation pose on conformational changes within the backbone of peptide 28, we think that its backbone conformation in aqueous solution is very close to the conformation present in the receptor bound state.
The conformation of the peptide backbone also offers an explanation for the strong interference of DPhe7 N-methylation with hMCR affinity, as N-methylation goes in hand with an increased spatial requirement and increased hydrophobicity in comparison to the replaced amide proton. If DPhe7 is N-methylated, these hydrophobic and steric effects would prohibit the formation of the backbone conformation present in peptide 28, as close proximity between the Asp5 carbonyl oxygen and the DPhe7 N-methyl group is disfavored. Hence, the NMe-DPhe7 substitution would not simply displace a hydrogen bond donor, but also lead to an altered conformation of the peptide backbone, which would affect the presentation of the pharmacophore and prevent interaction of the aromatic ring with the 3rd and the 6th transmembrane binding domains aromatic groups.76
Side chain structure and dynamics
Investigation of side chain conformation about the χ1 angle requires a careful analysis of homo- and heteronuclear J-couplings as well as the consideration of NOE distances in stereospecifically assigned β protons.87 Extended MD simulations of a solution structure in explicit solvent can further clarify which structural flips of side chain dihedral angles are correlated.
The sums of the 3JHα–Hβ coupling constants of the individual amino acid side chains are in the order of 15 Hz, which excludes higher populations of the χ1=60° conformation (gauche+) for the L amino acid residues, and of the χ1=−60° conformation (gauche-) for the DPhe7 residue (Table 6). A strong difference between the two 3JHα-Hβ coupling constants together with a sum of both of about 15 Hz indicate however a preferred (χ1=−60°) conformation for Asp5 (3JHα-HβproS = 10.7 Hz) and NMe-Trp9 (3JHα-HβproR = 11.1 Hz), whereas NMe-His6 and NMe-Arg8 with identical 3JHα-HβproR and 3JHα-HβproS coupling constants populate the (χ1=−60°) and (χ1=180°) conformations. For DPhe7 the difference of the 3JHα-Hβ coupling constants is small indicating a less pronounced preference of the χ1=60° over the χ1=180° rotamer. For NMe-Lys10 the 3JHα-Hβ coupling constants indicate populations of 70 to 30 % for the χ1=−60° and χ1=180° rotamers or vice versa. It is not clear, which of both is higher populated as the chemical shifts of the two β protons are degenerated. The NMe-Lys10 Hζ signal is slightly broadened to a singlet of 18 Hz linewidth which has weak shoulders (see supporting information) but lacks the expected double duplet or triplet structure expected from the coupling to the two δ-Protons when the NMR data is processed for signal intensity. Optimization of the linewidth by gaussian multiplication of the FID prior to Fourier transformation led to two similar 3JHε–Hζ coupling constants of 6.5 to 7.0 Hz. The similar 3JHα–Hβ coupling constants indicate an equilibrium of different conformers within the amide bridged Asp5 and NMe-Lys10 side chains. The slightly broadened NMe-Lys10 Hζ signal further suggests that the underlying conformers exchange on a timescale slower than the ns regime what prohibits their sampling by MD simulation in explicit water.
Table 6.
3JHα-Hβ Coupling Constants as Experimentally Determined from E.COSY. The According χ1 Populations were Derived by Linear Combination of 3JHα–Hβ(ap) =12 Hz, 3JHα–Hβ(ga)=3.5 Hz.
| 3JHα-Hβ [Hz] | p(χ1 =−60°) [%] | p(χ1 = 180°) [%] | p(χ1 = 60°) [%] | ||
|---|---|---|---|---|---|
| H&agr;–Hβ proR | Hα–Hβ proS | ||||
| Nle4 | 8.2; 6.2 | 84 | 16 | ||
| Asp5 | 3.8 | 10.7 | 74 | 11 | 15 |
| NMe-His6 | 7.9 | 7.9 | 48 | 48 | 4 |
| DPhe7 | 5.3 | 9.2 | 14 | 25 | 61 |
| NMe-Arg8 | 7.0 | 7.0 | 40 | 40 | 20 |
| NMe-Trp9 | 11.1 | 4.7 | 78 | 19 | 3 |
| NMe-Lys10 | 9.5, 6 Hz | 100 | 0 | ||
Dynamics of the side chains that are not involved in cyclization were further investigated by 30 ns molecular dynamics simulations in explicit water (Figure 5). For analyzing the side chain dynamics based on the structure obtained from the rMD calculation, position restraints were applied on the carbon and nitrogen atoms of the peptide backbone from Asp5 Cα to NMe-Lys10 Cα. Analysis of the MD simulation performed with such position restraints revealed that all χ1 populations except of the χ1 populations of NMe-Arg8 and of NMe-Lys10 were well consistent with the 3JHα-Hβ coupling constants (Table 6, Figure 5). The strong preference of χ1=−60° for Asp5 and for NMe-Trp9 as well as the evenly populated χ1=−60° and χ1=180° rotamers for Nle4 and NMe-His6 are indicated by 3JHα-Hβ coupling constants and well reproduced by the MD simulation. For DPhe7, the preference of the χ1=60° rotamer with respect to the χ1=180° rotamer, that is indicated by the 3JHα-Hβ coupling constants, is not reflected by the MD trajectory, which suggests similar populations of the two rotamers.
Figure 5.

χ dihedral angles as observed during the unrestrained 30 ns MD simulation. The column number (from the left to the right) corresponds to the χ dihedral angle positions within the sidechains.
As indicated by the MD simulation, by the ROEs and by the upfield or downfield chemical shifts, hydrophobic clustering is crucial for the different side chain conformations of peptide 28 in aqueous solution. Stacking of the NMe-His6 and DPhe7 side chains, that was also reported in other structural investigations of α-MSH analogs is well observed within the MD trajectory and is also present in the exemplary conformation shown in Figure 4A. Hydrophobic contacts between the DPhe7 and NMe-Arg8 side chains that agree with strong upfield shifts of the NMe-Arg8 β and γ protons are also observed within the MD trajectory and depicted in the conformation shown in Figure 4B. Overall side chain dynamics as indicated by 3JHα-Hβ coupling constants, is reflected by the rotamers shown for NMe-His6, DPhe7 and NMe-Arg8 in Figure 4A and 4B, respectively. The clustering of the NMe-Trp9 indolyl ring with the N-methyl group of NMe-Arg8, which is observed in Figures 4A and 4B, is confirmed by ROE contacts between methyl protons and all indolyl protons as well as by a strongly upfield shifted HMe resonance at 1.559 ppm. ROEs between the NMe-His6 methyl protons and the NMe-Trp9 δ1 and ε1 protons as well as between DPhe7 Hα and NMe-Trp9 Hε1 are well consistent with the orientation of the indolyl ring given in Figure 4A and 4B. An additional ROE between the NMe-His6 methyl protons and the NMe-Trp9 ε3 proton indicates another orientation of the indolyl group that was not sampled during 30 ns unrestrained MD simulation. In this orientation the indolyl group also seems to stack on top of the NMe-Arg8 N-methyl group as shown in Figure 4 but with the six membered ring of the indolyl group pointing to the left and the five membered ring pointing to the right.
A dispersion of chemical shifts of peptide 28 in DMSO-d6 (Hα: 4.245 to 5.824 ppm (Figure 3), HMe: 1.893 to 3.058 ppm), which is similar to the dispersion in the aqueous buffer (Ha: 4.231 to 6.021 ppm (Figure 3), HMe: 1.559 to 3.115 ppm), indicates that hydrophobic interactions observed in aqueous buffer are also present in the slightly more hydrophobic DMSO. This suggests that such hydrophobic interactions might also be present in hMC1R bound state.
In addition it is often found that stronger conformational preference which is accompanied by stronger biological activity indicates a closer similarity to the bioactive conformation. To our experience these effects seem to be stronger than calculated from population of conformations. Maybe this can be attributed to the importance of entropic effects in the binding process.
Comparison with structures of other peptidic GPCR effectors
According to our knowledge, no inverted γ-turn centered at His6 has been described so far in structures of similar, but not N-methylated α-MSH analogs, like MT-II. β-turns ranging from Asp5 to Arg8 have more frequently been described than β-turns ranging from His6 to Trp9 which agrees with the structure of 28 to such a degree as the distance between the Asp5 and NMe-Arg8 α carbon atoms (6.4 Å) is smaller than the distance between the NMe-His6 to NMe-Trp9 α carbon atoms (7.6 Å).88 In spite of certain similarities with respect to the occurrence of β-turns, the presentation of the side chains of residues NMe-Arg8 and NMe-Trp9 differs strongly from MT-II. A structure investigation of MT-II suggested that the Arg8 side chain is pointing up from the peptide plane and the Trp9 side chain is below the peptide plane, when the backbone is oriented as shown in Figure 4.88 In contrast, the NMe-Arg8 side chain of compound 28 is pointing down from the peptide plane and the NMe-Trp9 side chain of compound 28 is pointing up. This provides further evidence that positively charged Arg8 is critical for hMC3R and hMC4R binding and functional activities both for the ligand and for the receptor.76,89,90 Our earlier chimeric and multiple receptor mutation work demonstrated that the positive charged Arg8 plays an important role in binding to the 3rd transmembrane domain using the Asp122 and Asp126 residues of the hMC3R and hMC4R to achieve binding and functional activities. Multiple N-methylation increases the hydrophobicity of the ligand and further reduces the ability of the guanidyl group to interact with the Asp122, Asp126 to activate the hMCRs,76 leading to a loss in the binding and functional activity use. These strong structural differences suggest that the side chain presentation required for MCR activity varies strongly for the individual receptor subtypes. Inherent MT-II flexibility,88,89 seems to be high enough for the adaptation to the binding regions of the individual receptor subtypes, whereas 28 possesses a rigid peptide backbone that hinders the pharmacophore of MT-II from interacting properly with the binding pockets of the hMC3R, hMC4R and hMC5R therefore leading to hMC1R selectivity.
Backbone dihedral angles of 96°, −126°, −135° and 80° for DPhe7 Φ, DPhe7 Ψ, NMe-Arg8 Φ, and NMe-Arg8 Ψ, respectively, resemble type II’ β-turn positions i+1 and i+2. This directed our attention on obvious similarities between peptide 28 and potent somatostatin analogs,91–93 which possess a type II’ β-turn with an aromatic D amino acid residue in position i+1 and a positively charged L amino acid residue in position i+2.6 Both bind selectively to distinct GPCR subtypes of individual GPCR families (peptide 28 to hMC1R, somatostatin to sst2 and sst5 of the SRIF receptors). Hydrophobic interactions between aromatic side chains stabilize the turn structure of somatostatin analogs, while hydrophobic interactions of the NMe-Trp9 methyl group with Asp5 and NMe-Lys10 side chain protons are present in peptide 28. These similarities in peptide structure and receptor selectivity suggest further similarities between α-MSH analogs and somatostatin analogs with respect to their receptor subtype preference. For somatostatin the empirical correlation of Lys Hγ upfield shifts with the potency of the ligands have been extensively used to predict activities.93 Possibly, comparable correlations between Arg Hγ upfield shifts and potency could be found in α-MSH analogs as well.
DG calculations
DG calculations resulted in a very homogeneous ensemble of compound 28 conformers that were in good agreement with the distance restraints and very similar to the structure shown in Figure 4. The best 30 out of 50 calculated structures possessed an RMSD of 0.27 +/− 0.10 Å with respect to the backbone nitrogen and carbon atoms of residues Asp5 to NMe-Lys10. Although 3J coupling constants and broadening of the NMe-Lys10 Hζ signal indicated dynamics in the amide linked side chains, the structures obtained from DG possessed very similar homogeneous NMe-Lys10 χ1and χ2 dihedral angles (χ1= −55.2° +/− 2.01 Å, χ2= −109° +/− 3.73°). The observation of only one preferred χ1 dihedral angle did not reflect the 3JHα–Hβ coupling constants (Table 6) and the only populated eclipsed χ2 rotamer was thermodynamically unfavorable. Apparently, conformation averaging in the NMe-Lys10 side chain led to a number of conformation averaged distance restraints that forced the lactam bridged NMe-Lys10 side chain into conformations during DG calculations that could not be highly populated at equilibrium.
Restrained Molecular Dynamics calculations
The best DG structure in respect of violations of experimental distance restraints was subjected to refinement by restrained molecular dynamics (rMD) calculation in explicit water. The average structure of the rMD calculation was very similar to the DG starting structure (RMSD=0.69 Å with respect to backbone heavy atoms from Asp5 to NMe-Lys10). Within the cyclic core structure significant changes occurred only in the NMe-Lys10 side chain, that was strained in the starting conformer as side chain dihedral angles were eclipsed (χ2= −111° and χ3= −137°). As expected, the force field applied during restrained MD calculation rotated the dihedral angles within the NMe-Lys10 side chain to thermodynamically feasible values (χ2= −169° and χ3= 163°).
Unrestrained Molecular Dynamics calculations
The average structure obtained from the rMD calculation was further refined by extended (30 ns) unrestrained MD simulation in explicit water. This lead to significant changes of some backbone dihedral angles (Table 5) that were accompanied by further conformational changes in the lactam bridged NMe-Lys10 sidechain. The changes of backbone dihedral angles led to significant violations of distance restraints and most 3JHα–Hβ coupling constants were not consistent with the χ1 populations extracted from the MD trajectory. Consistency with 3JHα–Hβ coupling constants and with distance restraints was much higher in a trajectory of an MD simulation in which the conformation of the peptide backbone obtained from rMD was conserved by applying position restraints on all backbone carbon and nitrogen atoms from Asp5 Cα to NMe-Lys10 Cα.
We believe that the artificial strains in the lactam bridged side chains, that were described above, induced the conformational changes that were observed in the peptide backbone during 30 ns non-position restrained MD simulation. The slight variations in Φ and Ψ dihedral angles seemed to be energetically less demanding than flips between individual staggered conformations within the NMe-Lys10 side chain. Apparently, the backbone compensated for the strains within the lactam bridged side chains during the unrestrained MD simulation and position restraining of the backbone during the unrestrained MD simulation seems to be a legitimate way to avoid faulty changes of backbone dihedral angles.
Conclusions
Multiple N-methylation in MT-II led to several derivatives in which the affinity and activity for hMC1R could be retained, but simultaneously the binding affinity to the other 3 melanocortin receptors was completely lost. Hence, selectivity and retained activity was achieved in this way. A careful analysis of the conformation of the most active (13 nM) compound reveals a strong preference of one backbone conformation, which indicates a strongly reduced conformational space in comparison to the non-methylated MT-II. Hence, this work again shows that restriction of peptide backbone conformation resulted in strong receptor subtype selectivity with retained or even improved activity for an individual receptor subtype. This approach, discussed by us already a quarter of a century ago94,95 is not achievable only by cyclization but can be further increased by multiple N-methylation.48 With an increasing extent of introduction of N-methyl groups into its backbone, MT-II derivatives lose their ability to bind to particular hMCRs out of the total family of melanocortin receptors. This leads in two cases (peptides 24 and 28) to highly selective and potent agonists for the hMC1R, but also to antagonists and compounds with both, agonist and antagonist activities. These compounds possess multiple amide bonds that are N-methylated which was recently reported to be a possible key towards bioavailability5 and maybe also enhanced oral bioavailability6 of peptides. Whereas the effect of N-methylation on receptor subtype selectivity is strongly supported by our results, the effect on pharmacological properties that has been improved in other examples4–6 has to be demonstrated for the N-methylated MT-II derivatives. These investigations are in progress.
Experimental Section
Materials
Nα-Fmoc-amino acids, peptide coupling reagents, Rink amide MBHA resin and solvents were reagent grade and used without further purification unless otherwise noted. These chemicals were obtained from Aldrich, Novabiochem, Iris Biotech GmbH, Merck, NeoMPS, ORPEGEN Pharma and GLS. The following amino acids were used: Fmoc-Lys(Alloc)-OH, Fmoc-Trp(Boc)-OH, Fmoc-Arg(Pbf)-OH, Fmoc-DPhe-OH, Fmoc-His(Trt)-OH, Fmoc-Asp(OAllyl)-OH and Fmoc-Nle-OH. The polypropylene reaction vessels (syringes with frits) were purchased from B. Braun Melsungen AG. The purity of the peptides was checked by analytical reverse-phase HPLC using an Amersham Pharmacia Biotech Äkta Basic 10F with a ODS-A C18 (120 W, 5 Xm, 250 mm × 4.6 mm) column (Omnicrom YMC Europe GmbH) monitored at 220 and 254 nm and by high resolution mass spectral analysis.
Synthesis
The MT-II analogues were synthesized manually by the Fmoc-SPPS using methods described in refs. 96 and 97.
0.3 g Rink amide MBHA resin (4-(2′,4′-dimethoxyphenyl-Fmoc-aminomethyl)-phenoxyacetamido-norleucyl-MBHA resin) in a 10 mL syringe with a frit on the bottom was swollen in NMP (3 mL) for 10 min. The Fmoc protecting group on the Rink linker was removed by 20% piperidine in NMP (1 × 8 min, 1 × 12 min). The resin was washed with NMP (5 × 3 mL). The first Nα-Fmoc-amino acid was coupled using preactivated ester (3 eq. Nα-Fmoc-amino acid, 3 eq. TBTU, 3 eq. HOBt and 8 eq. DIEA) in NMP. The coupling mixture was transferred into the syringe with the resin and shaken for 3 h. Afterwards the resin was washed with NMP (5 × 3 mL). Complete coupling was confirmed by HPLC monitoring. Therefore the resin was washed with DCM (2 × 3 mL). A small amount of resin was taken out of the syringe and three drops of a mixture of trifluoracetic acid/triethylsilane/water (95:2.5:2.5, v/v/v) was added. After 10 min, acetonitrile and water were added, and after filtration, HPLC and ESI-MS showed complete coupling. The peptide sequences were continued by consecutively coupling of the particular amino acids. N-Methylation was carried out after coupling of the corresponding amino acid. This procedure includes three steps. First, the N-terminal Nα-Fmoc-deprotected amino acid was protected as an Nα-o-nitrobenzene sulfonamide by reaction with 5 eq. of o-nitrobenzenesulfonyl chloride (o-NBS) and 10 eq. of collidine in NMP (15 minutes). The methylation was done under Mitsunobu conditions98 with 5 eq. of triphenylphosphine, 10 eq. of methanol and 5 eq. of diisopropyl azodicarboxylate (DIAD) in THF (10 minutes). Then the o-NBS group was removed with 10 eq. of 2-mercaptoethanol and 5 eq. of DBU in NMP (5 minutes). This deproctection procedure was repeated once. For the coupling of the next amino acid, 3 eq. HATU, 3 eq. HOAt and 8 eq. DIEA were used. After coupling of the last amino acid Fmoc-Nle-OH and Fmoc deprotection as described above, acetylation was achieved by treatment of the DCM washed resin (3 × 3 mL) with acetic anhydride/DCM/DIEA (25:75:2.5, v/v/v, 3mL) for 30 min and washing of the resin with DCM (5 × 3 mL). The next step was the removal of the N3-Alloc group of Lys and the β-allyl group of Asp.96,97,99 For this, a solution of PhSiH3 (24 eq.) in 1 mL DCM was added to the peptide resins. Then a solution of Pd(PPh3)4 (0.25 eq.) in 2 mL DCM was added and the reaction allowed to proceed for 30 min. The peptide resins were washed with DCM (3 × 3 mL), NMP (2 × 3 mL), afterwards twice with 0.5% DIEA in DMF (v/v) and 0.5% sodium diethyldithiocarbamate trihydrate (DEDTC) in DMF (w/w) to remove any remaining traces of the Pd catalyst,100 followed by NMP (5 × 3 mL). The macrocyclic lactam ring formation was then mediated by addition of TBTU (3 eq.), HOBt (3 eq.) and DIEA (8 eq.) for 3 h. The cyclized peptides were cleaved off solid support using 3 mL trifluoracetic acid/triethylsilane/water (95:2.5:2.5, v/v/v, 1 × 3 h, 1 × 15 min), and the crude peptides were precipitated by dropping the solution in chilled diethyl ether to give white to light yellow precipitates. The resulting peptide suspensions were centrifuged for 5 min at 4000 rpm, and the liquid was decanted. The crude peptides were washed with chilled diethyl ether (2 × 30 mL), and after the final centrifugation, dried under vacuum over night. Purification was performed by HPLC using a ODS-A C18 (120 Ǻ, 5 μm, 250 mm × 20mm) column (Omnicrom YMC Europe GmbH) eluting with a linear gradient of acetonitrile and water containing 0.1% TFA. The products were obtained by lyophilisation of the appropriate fractions after removal of the acetonitrile by rotary evaporation. Analysis by analytical HPLC and HPLC-MS (Supporting Information) showed the peptides to be pure (>95%). The purified peptides were isolated in 2–14% overall yields.
Biological Activity Assays
Receptor Binding Assay
Competition binding experiments were carried out using whole HEK293 cells stably expressing human MC1, MC3, MC4, and MC5 receptors as described before.101–103 HEK293 cells transfected with hMCRs101–103 were seeded on 96-well plates 48 hours before assay (50,000 cells/well). For the assay, the cell culture medium was aspirated and the cells were washed once with a freshly prepared MEM buffer containing 100% minimum essential medium with Earle’s salt (MEM, GIBCO), and 25 mM sodium bicarbonate. Next, the cells were incubated for 40 min at 37°C with different concentrations of unlabeled peptide and labeled [125I]-[Nle4,DPhe7]-α-MSH (Perkin-Elmer Life Science, 20,000 cpm/well, 33.06 pM) diluted in a 125 μL of freshly prepared binding buffer containing 100% MEM, 25 mM HEPES (pH 7.4), 0.2% bovine serum albumin, 1 mM 1,10-phenanthrolone, 0.5 mg/L leupeptin, 200 mg/L bacitracin. The assay medium was subsequently removed, the cells were washed once with basic medium, and then lysed by the addition of 100 μL of 0.1M NaOH and 100 μL of 1% Triton X-100. The lysed cells were transferred to 12×75 mm borosilicate glass tubes, and the radioactivity was measured by a Wallac 1470 WIZARD Gamma Counter. The results are shown in Table 1.
Adenylate Cyclase Assay
HEK 293 cells transfected with human melanocortin receptors103 were grown to confluence in MEM medium (GIBCO) containing 10% fetal bovine serum, 100 units/mL penicillin and streptomycin, and 1 mM sodium pyruvate. The cells were seeded on 96-well plates 48 hours before assay (50,000 cells/well). For the assay, the cell culture medium was removed and the cells were rinsed with 100 μL of MEM buffer (GIBCO). An aliquot (100 μL) of the Earle’s balanced salt solution with 5 nM isobutylmethylxanthine (IBMX) was placed in each well along for 1 min at 37°C. Next, aliquots (25 μL) of melanotropin peptides of varying concentration were added, and the cells were incubated for 3 min at 37°C. The reaction was stopped by aspirating the assay buffer and adding 60 μL ice-cold Tris/EDTA buffer to each well, then placing the plates in a boiling water bath for 7 min. The cell lysates were then centrifuged for 10 min at 2,300 x g. A 50 μL aliquot of the supernatant was transferred to another 96-well plate and placed with 50 μL [3H] cAMP and 100 μL protein kinase A (PKA) buffer in an ice bath for 2–3 hours. The PKA buffer consisted of Tris/EDTA buffer with 60 μg/mL PKA and 0.1% bovine serum albumin by weight. The incubation mixture was filtered through 1.0 μm glass fiber filters in MultiScreen™-FB 96-well plates (Millipore, Billerica, MA). The total [3H] cAMP was measured by a Wallac MicroBeta TriLux 1450 LSC and Luminescence Counter (PerkinElmer Life Science, Boston, MA). The cAMP accumulation data for each peptide analogue was determined with the help of a cAMP standard curve generated by the same method as described above. IC50 and EC50 values represent the mean of two experiments performed in triplicate. IC50 and EC50 estimates and their associated standard errors were determined by fitting the data using a nonlinear least squares analysis, with the help of GraphPad Prism 4 (GraphPad Software, San Diego, CA). The maximal cAMP produced at 10 μM concentration of each ligand was compared to the amount of cAMP produced at 10 μM concentration of the standard agonist MT-II, and is expressed in per cent (as % max effect) in the Table 2. The antagonist properties of the lead compounds were evaluated by their ability to competitively displace the MT-II agonist in a dose-dependent manner, at up to 10 μM.
Data Analysis
IC50 and EC50 values represent the mean of two experiments performed in triplicate. IC50 and EC50 estimates and their associated standard errors were determined by fitting the data using a nonlinear least squares analysis, with the help of GraphPad Prism 4 (GraphPad Software, San Diego, CA).
NMR Spectroscopy
DQF-COSY, E.COSY, TOCSY, ROESY, 13C-HMBC and 13C-HSQC spectra were recorded at 298 K on a Bruker DMX spectrometer operating at 600 MHz. A COLOC spectrum104 was recorded at 298 K on a Bruker Avance III spectrometer operating at 600 MHz. A phase sensitive HMBC and a reference HSQC spectrum with offset and rf-amplitude-compensated BEBOP105,106 and BIBOP pulses107,108 were detected at 298K on a Bruker Avance III spectrometer operating at 750 MHz.109,110 Samples were prepared in 50 mM Sodium acetate-d4 buffer (pH 4.5, 10% D2O, 0.05 % NaN3) at concentrations of 12-38 mM. This low pH was chosen to keep hydrogen exchange induced line broadening small and to enhance comparability with numerous earlier structural studies on MCR effectors that were performed under the same conditions. Sodium 3-(trimethylsilyl)propionate-2,2,3,3-d4 (1H at 0.000 ppm) was used as internal standard. Data were processed with Topspin 1.3 software from Bruker. The homo- and heteronuclear experiments DQF-COSY, E.COSY, TOCSY, ROESY, and magnitude mode 13C-HMBC were performed with a spectral width of 11 ppm for 1H and 180 ppm for 13C. Individual HSQC spectra covering aliphatic (13C offset = 35 ppm, spectral width = 50 ppm) and aromatic 13C resonances (13C offset = 120 ppm, spectral width = 30 ppm) were detected. The phase sensitive HMBC spectrum and the reference HSQC spectrum were detected with a spectral width of 9 ppm for 1H and 190 ppm for 13C. A COLOC spectrum was detected with spectral widths of 9.5 ppm for 1H and 190 ppm for 13C. The increments in t1 and t2 were adjusted to the information extracted from the individual experiments, ranging from 384 to 2048 increments in t1 and from 4096 to 16384 complex data points in t2. Depending on the sample concentration and the individual experiments, 16 to 48 transients were averaged for each t1 value. A mixing time of 80 ms was used for TOCSY (spin lock field: 6 kHz; mixing sequence MLEV-17). Water signal suppression was achieved by WATERGATE techniques.111 The sequential assignment was obtained from heteronuclear J correlations that were extracted from HSQC and HMBC spectra. A compensated ROESY experiment, which was used for the extraction of inter proton distances, was performed with 150 ms mixing time and a spin lock field of 4000 kHz.112 The volume integrals of the individually assigned cross-peaks were compensated for offset effects and converted into distance constraints using the isolated spin pair approximation.113 In order to compensate for watergate solvent signal suppression artifacts, of any two cross peaks representing an internuclear distance, the peak with the higher water resonance offset in the direct dimension was preferably considered for distance calculations. The ROESY cross-peak volumes were calibrated against the distance (1.78 Å) between the prochiral NMe-Lys10 Hε protons. Upper and lower distance limits were set to plus and minus 10% of the calculated distances, respectively. For distance restraints referring to non-stereospecifically assigned methylene protons, another 0.7 Å was added on upper bounds as pseudoatoms and multiplicity corrections. For distance restraints referring to methyl protons, subtraction of 10% for the lower bounds was omitted and another 0.9 Å was added on upper bounds as pseudoatoms and multiplicity corrections. 18 intraresidual, 36 sequential interresidual and 9 non-sequential interresidual ROE derived distance restraints were used for structure calculations, with restraints between the lactam bridged residues Asp5 and NMe-Lys10 counted as sequential. 3JHN-Hα coupling constants were determined from 1D 1H NMR spectra, 3JHα-Hβ coupling constants from E.COSY and heteronuclear 3JC-H coupling constants from HMBC and reference HSQC spectra.109,110,114,115
Structure calculations for 28
The structural NMR refinement protocol included distance geometry (DG) calculations, energy minimizations, and molecular dynamics (MD) simulations. The DG program DISGEO was used to generate structures consistent with the 63 distance restraints derived from the ROEs.116,117 The DG procedure started with the embedding of 50 structures using random metrization. The structures obtained from DG were evaluated according to the lowest total restraint violations and additionally counterchecked for close contacts of protons that did not show corresponding ROESY crosspeaks. Among 30 structures that were in best accordance with experimentally derived restraints, all had identical peptide backbone conformations.
Restrained MD calculations (rMD) were carried out employing the module DISCOVER of the INSIGHT II 2001 program (Biosym/MSI Inc.) with the CVFF force field. The ROE restraints were included with a force constant of 10 kcal mol−1 Å−2. The calculations were done with the explicit-image model of periodic boundary conditions. The best structure resulting from the DG calculation was placed in a cubic box of length 30 Å and soaked with water. After energy minimization using steepest descent and conjugate gradient, the system was heated gradually starting from 10° K and increasing to 50, 100, 150, 200, 250 and 300° K in 1 ps steps, each by direct scaling of velocities. The system was equilibrated for 50 ps with temperature bath coupling. Coordinates were saved every 100 fs for another 150 ps. The average structure of the 150 ps rMD calculation was subjected to energy minimization and further investigated by unrestrained MD simulations.
The GROMACS 4.0 software package (www.gromacs.org)118–120 was used to perform unrestrained MD calculations. Visualization of the simulation trajectories was performed using the software packages VMD121 and SYBYL 8.0.122 The scripts g_cluster, g_dist and g_angle, that were used for analysis of the MD trajectory, were all packaged with GROMACS. The 53a6 united atom (CH, CH2 and CH3 groups represented as a single atom) forcefield, one of the GROMOS96 force fields,123 was used for the molecular dynamic simulations. Rigid SPC water which was constrained using SETTLE124 served as water model. Solute bonds were constrained by the SHAKE algorithm125 and temperature and pressure control was executed by Berendsen coupling.126 Periodic boundary conditions were employed on a octahedral simulation box, which was built with a distance of 1.4 nm for the solute. Cut off distances of 1.4 nm for electrostatic and Lennard-Jones non-bonding interactions were applied. Simulation time steps were set to 2 fs. Upon addition of two acetate counter ions and soaking of the box with water, the system was equilibrated by an initial minimization and subsequent 50 ps MD simulations at 50, 100, 150, 200, 250 and 298 K using position restraints. Within the individual MD steps, the temperature was gradually increased, while the force constants of the position restraints were decreased exponentially from 250000 KJmol−1nm−2 at 50° K to 25° KJmol−1nm−2 at 250° K. At 298 K no position restraints were applied. For adjacent pressure equilibration a 100 ps MD simulation was performed at 298 K. The final 30 ns MD simulation was carried out at 298° K. Coordinates were saved every 10 ps. For the MD simulation with strict conservation of the peptide backbone obtained from the restrained MD simulation, position restraints of 250000 KJmol−1nm−2 were applied on all backbone carbon and nitrogen atoms between Asp5 Cα and NMe-Lys10 Cα.
Supplementary Material
Acknowledgments
These studies were supported in parts by grants from the U.S. Public Health Service, National Institutes of Health, DK017420 and DA06284. We also thank the Humboldt Foundation for the support via the Max-Planck Award, the Deutschen Forschungsgemeinschaft (DFG) for financial support and Prof. Eberle in Basel for initial measurements of activities. JGB thanks Oliver Demmer for help with GROMACS and Dr. Burkhard Luy for help with HMBC and reference HSQC spectra.
Footnotes
Supporting Information Available: Description of the material included. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Victor J. Hruby, Email: hruby@email.arizona.edu.
Horst Kessler, Email: kessler@ch.tum.de.
References
- 1.Lindenberg H. J Prakt Chem. 1875;12:244–259. [Google Scholar]
- 2.Fischer E, Bergmann M. Liebigs Ann Chem. 1913;398:96–124. [Google Scholar]
- 3.Fischer E, Lipschitz W. Ber Dtsch Chem Ges. 1915;48:360–378. [Google Scholar]
- 4.Gilon C, Dechantsreiter MA, Burkhart F, Frieder A, Kessler H. Houben-Weyl Methods of Organic Chemistry. E22c. Georg Theime Verlag; Stuttgart: 2003. pp. 215–271. [Google Scholar]
- 5.Chatterjee J, Gilon C, Hoffman A, Kessler H. Acc Chem Res. 2008;41:1331–1342. doi: 10.1021/ar8000603. [DOI] [PubMed] [Google Scholar]
- 6.Biron E, Chatterjee J, Ovadia O, Langenegger D, Brueggen J, Hoyer D, Schmid HA, Jelinek R, Gilon C, Hoffman A, Kessler H. Angew Chem, Int Ed. 2008;47:2595–2599. doi: 10.1002/anie.200705797. [DOI] [PubMed] [Google Scholar]
- 7.Smith JM, Hows JM, Gordon-Smith EC. J Clin Pathol. 1983;36:41–43. doi: 10.1136/jcp.36.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sangalli L, Bortolotti A, Jiritano L, Bonati M. Drug Metab Dispos. 1988;16:749–753. [PubMed] [Google Scholar]
- 9.Al-Obeidi F, Hadley ME, Pettitt BM, Hruby VJ. J Am Chem Soc. 1989;111:3413–3416. [Google Scholar]
- 10.Al-Obeidi F, Castrucci AM, Hadley ME, Hruby VJ. J Med Chem. 1989;32:2555–2561. doi: 10.1021/jm00132a010. [DOI] [PubMed] [Google Scholar]
- 11.The Melanotropic Peptides. Ann N Y Acad Sci. 1993;680:1–687. [PubMed] [Google Scholar]
- 12.Chhajlani V, Wikberg JE. FEBS Lett. 1992;309:417–420. doi: 10.1016/0014-5793(92)80820-7. [DOI] [PubMed] [Google Scholar]
- 13.Chhajlani V, Muceniece R, Wikberg JE. Biochem Biophys Res Commun. 1993;195:866–873. doi: 10.1006/bbrc.1993.2125. [DOI] [PubMed] [Google Scholar]
- 14.Gantz I, Konda Y, Tashiro T, Shimoto Y, Miwa H, Munzert G, Watson SJ, DelValle J, Yamada T. J Biol Chem. 1993;268:8246–8250. [PubMed] [Google Scholar]
- 15.Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, DelValle J, Yamada T. J Biol Chem. 1993;268:15174–15179. [PubMed] [Google Scholar]
- 16.Gantz I, Shimoto Y, Konda Y, Miwa H, Dickinson CJ, Yamada T. Biochem Biophys Res Commun. 1994;200:1214–1220. doi: 10.1006/bbrc.1994.1580. [DOI] [PubMed] [Google Scholar]
- 17.Labbe O, Desarnaud F, Eggerickx D, Vassart G, Parmentier M. Biochemistry. 1994;33:4543–4549. doi: 10.1021/bi00181a015. [DOI] [PubMed] [Google Scholar]
- 18.The Melanocortin System. Ann N Y Acad Sci. 2003;994:1–367. provides the proceedings of a recent symposium discussing the latest developments in this area. [Google Scholar]
- 19.Hadley ME. The Melanotropin Peptides. CRC Press; Boca Raton, FL: 1989. [Google Scholar]
- 20.Eberle AN. The Melanotropins: Chemistry, Physiology and Mechanisms of Action. Karger; Basel: 1988. [Google Scholar]
- 21.Manna SK, Aggarwal BB. J Immunol. 1998;161:2873–2880. [PubMed] [Google Scholar]
- 22.Maaser C, Kannengiesser K, Specht C, Lugering A, Brzoska T, Luger TA, Domschke W, Kucharzik T. Gut. 2006;55:1415–1422. doi: 10.1136/gut.2005.083634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia Y, Wikberg JE. Cell Tissue Res. 1996;286:63–68. doi: 10.1007/s004410050675. [DOI] [PubMed] [Google Scholar]
- 24.Buckley DI, Ramachandran J. Proc Natl Acad Sci U S A. 1981;78:7431–7435. doi: 10.1073/pnas.78.12.7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Low MJ. J Endocrinol Invest. 2004;27:95–100. [PubMed] [Google Scholar]
- 26.Li SJ, Varga K, Archer P, Hruby VJ, Sharma SD, Kesterson RA, Cone RD, Kunos G. J Neuroscience. 1996;16:5182–5188. doi: 10.1523/JNEUROSCI.16-16-05182.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gantz I, Fong TM. Am J Physiol Endocrinol Metab. 2003;284:E468–474. doi: 10.1152/ajpendo.00434.2002. [DOI] [PubMed] [Google Scholar]
- 28.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 29.Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. N Engl J Med. 2003;348:1085–1095. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 30.Branson R, Potoczna N, Kral JG, Lentes KU, Hoehe MR, Horber FF. N Engl J Med. 2003;348:1096–1103. doi: 10.1056/NEJMoa021971. [DOI] [PubMed] [Google Scholar]
- 31.Wessells H, Gralnek D, Dorr R, Hruby VJ, Hadley ME, Levine N. Urology. 2000;56:641–646. doi: 10.1016/s0090-4295(00)00680-4. [DOI] [PubMed] [Google Scholar]
- 32.Wessells H, Levine N, Hadley ME, Dorr R, Hruby V. Int J Impot Res. 2000;12(Suppl 4):S74–79. doi: 10.1038/sj.ijir.3900582. [DOI] [PubMed] [Google Scholar]
- 33.King SH, Mayorov AV, Balse-Srinivasan P, Hruby VJ, Vanderah TW, Wessells H. Curr Top Med Chem. 2007;7:1098–1106. [PMC free article] [PubMed] [Google Scholar]
- 34.Eberle A, Hübscher W. Helv Chim Acta. 1979;62:2460–2483. [Google Scholar]
- 35.Eberle A, Schwyzer R. Helv Chim Acta. 1979;62:2452–2459. [Google Scholar]
- 36.Medzihradszky K. Recent Developments in the Chemistry of Natural Carbon Compounds. Hungarian Academy of Science; Budapest: 1976. pp. 207–250. [Google Scholar]
- 37.Schwyzer R, Eberle A. Frontiers of Hormone Research. 4. Karger; Basel: 1977. pp. 18–25. [PubMed] [Google Scholar]
- 38.Hruby VJ, Wilkes BC, Hadley ME, Al-Obeidi F, Sawyer TK, Staples DJ, de Vaux AE, Dym O, Castrucci AM, Hintz MF, Riehm JP, Rao KR. J Med Chem. 1987;30:2126–2130. doi: 10.1021/jm00394a033. [DOI] [PubMed] [Google Scholar]
- 39.Castrucci AM, Hadley ME, Sawyer TK, Wilkes BC, Al-Obeidi F, Staples DJ, de Vaux AE, Dym O, Hintz MF, Riehm JP, Rao KR, Hruby VJ. Gen Comp Endocrinol. 1989;73:157–163. doi: 10.1016/0016-6480(89)90066-x. [DOI] [PubMed] [Google Scholar]
- 40.Hruby VJ, Cai M, Grieco P, Han G, Kavarana M, Trivedi D. Ann N Y Acad Sci. 2003;994:12–20. doi: 10.1111/j.1749-6632.2003.tb03157.x. [DOI] [PubMed] [Google Scholar]
- 41.Hruby VJ, Cai M, Cain JP, Mayorov AV, Dedek MM, Trivedi D. Curr Top Med Chem. 2007;7:1107–1119. doi: 10.2174/156802607780906645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rajeswaran WG, Hocart SJ, Murphy WA, Taylor JE, Coy DH. J Med Chem. 2001;44:1416–1421. doi: 10.1021/jm000361p. [DOI] [PubMed] [Google Scholar]
- 43.Laufer R, Gilon C, Chorev M, Selinger Z. J Biol Chem. 1986;261:10257–10263. [PubMed] [Google Scholar]
- 44.Laufer R, Wormser U, Friedman ZY, Gilon C, Chorev M, Selinger Z. Proc Natl Acad Sci U S A. 1985;82:7444–7448. doi: 10.1073/pnas.82.21.7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawasaki AM, Knapp R, Wire W, Kramer T, Yamamura HI, Burks TF, Hruby VJ. Eleventh American Peptide Symposium; 1990. [Google Scholar]
- 46.Hruby VJ, Fang SA, Knapp R, Kazmierski W, Lui GK, Yamamura HI. Int J Pept Protein Res. 1990;35:566–573. doi: 10.1111/j.1399-3011.1990.tb00263.x. [DOI] [PubMed] [Google Scholar]
- 47.Knapp RJ, Vaughn LK, Fang SN, Bogert CL, Yamamura MS, Hruby VJ, Yamamura HI. J Pharmacol Exp Ther. 1990;255:1278–1286. [PubMed] [Google Scholar]
- 48.Chatterjee J, Ovadia O, Zahn G, Marinelli L, Hoffman A, Gilon C, Kessler H. J Med Chem. 2007;50:5878–5881. doi: 10.1021/jm701044r. [DOI] [PubMed] [Google Scholar]
- 49.Cody WL, He JX, Reily MD, Haleen SJ, Walker DM, Reyner EL, Stewart BH, Doherty AM. J Med Chem. 1997;40:2228–2240. doi: 10.1021/jm970161m. [DOI] [PubMed] [Google Scholar]
- 50.Haviv F, Fitzpatrick TD, Swenson RE, Nichols CJ, Mort NA, Bush EN, Diaz G, Bammert G, Nguyen A, Rhutasel NS, Nellans HN, Hoffman DJ, Johnson ES, Greer J. J Med Chem. 1993;36:363–369. doi: 10.1021/jm00055a007. [DOI] [PubMed] [Google Scholar]
- 51.Fairlie DP, Abbenante G, March DR. Curr Med Chem. 1995;2:654–686. [Google Scholar]
- 52.Tonelli AE. Biopolymers. 1976;15:1615–1622. doi: 10.1002/bip.1976.360150814. [DOI] [PubMed] [Google Scholar]
- 53.Manavalan P, Momany FA. Biopolymers. 1980;19:1943–1973. doi: 10.1002/bip.1980.360191103. [DOI] [PubMed] [Google Scholar]
- 54.Ron D, Gilon C, Hanani M, Vromen A, Selinger Z, Chorev M. J Med Chem. 1992;35:2806–2811. doi: 10.1021/jm00093a013. [DOI] [PubMed] [Google Scholar]
- 55.Dechantsreiter MA, Planker E, Mathä B, Lohof E, Hölzemann G, Jonczyk A, Goodman SL, Kessler H. J Med Chem. 1999;42:3033–3040. doi: 10.1021/jm970832g. [DOI] [PubMed] [Google Scholar]
- 56.Ali FE, Bennett DB, Calvo RR, Elliott JD, Hwang SM, Ku TW, Lago MA, Nichols AJ, Romoff TT, Shah DH, Vasko JA, Wong AS, Yellin TO, Yuan CK, Samanen JM. J Med Chem. 1994;37:769–780. doi: 10.1021/jm00032a009. [DOI] [PubMed] [Google Scholar]
- 57.Mazur RH, James PA, Tyner DA, Hallinan EA, Sanner JH, Schulze R. J Med Chem. 1980;23:758–763. doi: 10.1021/jm00181a012. [DOI] [PubMed] [Google Scholar]
- 58.Vitoux B, Aubry A, Cung MT, Marraud M. Int J Pept Protein Res. 1986;27:617–632. doi: 10.1111/j.1399-3011.1981.tb02016.x. [DOI] [PubMed] [Google Scholar]
- 59.Sugano H, Higaki K, Miyoshi M. Bull Chem Soc Jpn. 1973;46:226–230. [Google Scholar]
- 60.Aumailley M, Gurrath M, Müller G, Calvete J, Timpl R, Kessler H. FEBS Lett. 1991;291:50–54. doi: 10.1016/0014-5793(91)81101-d. [DOI] [PubMed] [Google Scholar]
- 61.Dechantsreiter MA, Mathä B, Jonczyk A, Goodman SL, Kessler H. Peptides 1996. Mayflower Scientific; Kingswinford: 1996. p. 329. [Google Scholar]
- 62.Shemyakin MM, Ovchinnkov YA, Ivanov VT, Kiryushkin AA. Tetrahedron. 1963;19:581–591. doi: 10.1016/s0040-4020(01)98545-x. [DOI] [PubMed] [Google Scholar]
- 63.Bevan K, Davies JS, Hall MJ, Hassall CH, Morton RB, Phillips DA, Ogihara Y, Thomas WA. Experientia. 1970;26:122–123. doi: 10.1007/BF01895528. [DOI] [PubMed] [Google Scholar]
- 64.Corbaz R, Ettlinger L, Gaumann E, Keller-Schierlein W, Kradolfer F, Neipp L, Prelog V. Helv Chim Acta. 1957;23:199. [Google Scholar]
- 65.Jolad SD, Hoffmann JJ, Torrance SJ, Wiedhopf RM, Cole JR, Arora SK, Bates RB, Gargiulo RL, Kriek GR. J Am Chem Soc. 1977;99:8040–8044. doi: 10.1021/ja00466a043. [DOI] [PubMed] [Google Scholar]
- 66.Pettit GR, Kamano Y, Dufresne C, Cerny RL, Herald CL, Schmidt JM. J Org Chem. 1989;54:6005. [Google Scholar]
- 67.Pettit GR, Kamano Y, Herald CL, Tuinman AA, Boettner FE, Kizu H, Schmidt JM, Baczynskyj L, Tomer KB, Bontems RJ. J Am Chem Soc. 1987;109:6883–6885. [Google Scholar]
- 68.Ruegger A, Kuhn M, Lichti H, Loosli HR, Huguenin R, Quiquerez C, von Wartburg A. Helv Chim Acta. 1976;59:1075. doi: 10.1002/hlca.19760590412. [DOI] [PubMed] [Google Scholar]
- 69.Biron E, Kessler H. J Org Chem. 2005;70:5183–5189. doi: 10.1021/jo050477z. [DOI] [PubMed] [Google Scholar]
- 70.Miller SC, Scanlan TS. J Am Chem Soc. 1997;119:2301–2302. [Google Scholar]
- 71.Biron E, Chatterjee J, Kessler H. J Pept Sci. 2006;12:213–219. doi: 10.1002/psc.711. [DOI] [PubMed] [Google Scholar]
- 72.Teixido M, Albericio F, Giralt E. J Pept Res. 2005;65:153–166. doi: 10.1111/j.1399-3011.2004.00213.x. [DOI] [PubMed] [Google Scholar]
- 73.Haskell-Luevano C, Miwa H, Dickinson C, Hruby VJ, Yamada T, Gantz I. Biochem Biophys Res Commun. 1994;204:1137–1142. doi: 10.1006/bbrc.1994.2581. [DOI] [PubMed] [Google Scholar]
- 74.Haskell-Luevano C, Sawyer TK, Hendrata S, North C, Panahinia L, Stum M, Staples DJ, Castrucci AM, Hadley MF, Hruby VJ. Peptides. 1996;17:995–1002. doi: 10.1016/0196-9781(96)00141-6. [DOI] [PubMed] [Google Scholar]
- 75.Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel EH, Heward CB, Burnett JB, Hadley ME. Proc Natl Acad Sci USA. 1980;77:5754–5758. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen M, Cai M, Aprahamian CJ, Georgeson KE, Hruby V, Harmon CM, Yang Y. J Biol Chem. 2007;282:21712–21719. doi: 10.1074/jbc.M702285200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grieco P, Cai M, Han G, Trivedi D, Campiglia P, Novellino E, Hruby VJ. Peptides 2007. 28:1191–1196. doi: 10.1016/j.peptides.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mogil JS, Wilson SG, Chesler EJ, Rankin AL, Nemmani KVS, Lariviere WR, Groce MK, Wallace MR, Kaplan L, Staud R, Ness TJ, Glover TL, Stankova M, Mayorov A, Hruby VJ, Grisel JE, Fillingim RB. Proc Natl Acad Sci USA. 2003;100:4867–4872. doi: 10.1073/pnas.0730053100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dorr RT, Dvorakova K, Brooks C, Lines R, Levine N, Schram K, Miketova P, Hruby VJ, Alberts DS. Protochem Photobiol. 2000;72:526–532. doi: 10.1562/0031-8655(2000)072<0526:ieeati>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 80.Hadley ME, Hruby VJ, Sharma SD, Dorr RT, Levine N. Ann New York Acad Sci. 1993;680:424–439. doi: 10.1111/j.1749-6632.1993.tb19700.x. [DOI] [PubMed] [Google Scholar]
- 81.Sharma SD, Jiang J, Hadley ME, Bentley DL, Hruby VJ. Proc Natl Acad Sci USA. 1996;93:13715–13720. doi: 10.1073/pnas.93.24.13715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiang J, Sharma SD, Fink JL, Hadley ME, Hruby VJ. Experimental Dermatol. 1996;5:325–333. doi: 10.1111/j.1600-0625.1996.tb00136.x. [DOI] [PubMed] [Google Scholar]
- 83.Kessler H, Griesinger C, Lautz J, Müller A, van Gunsteren WF, Berendsen HJC. J Am Chem Soc. 1988;110:3393–3396. [Google Scholar]
- 84.Heller M, Sukopp M, Tsomaia N, John M, Mierke DF, Reif B, Kessler H. J Am Chem Soc. 2006;128:13806–13814. doi: 10.1021/ja063174a. [DOI] [PubMed] [Google Scholar]
- 85.Matthews BW. Macromolecules. 1972;5:818–819. [Google Scholar]
- 86.Venkatachalam CM. Biopolymers. 1968;6:1425–1436. doi: 10.1002/bip.1968.360061006. [DOI] [PubMed] [Google Scholar]
- 87.Kessler H, Griesinger C, Wagner K. J Am Chem Soc. 1987;109:6927–6933. [Google Scholar]
- 88.Ying J, Kover KE, Gu X, Han G, Trivedi DB, Kavarana MJ, Hruby VJ. Biopolymers. 2003;71:696–716. doi: 10.1002/bip.10596. [DOI] [PubMed] [Google Scholar]
- 89.Frändberg P-A, Xu X, Chhajlani V. Biochem Biophy Res Commun. 1997;236:489–492. doi: 10.1006/bbrc.1997.6994. [DOI] [PubMed] [Google Scholar]
- 90.Grieco P, Balse PM, Weinberg D, MacNeil T, Hruby VJ. J Med Chem. 2000;43:4998–5002. doi: 10.1021/jm000211e. [DOI] [PubMed] [Google Scholar]
- 91.Veber DF, Holly FW, Paleveda WJ, Nutt RF, Bergstrand SJ, Torchiana M, Glitzer MS, Saperstein R, Hirschmann R. Proc Natl Acad Sci U S A. 1978;75:2636–2640. doi: 10.1073/pnas.75.6.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sukopp M, Schwab R, Marinelli L, Biron E, Heller M, Varkondi E, Pap A, Novellino E, Keri G, Kessler H. J Med Chem. 2005;48:2916–2926. doi: 10.1021/jm049500j. [DOI] [PubMed] [Google Scholar]
- 93.Arison BH, Hirschmann R, Veber DF. Bioorg Chem. 1978;7:447–451. [Google Scholar]
- 94.Kessler H. Angew Chem, Int Ed. 1982;21:512–523. [Google Scholar]
- 95.Hruby VJ. Life Sci. 1982;31:189–199. doi: 10.1016/0024-3205(82)90578-1. [DOI] [PubMed] [Google Scholar]
- 96.Mayorov AV, Han SY, Cai M, Hammer MR, Trivedi D, Hruby VJ. Chem Biol Drug Des. 2006;67:329–335. doi: 10.1111/j.1747-0285.2006.00383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Heckmann D, Kessler H. Methods Enzymol. 2007;426:463–503. doi: 10.1016/S0076-6879(07)26020-3. [DOI] [PubMed] [Google Scholar]
- 98.Mitsunobu O. Synthesis. 1981;1:1–28. [Google Scholar]
- 99.Thieriet N, Alsina J, Giralt E, Guibe F, Albericio F. Tetrahedron Lett. 1997;38:7275–7278. [Google Scholar]
- 100.Mayorov AV, Cai M, Chandler KB, Petrov RR, VanScoy AR, Yu Z, Tanaka DK, Trivedi D, Hruby VJ. J Med Chem. 2006;49:1946–1952. doi: 10.1021/jm0510326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cai M, Mayorov AV, Ying J, Stankova M, Trivedi D, Cabello C, Hruby VJ. Peptides. 2005;26:1481–1485. doi: 10.1016/j.peptides.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 102.Cai M, Mayorov AV, Cabello C, Stankova M, Trivedi D, Hruby VJ. J Med Chem. 2005;48:1839–1848. doi: 10.1021/jm049579s. [DOI] [PubMed] [Google Scholar]
- 103.Cai M, Cai C, Mayorov AV, Xiong C, Cabello CM, Soloshonok VA, Swift JR, Trivedi D, Hruby VJ. J Pept Res. 2004;63:116–131. doi: 10.1111/j.1399-3011.2003.00105.x. [DOI] [PubMed] [Google Scholar]
- 104.Kessler H, Griesinger C, Zarbock J, Loosli HR. J Magn Reson. 1984;57:331–336. [Google Scholar]
- 105.Skinner TE, Reiss TO, Luy B, Khaneja N, Glaser SJ. Journal of Magnetic Resonance. 2003;163:8–15. doi: 10.1016/s1090-7807(03)00153-8. [DOI] [PubMed] [Google Scholar]
- 106.Skinner TE, Reiss TO, Luy B, Khaneja N, Glaser SJ. Journal of Magnetic Resonance. 2004;167:68–74. doi: 10.1016/j.jmr.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 107.Kobzar K, Skinner TE, Khaneja N, Glaser SJ, Luy B. Journal of Magnetic Resonance. 2004;170:236–243. doi: 10.1016/j.jmr.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 108.Luy B, Kobzar K, Skinner TE, Khaneja N, Glaser SJ. Journal of Magnetic Resonance. 2005;176:179–186. doi: 10.1016/j.jmr.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 109.Verdier L, Sakhaii P, Zweckstetter M, Griesinger C. J Magn Reson. 2003;163:353–359. doi: 10.1016/s1090-7807(03)00063-6. [DOI] [PubMed] [Google Scholar]
- 110.Kobzar K, Luy B. J Magn Reson. 2007;186:131–141. doi: 10.1016/j.jmr.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 111.Piotto M, Saudek V, Sklenár V. J Biomol NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 112.Griesinger C, Ernst R. J Magn Reson. 1987;75:261–271. [Google Scholar]
- 113.Kumar A, Wagner G, Ernst R, Wuethrich K. J Am Chem Soc. 1981;103:3654–3658. [Google Scholar]
- 114.Eberstadt M, Gemmecker G, Mierke DF, Kessler H. Angew Chem, Int Ed. 1995;34:1671–1695. [Google Scholar]
- 115.Richardson JM, Titman JJ, Keeler J, Neuhaus D. J Magn Reson. 1991;93:533–553. [Google Scholar]
- 116.Mierke DF, Kessler H. Biopolymers. 1993;33:1003–1017. doi: 10.1002/bip.360330703. [DOI] [PubMed] [Google Scholar]
- 117.Havel TF. Prog Biophys Mol Biol. 1991;56:43–78. doi: 10.1016/0079-6107(91)90007-f. [DOI] [PubMed] [Google Scholar]
- 118.Lindahl E, Hess B, van der Spoel D. J Mol Model. 2001;7:306–317. [Google Scholar]
- 119.van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 120.van der Spoel D, Lindahl E, Hess B, van Buuren AR, Apol PJ, Meulenhoff PJ, Tieleman DP, Sijbers ALTM, Feenstra KA, van Drunen R, Berendsen HJC. Gromacs User Manual version 40. 2005. [Google Scholar]
- 121.Humphrey W, Dalke A, Schulten K. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 122.SYBYL 7.3. Tripos International; 1699 South Hanley Rd., St. Louis, Missouri, 63144, USA: [Google Scholar]
- 123.van Gunsteren WF, Billeter SR, Eising AA, Hunenberger PH, Krueger P, Mark AE, Scott WRP, Tironi IG. Biomolecular simulation: the GROMOS96 manual and user guide. 1. Hochschulverlag AG an der ETH Zürich; Zürich: 1996. [Google Scholar]
- 124.Miyamoto S, Kollman PA. J Comput Chem. 1992;13:952–962. [Google Scholar]
- 125.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 126.Berendsen HJC, Postma JPM, van Gunsteren WF, Dinola A, Haak JR. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.