

Abstract
Many protein functions can be directly linked to conformational changes. Inside cells, the equilibria and transition rates between different conformations may be affected by macromolecular crowding. We have recently developed a new approach for modeling crowding effects, which enables an atomistic representation of “test” proteins. Here this approach is applied to study how crowding affects the equilibria and transition rates between open and closed conformations of seven proteins: yeast protein disulfide isomerase (yPDI), adenylate kinase (AdK), orotidine phosphate decarboxylase (ODCase), Trp repressor (TrpR), hemoglobin, DNA β-glucosyltransferase, and Ap4A hydrolase. For each protein, molecular dynamics simulations of the open and closed states are separately run. Representative open and closed conformations are then used to calculate the crowding-induced changes in chemical potential for the two states. The difference in chemical-potential change between the two states finally predicts the effects of crowding on the population ratio of the two states. Crowding is found to reduce the open population to various extents. In the presence of crowders with a 15 Å radius and occupying 35% of volume, the open-to-closed population ratios of yPDI, AdK, ODCase and TrpR are reduced by 79%, 78%, 62% and 55%, respectively. The reductions for the remaining three proteins are 20–44%. As expected, the four proteins experiencing the stronger crowding effects are those with larger conformational changes between open and closed states (e.g., as measured by the change in radius of gyration). Larger proteins also tend to experience stronger crowding effects than smaller ones [e.g., comparing yPDI (480 residues) and TrpR (98 residues)]. The potentials of mean force along the open-closed reaction coordinate of apo and ligand-bound ODCase are altered by crowding, suggesting that transition rates are also affected. These quantitative results and qualitative trends will serve as valuable guides for expected crowding effects on protein conformation changes inside cells.
Author Summary
The biophysical properties of proteins inside cells can be expected to be quite different from those typically measured by in vitro experiments in dilute solutions. In particular, intracellular macromolecular crowding may significantly affect the equilibria and transition rates between different conformations of a protein, and hence its functions. What are the trends and magnitudes of such crowding effects? We address this question here by applying a recently developed approach for modeling crowding. Seven proteins, each with structures for both an open state and a closed state, are studied. Crowding exerts significant effects on the open-closed equilibria of four proteins and more modest effects on the remaining three. Potentials of mean force along the open-closed reaction coordinate, and hence transition rates, are similarly affected. The extent of conformational changes is the main determinant for the magnitudes of crowding effects, but the protein size also plays an important role. The effects of crowding become stronger as the protein size increases. Conformational transitions of the ribosome, an extremely large complex, during translation are predicted to experience particularly strong effects of intracellular crowding. We conclude that deduction of intracellular behaviors from in vitro experiments requires explicit consideration of crowding effects.
Introduction
It is increasingly recognized that protein dynamics serves the critical link between structure and function [1]–[6]. An important manifestation of protein dynamics is the sampling of alternative conformations. These conformational changes can be triggered by substrate (or ligand) binding [7] and post-translational modifications such as phosphorylation [8]. Increasingly, structures of the same proteins at different functional states are becoming available. These structures provide atomistic details of conformational changes. For example, adenylate kinase (AdK), an enzyme that catalyzes the phosphoryl transfer from ATP to AMP, undergoes a significant conformational transition, from an “open” conformation in the apo form and to a “closed” conformation in the ligand-bound form [9]–[11]. The main differences between these two conformations occur in the ATP- and AMP-binding domains, with the CORE domain relatively rigid (Figure 1). Other examples with well-characterized conformational transitions include proteins responsible for signal transduction across cell membranes [12]–[14] and ion channels [15].
Figure 1. Structures of AdK in the open and closed states.

(A) The substrate-free open state (PDB code 4AKE). (B) The closed state (PDB code 1AKE) in the presence of inhibitor Ap5A. The ATP-binding domain is in red, the AMP-binding domain is in green, the inhibitor is in yellow, and the CORE domain is in blue.
Biophysical characterizations of protein conformational changes have mostly been carried out under dilute conditions. However, the environments where proteins perform their biological functions, i.e., extracellular space, cell membrane, and cytoplasm, are crowded with macromolecules. For example, the cytoplasm of Escherichia coli contains about 300–400 g/l of macromolecules [16], which are estimated to occupy over 30% of the total volume. In cell membranes, membrane proteins occupy a similar level of the total surface area [17]. How the crowded cellular environments affect the equilibria and transition rates between different conformations of proteins is still poorly understood. Qualitatively, one expects that macromolecular crowding will significantly modify the energy landscapes of conformational changes, favoring more compact structures over more open ones [18]. Such effects of crowding have been scrutinized experimentally (see [18] for a recent review). Molecular dynamics simulations have also been carried out to investigate the energy landscapes of a number of proteins under crowding, in the context of either conformational change [19] or folding-unfolding transition [20]–[22]. To speed up conformational sampling, the proteins in these studies were represented at a coarse-grained level.
Recently we have developed an approach [23], [24], referred to as postprocessing, which opens the door to atomistic modeling of proteins under crowding. In this approach, the motions of a test protein are simulated in the absence of crowders. Conformations from this simulation are then used to calculate the change in chemical potential if they are transferred to a crowded solution. The dependence of the change in chemical potential on reaction coordinates then captures the influence of crowding on the energy landscape. The postprocessing approach has been applied to study effects of crowding on protein folding and binding stability [23], [25], [26] and on the open-closed equilibrium of the HIV-1 protease dimer [24]. In the latter application it has been shown that the postprocessing approach yields results identical to those obtained from direct simulations of the protein in the presence of crowders [19], [24].
Here we apply the postprocessing approach to investigate the impact of macromolecular crowding on the open-closed equilibria of seven proteins: AdK [9], [10], yeast protein disulfide isomerase (yPDI) [27], [28], orotidine phosphate decarboxylase (ODCase; functioning as a dimer) [29], Trp repressor (TrpR) [30], hemoglobin (Hb) [31], DNA β-glucosyltransferase (BGT) [32], and Ap4A hydrolase (Ap4Aase) [33], [34]. The biological functions and subcellular locations of these proteins are listed in Table 1. We find crowding to reduce the open-to-closed population ratios to various extents; the potentials of mean force (PMFs) along the open-closed reaction coordinate of apo and ligand-bound ODCase, and hence the transition rates, are similarly affected. The biological implications of these results are discussed below.
Table 1. Biological functions and subcellular locations of seven proteins.
| Protein | Organism | Subcellular location | Biological Function | PDB code |
| AdK | Escherichia coli | cytoplasm | ATP-AMP transphosphorylase | 4AKE & 1AKE |
| yPDI | Saccharomyces cerevisiae | lumen of endoplasmic reticulum | catalyses rearrangement of protein disulfide bonds | 3BOA & 2B5E |
| ODCase | Saccharomyces cerevisiae | cytoplasm | converts orotidine mono- phosphate to uridine mono-phosphate, the last step in biosynthesis of pyrimidine nucleotides | 1DQW & 1DQX |
| TrpR | Escherichia coli | cytoplasm | regulates transcription in tryptophan biosynthesis | 1WRP & 3WRP |
| Hb | Homo sapiens | cytoplasm | oxygen transport | 2DN1 & 2DN2 |
| BGT | Enterobacteria phage T4 | cytoplasm of host cell | catalyzes transfer of glucose from uridine diphosphoglucose to hydroxymethylcytosine in DNA | 1JEJ & 1JG6 |
| Ap4Aase | Lupinus angustifolius | cytoplasm | catalyses cleavage of diadenosine tetraphosphate into ATP and AMP | 1F3Y & 1JKN |
Results/Discussion
The X-ray structures of AdK in the open and closed states are shown in Figure 1; comparison of open and closed structures for all the seven proteins is found in Figure S1. For each protein, molecular dynamics simulations of the open and closed states are separately run. Representative open and closed conformations are then used to calculate the crowding-induced changes in chemical potential for the two states. The difference in chemical-potential change between the two states finally predicts the effects of crowding on the population ratio of the two states.
We study a range of crowding conditions, as defined the radius, R c, and the volume fraction, φ, of the crowders. The value of R c ranges from 15 to 50 Å and the value of φ ranges from 5 to 35%. These ranges of crowder sizes and volume occupancies likely cover the conditions of cytoplasmic milieus [35]. Among the seven proteins studied, four are found to experience significant crowding effects on the open-to-closed population ratios while the remaining three experiencing more modest effects.
Proteins experiencing significant crowding effects
Relatively strong effects of crowding are found on the open-to-closed population ratios of four proteins: AdK, yPDI, ODCase, and TrpR. In Figure 2 we show the relative changes in the open-to-closed population ratios of the four proteins by 15-Å crowders at various volume fractions. Corresponding results for 30-Å crowders are shown in Figure S2.
Figure 2. Effects of crowding on the open-to-closed population ratios of seven proteins.

The crowder radius is 15 Å.
We describe the calculation results on AdK in some details. Previous computational and experimental studies have determined the pathways and rates of the open-closed transitions for this protein [36]–[39]. In dilute solutions, the open-to-closed population ratio is ∼3 in the ligand-free form and ∼1/5 in the ligand-bound form [36], [39]. In the presence of 15-Å crowders occupying 35% of volume, the open-to-closed population ratio is reduced by 4.6-fold, or, equivalently, 78%, making the open state less stable than the closed one even in the ligand-free form. The reduction in the open-to-closed population ratio comes about because it is harder to accommodate the open conformations than the closed conformations in the crowded solution. The probabilities of successful placement for the open and closed states are 2.96×10−8 and 1.36×10−7, respectively (see Figure S3).
At a given volume fraction, the effects of crowding are larger for smaller crowders than for larger crowders. For example, at 35% volume fraction, crowding reduces the open-to-closed population ratio of AdK by 78%, 59%, 35%, and 17%, respectively, when the crowder radii are 15, 20, 30, and 50 Å. Comparison of the results for 15-Å crowders in Figure 2 and the counterparts for 30-Å crowders in Figure S2 shows the same trend. This dependence on crowder size has been seen previously in studies of protein folding, binding, and conformational change [23]–[26]. As we noted [23], only a small number of large crowders are needed to occupy the same volume as a large number of small crowders. Even though the “obstacles” amount to the same total volume, the former arrangement is more compact than the latter, and hence easier to accommodate a test protein. In other words, the latter arrangement is more discriminating between the open conformations and closed conformations of a protein, and therefore produces a stronger effect on the open-to-closed population ratio.
Qualitatively similar effects of crowding are found for the other three proteins. In the presence of 15-Å crowders occupying 35% of volume, the open-to-closed population ratios of yPDI, ODCase and TrpR are reduced by 79%, 62% and 55%, respectively. These are to be compared with the 78% reduction reported above. Clearly, the open-closed transitions of all these four proteins are prone to crowding effects expected of their intracellular environments.
Proteins experiencing modest crowding effects
As shown in Figure 2, the effects of crowding on the open-to-closed ratios of the other three proteins, Hb, BGT, and Ap4Aase, are modest. In the presence of 15-Å crowders occupying 35% of volume, the open-to-closed population ratios of these proteins are reduced by 44%, 39%, and 20%, respectively.
It should be noted that the modest effects on the open-to-closed population ratios come about not because crowding does not exert significant effects on the open or closed state, but rather because the effects exerted on the open and closed states are similar. For example, at R c = 15 Å and φ = 35%, the probabilities of successful placements for the open and closed states of Hb are 3.26×10−15 and 5.83×10−15, respectively. These values are seven orders of magnitude smaller than the corresponding quantities for AdK reported above (due to the much larger size of Hb). However, here the values for the open and closed states are very similar, leading to a modest effect on the open-to-closed population ratio.
Determinants for the magnitudes of crowding effects
What explains the large variations in crowding effects reported above? For a given protein, what matters is the difference in the effects exerted on the open state and the closed state. Therefore we expect the extent of conformational changes between the two states to be a main determinant. We use two quantities to measure the conformational changes: the solvent accessible surface area (SASA) and the radius of gyration, R g. In Table 2 we list the relative differences in SASA and in R g for the seven proteins. It is clear that the four proteins that experience significant crowding effects (AdK, yPDI, ODCase, and TrpR) have large differences in SASA and R g between the open and closed states. On the other hand, relatively small differences in SASA and R g are found for the remaining three proteins (Hb, BGT, and Ap4Aase), which experiences more modest crowding effects. In one extreme, in the case of AdK, both measures differ by over 10% between the open and the closed states, whereas in the other extreme, in the case of Hb, both measures differ by less than 2%. We may thus conclude that the extent of conformational changes is the main determinant for the magnitudes of crowding effects.
Table 2. Number of residues (N res) for each of seven proteins and solvent accessible surface area (SASA) and radius of gyration (R g) in either open or closed state.
| N res | SASA (Å2) | R g (Å) | |||||
| Open state | Closed State | Rel. Diff. (%) | Open state | Closed State | Rel. Diff. (%) | ||
| AdK | 214 | 12441 | 11179 | 11.3 | 19.1 | 16.7 | 14.1 |
| yPDI | 480 | 29080 | 27484 | 5.8 | 23.8 | 21.7 | 9.6 |
| ODCase | 534 | 22972 | 21544 | 6.0 | 24.6 | 23.8 | 3.4 |
| TrpR | 98 | 7795 | 7211 | 8.1 | 14.9 | 14.0 | 6.6 |
| Hb | 574 | 24631 | 24522 | 0.4 | 23.7 | 23.3 | 1.7 |
| BGT | 351 | 17609 | 16918 | 4.1 | 22.5 | 21.8 | 2.9 |
| Ap4Aase | 165 | 9743 | 9712 | 0.3 | 16.4 | 15.8 | 3.5 |
N res is the total number of residues identical in the open and closed structures. For each protein except ODCase in either the open or closed state, averages of SASA and R g calculated over 100 representative conformations are shown; variance for each entry is <1.5% of the average. For ODCase, further averaging over four independent trajectories is taken.
We have emphasized the fact that, for a given protein, the effect of crowding depends on the size of the crowders (when the volume fraction is kept constant). Conversely, for the same crowder size and crowder volume fraction, it can be expected that larger proteins will experience stronger crowding effects. This expectation is borne out by our calculation results. In particular, yPDI and TrpR show similar differences in SASA and R g between their open and closed states but differ in size. yPDI has ∼5 times more residues than TrpR. Correspondingly the crowding effects experienced by yPDI is ∼2-fold higher than those by TrpR (Figure 2). Similarly the difference in crowding effects between Hb and Ap4Aase can be partly attributed to their size difference (574 vs. 165 residues).
Crowding effects on conformational transition rates
Crowding not only affects the equilibria of open and closed states but is also expected to affect their transition rates. We study the latter problem on ODCase. The open and closed structures of this protein were solved in the absence and presence of a transition-state analogue, 6-hydroxyuridine 5′-phosphate (BMP), respectively [29]. The two structures differ significantly in the distance between two loops around the active site (Figure S1C), which is ∼20 and 12.5 Å, respectively. The PMFs of the apo and BMP-bound forms along the loop distance are calculated in the absence of crowding and shown in Figure 3. In the apo form, the transition from the open state (at loop distance ∼20.5 Å) to the closed state (at loop distance ∼12.5 Å) encounters an energy barrier ∼6.7 k B T, where k B is Boltzmann's constant and T is the absolute temperature. In the BMP-bound form, the transition from the closed state to the open state encounters a series of barriers, the most significant of which is at ∼1.5 k B T.
Figure 3. Potentials of mean force along the open-closed reaction coordinate of ODCase in the absence and presence of crowding.

(A) The apo form. (B) The BMP-bound form.
Using the postprocessing approach we calculate the PMFs in the presence of crowding. The results for the crowding condition of R c = 15 Å and φ = 35% are also shown in Figure 3. It can be seen that the energy barrier for the open-to-closed transition in the apo form is lowered, by ∼0.5 k B T, which corresponds to a 65% increase in transition rate. On the other hand, the most significant energy barrier for the closed-to-open transition is raised, by ∼0.2 k B T, corresponding to a 22% decrease in transition rate. These effects of crowding on the energy barriers, while relatively moderate, are nevertheless statistically significant (see error bars in Figure 3).
Biological implications
Proteins dynamics provides the critical link between structure and function. In present study, we have investigated how the crowded environments inside cells might affect protein dynamics. Our results have important implications for the functions of the proteins studied here and for in vivo biological functions in general.
In catalyzing the phosphoryl transfer from ATP to AMP, AdK undergoes large conformational changes. In dilute solutions, the open conformation is favored in the apo form whereas the closed conformation is favored when the substrates are bound. However, open-closed transitions occur in both the apo form and the substrate-bound form [36], [39]. Moreover, the closed-to-open transition in the substrate-bound form is thought to limit the overall catalytic rate (under substrate saturation) [36]. Our calculations show that crowding will significantly shift the open-closed equilibrium toward the closed state, such that the closed state may become favored even in the apo form. Qualitatively it can be expected that, under crowding, the rate of the closed-to-open transition in the substrate-bound form, i.e., the putative rate-limiting step of the catalytic reaction, will be reduced. Intracellular crowding will thus have a significant impact on the catalysis activity of AdK. Future work on the transition state of the open-closed transitions will allow us to quantitatively assess the effects of crowding on the transition rates.
The case of ODCase is similar. Conformational changes, especially closure and opening of the loops around the active site, are essential for substrate binding and product dissociation. A recent kinetic study by Wood et al. [40] indicates that loop closure and opening are partially rate-limiting for k cat/K M and k cat, respectively, in dilute solutions. Our calculations show that crowding stabilizes the closed state. Furthermore, calculations of the PMFs along the loop distance show that crowding increases the rate of loop closure in the apo form and hence k cat/K M but decreases the rate of loop opening in the ligand-bound form and hence k cat. Interestingly, these predicted trends were actually observed by Wood et al. when the kinetic study was done in the presence of Ficoll, a common crowding agent.
The considerable conformational flexibility of yPDI, which is essential for the formation of the correct pattern of disulfide bonds in substrate proteins, has been demonstrated by two structures determined from crystals grown at two different temperatures, 4°C and 22°C [27], [28]. The protein consists of four domains, labeled as a, b, b′, and a′ (see Figure S1B). Both the a and a′ domains harbor an active site. The 4-°C structure is more compact, forming a twisted U shape with the b and b′ as the base and the a and a′ domains as the arms. The 22-°C structure is more open, with the a and a′ domains showing significant rearrangement relative to the b and b′ domains, resulting in a boat shape. The conformational flexibility may be important in affording yPDI the ability to accommodate substrate proteins with different sizes. It may also be important in promoting the activity of yPDI on each substrate. Indeed, with ether reduced or scrambled ribonuclease as substrate in dilute solutions, the activity of yPDI is reduced when the arrangement of the a and a′ domains relative to the b and b′ domains is locked in that of the twisted U shape by new disulfide bonds [28]. Moreover, the same restriction on domain rearrangement also reduces the activity of yPDI in vivo. Our calculations show that crowding favors the compact U conformation over the open boat conformation, thereby reducing the conformational flexibility. In addition, crowding promotes protein oligomerization [23], [25]. yPDI forms a weaker dimer in dilute solutions [28], but the propensity to dimerize may be significantly enhanced in the crowded environment of its cellular location, the lumen of the endoplasmic reticulum. (In a model for the dimer as present in the 22°C crystal structure, the a′ domain active site becomes buried whereas the a domain active site is still solvent exposed [28].) Dimer formation has indeed been detected in the endoplasmic reticulum [28]. yPDI exhibits a difference between in vitro and in vivo activities: the a′ domain active site is more potent than the a domain active site for a native substrate in vitro, but the reserve is true in vivo [41]. In vivo dimer formation suggests a plausible explanation for this difference. Intrinsically the a′ domain active site may be more potent, but in the endoplasmic reticulum yPDI may most exist as dimer, in which the a′ domain active site is buried and hence inaccessible to the substrate whereas the a domain active site is accessible to the substrate.
The transition between the R state and the T state is essential for Hb to carry out its function of transporting oxygen from the lungs to the tissues. The R state is slightly more “open” than the T state and has high oxygen affinity; the T state has low oxygen affinity. In red blood cells, Hb has concentrations ∼300 g/l, amounting to ∼35% of the cell volume. Modeling Hb molecules as spheres with a radius ∼30 Å, our calculations show that intracellular crowding leads to a modest 10% increase in the T-to-R population ratio and correspondingly a small decrease in the oxygen affinity of Hb. Given that the oxygen affinity of Hb is strongly regulated by pH, CO2, Cl−, and D-2,3-bisphosphoglycerate [42], the small decrease in oxygen affinity expected of intracellular crowding is probably not of physiological significance. We note in passing that a variant of normal Hb, sickle Hb, can polymerize when deoxygenated, leading to deformation of red blood cells, vascular occlusion, and anemia. It has been demonstrated that macromolecular crowding has a dramatic effect on sickle Hb polymerization [43].
Our calculations show that the extent of conformational changes is the main determinant for the magnitudes of crowding effects. The study here has focused on structured proteins. We note that intrinsically disordered proteins should undergo even greater conformational changes, and are thus expected to experience even stronger effects of macromolecular crowding. Indeed, the intrinsically disordered α-synuclein undergoes a temperature-induced collapsed-to-expanded transition in dilute solutions, but such a transition is prevented in a solution crowded by a bystander protein and in living Escherichia coli cells [44]. α-synuclein has a tendency to aggregate into fibrils, which is the underlying cause for Parkinson's disease; the delay time before fibrillation is considerably shortened by macromolecular crowding [45], [46].
Our calculations also show that protein size plays an important role in the magnitudes of crowding effects. Larger proteins are expected to experience stronger crowding effects. One of the largest systems in the Protein Data Bank (PDB) is that of the ribosome, which is the machinery for protein translation. During translation, the ribosome undergoes a series of conformational changes; the structures of the ribosome in many of these functional states have now been determined [47]. Our preliminary calculation indicates that the relative stability between these states may be significantly changed by intracellular crowding. The largest change exerted by crowding in the relative stability between the open and closed states of the seven proteins studied here is 1.6 k B T. The same crowding condition favors the conformational change of the ribosome upon binding of a release factor by ∼5 k B T. The particularly strong crowding effects predicted for the conformational transitions of the ribosome perhaps partly explain why protein translation in an in vitro setting [48] is not as efficient as in vivo [49].
In conclusion, the preceding discussion makes it clear that intracellular crowding will significantly affect conformational changes and biological functions of proteins and molecular machines. Consequently, deduction of intracellular behaviors from in vitro experiments requires explicit consideration of crowding effects.
Methods
Postprocessing approach for modeling crowding
We briefly summarize our approach for atomistic modeling of crowding effects [23]. The aim here is to calculate the crowding-induced change in the free-energy difference, ΔΔG, between the open and closed states of a protein. If the change in chemical potential when the protein in the open state is transferred from a dilute solution to a crowded solution is Δμ o and the corresponding quantity in the closed state is Δμ c, then
As the measure of the effect of crowding, we report the relative change, κ, in the open-to-closed population ratio by crowding, given by
Here, like in previous molecular-dynamics studies of macromolecular crowding [19]–[22], we model the interactions between the test protein and crowders as hard-core repulsion. In that case, Δμ, the change in chemical potential in a given state due to crowding, is related to the probability, f, that the protein in that state can be successfully placed into a box of randomly distributed crowders:
Following previous studies [19]–[22], we further model the crowders as spherical particles. We have developed an algorithm for calculating f [23], hereafter referred to as the particle-insertion algorithm, since it is similar in spirit to Widom's particle-insertion method [50].
In our previous study of crowding effects on the open-closed equilibrium of the HIV-1 protease dimer [24], the aim was to demonstrate that the postprocessing approach yields results identical to those obtained from direct simulations of the protein in the presence of crowders [19]. The open and closed conformations in the absence of crowding, used for postprocessing, were obtained from a single simulation; we were able to simulate transitions between the two states because the protein was represented at a coarse-grained level. In the present study, we simulate the open state and closed state separately and thus avoid the slow transitions between them. (The separate simulations of end states follow our previous studies of protein folding and binding [23], [25], [26].) Postprocessing the conformations from the two separate simulations allows us to determine the effects of crowding on the open-to-closed population ratio.
The particle-insertion algorithm has been used to implement the postprocessing approach in all our previous studies [23]–[26]. More recently we have discovered that Δμ can be predicted theoretically [51], thus significantly speeding up the postprocessing approach. The gain in computational speed is especially important when the size of protein increases, whereupon the values of f become exceedingly small and hence difficult to obtain using the particle-insertion algorithm. In brief, the prediction of Δμ is based on generalizing the fundamental measure theory [52], which is designed for convex test particles and crowders, to atomistic proteins interacting with spherical crowders. The theory predicts Δμ as a linear function of the volume v p, surface area s p, and linear size l p of the test protein:
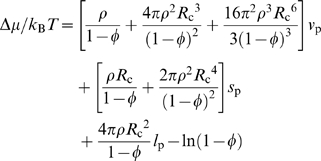 |
where ρ is the number density of the crowders. In the generalized fundamental measure theory (GFMT) [51], v p, s p, and l p are defined according to the so-called crowder-exclusion surface. This surface is similar to Richard's molecular surface [53], but with the probe radius set to the radius of the crowders. The average values of v p, s p, and l p calculated on the open and closed conformational ensembles of the seven proteins are listed in Table S1. We do not use these average values to predict the final Δμ result. Instead, we use the particular v p, s p, and l p for each conformation to calculate a Δμ value and then Boltzmann-average the individual Δμ values over the conformational ensemble to yield the final Δμ result. In Figure S3, we show that the GFMT predictions for Δμ o and Δμ c of AdK agree very well with the results obtained by the particle-insertion algorithm. Note that, for the crowding condition of R c = 15 Å and φ = 35%, convergent results could not be obtained by the particle-insertion algorithm. All the results reported in the main text are calculated by the GFMT.
Molecular dynamics simulations
For each protein, molecular dynamics simulations of the open and closed states are separately run using the Amber program. In each simulation, the protein molecule starts from the X-ray or NMR structure (with PDB code given in Table 1) and is solvated in TIP3P water molecules. The simulations are run at constant temperature (300 K) and constant pressure (1 bar), with the particle mesh Ewald method used to treat long-range electrostatic interactions. The total time of each simulation is 10 ns; 100 conformations are evenly selected from the last 8 ns as representatives of the conformational ensemble. Root-mean-square-deviations (RMSDs; measured on Cα atoms) of these representative conformations from the starting X-ray or NMR structure are typically ∼1.5 Å, but the open states of AdK and yPDI and the closed state of TrpR show higher RMSDs (see Figure S4).
To investigate how representative the conformational ensemble from a single trajectory is of a protein in a given state, four independent trajectories each of ODCase in the open state and the closed state are run. Crowding effects calculated on the four independent ensembles are similar. For example, the probabilities of successful placement for ODCase in the closed state calculated on the four ensembles are (1.89±0.34)×10−14 under the crowding condition of R c = 15 Å and φ = 35%.
For some proteins, there are small differences in the residues present in the starting structures of the open and closed states. Such differences would make an artificial contribution to ΔΔG; we eliminate this artifact by keeping only the residues that are present in both the open and closed conformations in calculations of Δμ.
Solvent accessible surface area
Solvent accessible surface area is calculated by using the NACCESS program [54], with a 1.4-Å probe radius.
Potentials of mean force
The PMFs along the open-closed reaction coordinate are calculated by umbrella sampling. The reaction coordinate, x, for ODCase is taken as the distance between the centers of mass of two loops, consisting of residues 151–161 and residues 203–218, respectively (Figure S1C). The umbrella sampling consists of 32 windows covering x values from 11.5 Å to 20.8 Å (with 0.3 Å increment). Harmonic restraints with force constants of 5 and 10 kcal/mol/Å2, respectively, are used for the apo form and the BMP-bound form, respectively. For the apo form, simulations in different restraint windows are independent; the simulation in each window consists of 0.2 ns of equilibration and 1.3 ns of production, with 1625 conformations used for PMF calculations. For the BMP-bound form, the simulations are carried out sequentially, with the conformation after 0.2 ns of equilibration in one window saved for the starting structure in the next window; 3500 subsequent conformations in the next 2.8 ns of simulation are used for PMF calculations.
The weighted histogram analysis method (WHAM) [55], [56] was used to obtain the PMFs. A PMF, W(x), is related to the probability density in x, P(x), via P(x) = exp[−W(x)/k B T]/C, where C is a normalization constant. In the standard WHAM, the probability density at discrete x values, xj, is obtained by iterating the following two equations to convergence:
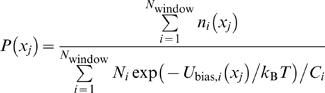 |
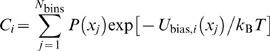 |
where i is the index for restraint windows; N window is the total number of such windows; U bias,i(x) is the bias potential in window i; Ni is the total number of sampled conformations from the window i simulation; Ci is a normalization constant for window i; N bin is the total number of x values for which P(x) is calculated; and ni(xj) is the number of conformations from the window i simulation which have x values belong to the xj bin. Errors are calculated by bootstrapping (http://membrane.urmc.rochester.edu).
From the conformations sampled in the absence of crowding, we use the postprocessing approach to obtain the PMF in the presence of crowding. Specifically, we assign each sampled conformation a statistical weight due to crowding; this statistical weight is f, the probability of successful placement into a box of crowders. Note that N i in the above equation can be viewed as the sum over a statistical weight of 1 for each conformation from the window i simulation; similarly ni(xj) can be viewed as a sum over a statistical weight of 1 for each conformation from the window i simulation with an x value in bin xj. For the PMF in the presence of crowding, Ni is replaced by the sum over the f values of the conformations from the window i simulation, and ni(xj) is replaced by the sum over the f values of the conformations from the window i simulation which have x values in bin xj.
ODCase functions as a homodimer but the two active sites are catalytically independent [57]. In our umbrella sampling, for each window the loop distances in the two subunits are restrained to the same value. For each sampled conformation, the actual loop distances of the two subunits were first averaged and then the result was used to find the corresponding bin xj. In addition, two independent sets of umbrella sampling simulations are carried out for both the apo form and the BMP-bound form; their averages are reported in Figure 3.
Supporting Information
Structural differences between open and closed states of seven proteins. (A) AdK. (B) yPDI. (C) ODCase. (D) TrpR. (E) Hb. (F) BGT. (G) Ap4Aase. For each protein, the open structure is in blue and the closed structure is in red; the two structures are superimposed on regions that exhibit relatively small changes. The PDB codes for these structures are listed in Table 1.
(3.02 MB TIF)
Effects of crowding on the open-to-closed population ratios of seven proteins. The crowder radius is 30 Å.
(0.54 MB TIF)
Comparison of GFMT predictions and results obtained by the particle-insertion algorithm. (A) Crowding-induced change, Δμo, in the chemical potential of AdK in the open state. (B) Corresponding quantity in the closed state. The crowder radius is 15 Å.
(0.52 MB TIF)
Root-mean-square-deviations of conformations during simulations from the starting X-ray or NMR structures. (A) AdK. (B) yPDI. (C) ODCase. (D) TrpR. (E) Hb. (F) BGT. (G) Ap4Aase. For each protein, Cα RMSDs of the open and closed states are displayed in blue and red, respectively. For ODCase, results averaged over four independent trajectories are shown.
(0.55 MB TIF)
Geometrical parameters of seven proteins for the prediction of Δμo and Δμc by the generalized fundamental measure theory
(0.07 MB DOC)
Footnotes
The authors have declared that no competing interests exist.
This work was supported in part by National Institutes of Health grant GM058187. The NIH had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Kohen A, Cannio R, Bartolucci S, Klinman JP. Enzyme dynamics and hydrogen tunnelling in a thermophilic alcohol dehydrogenase. Nature. 1999;399:496–499. doi: 10.1038/20981. [DOI] [PubMed] [Google Scholar]
- 2.Agarwal PK. Role of protein dynamics in reaction rate enhancement by enzymes. J Am Chem Soc. 2005;127:15248–15256. doi: 10.1021/ja055251s. [DOI] [PubMed] [Google Scholar]
- 3.Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, et al. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 4.Masgrau L, Roujeinikova A, Johannissen LO, Hothi P, Basran J, et al. Atomic description of an enzyme reaction dominated by proton tunneling. Science. 2006;312:237–241. doi: 10.1126/science.1126002. [DOI] [PubMed] [Google Scholar]
- 5.Wang L, Goodey NM, Benkovic SJ, Kohen A. Coordinated effects of distal mutations on environmentally coupled tunneling in dihydrofolate reductase. Proc Natl Acad Sci U S A. 2006;103:15753–15758. doi: 10.1073/pnas.0606976103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sytina OA, Heyes DJ, Hunter CN, Alexandre MT, van Stokkum IHM, et al. Conformational changes in an ultrafast light-driven enzyme determine catalytic activity. Nature. 2008;456:1001–1004. doi: 10.1038/nature07354. [DOI] [PubMed] [Google Scholar]
- 7.Qasba PK, Ramakrishnan B, Boeggeman E. Substrate-induced conformational changes in glycosyltransferases. Trends Biochem Sci. 2005;30:53–62. doi: 10.1016/j.tibs.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 8.Groban ES, Narayanan A, Jacobson MP. Conformational changes in protein loops and helices induced by post-translational phosphorylation. PLoS Comput Biol. 2006;2:238–250. doi: 10.1371/journal.pcbi.0020032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muller CW, Schulz GE. Structure of the complex between adenylate kinase from Escherichia coli and the inhibitor Ap5a refined at 1.9 Å resolution - a model for a catalytic transition-state. J Mol Biol. 1992;224:159–177. doi: 10.1016/0022-2836(92)90582-5. [DOI] [PubMed] [Google Scholar]
- 10.Muller CW, Schlauderer GJ, Reinstein J, Schulz GE. Adenylate kinase motions during catalysis: An energetic counterweight balancing substrate binding. Structure. 1996;4:147–156. doi: 10.1016/s0969-2126(96)00018-4. [DOI] [PubMed] [Google Scholar]
- 11.Aden J, Wolf-Watz M. NMR identification of transient complexes critical to adenylate kinase catalysis. J Am Chem Soc. 2007;129:14003–14012. doi: 10.1021/ja075055g. [DOI] [PubMed] [Google Scholar]
- 12.Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. 2000;69:373–398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- 13.Ferguson KM. Structure-based view of epidermal growth factor receptor regulation. Annu Rev Biophys. 2008;37:353–373. doi: 10.1146/annurev.biophys.37.032807.125829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tate CG, Schertler GF. Engineering G protein-coupled receptors to facilitate their structure determination. Curr Opin Struct Biol. 2009;19:386–395. doi: 10.1016/j.sbi.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 15.Zhou H-X, McCammon JA. The gates of ion channels and enzymes. Trends Biochem Sci. 2010;35:179–185. doi: 10.1016/j.tibs.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zimmerman SB, Trach SO. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J Mol Biol. 1991;222:599–620. doi: 10.1016/0022-2836(91)90499-v. [DOI] [PubMed] [Google Scholar]
- 17.Zhou H-X. Crowding effects of membrane proteins. J Phys Chem B. 2009;113:7995–8005. doi: 10.1021/jp8107446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou H-X, Rivas GN, Minton AP. Macromolecular crowding and confinement: Biochemical, biophysical, and potential physiological consequences. Annu Rev Biophys. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minh DDL, Chang CE, Trylska J, Tozzini V, McCammon JA. The influence of macromolecular crowding on HIV-1 protease internal dynamics. J Am Chem Soc. 2006;128:6006–6007. doi: 10.1021/ja060483s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheung MS, Klimov D, Thirumalai D. Molecular crowding enhances native state stability and refolding rates of globular proteins. Proc Natl Acad Sci U S A. 2005;102:4753–4758. doi: 10.1073/pnas.0409630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pincus DL, Thirumalai D. Crowding effects on the mechanical stability and unfolding pathways of ubiquitin. J Phys Chem B. 2009;113:359–368. doi: 10.1021/jp807755b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mittal J, Best RB. Dependence of protein folding stability and dynamics on the density and composition of macromolecular crowders. Biophys J. 2010;98:315–320. doi: 10.1016/j.bpj.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qin SB, Zhou H-X. Atomistic modeling of macromolecular crowding predicts modest increases in protein folding and binding stability. Biophys J. 2009;97:12–19. doi: 10.1016/j.bpj.2009.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin SB, Minh DDL, McCammon JA, Zhou H-X. Method to predict crowding effects by postprocessing molecular dynamics trajectories: application to the flap dynamics of HIV-1 protease. J Phys Chem Lett. 2010;1:107–110. doi: 10.1021/jz900023w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Batra J, Xu K, Qin SB, Zhou H-X. Effect of macromolecular crowding on protein binding stability: Modest stabilization and significant biological consequences. Biophys J. 2009;97:906–911. doi: 10.1016/j.bpj.2009.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tjong H, Zhou H-X. The folding transition-state ensemble of a four-helix bundle protein: helix propensity as a determinant and macromolecular crowding as a probe. Biophys J. 2010;98:2273–2280. doi: 10.1016/j.bpj.2010.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tian G, Xiang S, Noiva R, Lennarz WJ, Schindelin H. The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell. 2006;124:61–73. doi: 10.1016/j.cell.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 28.Tian G, Kober FX, Lewandrowski U, Sickmann A, Lennarz WJ, et al. The catalytic activity of protein-disulfide isomerase requires a conformationally flexible molecule. J Biol Chem. 2008;283:33630–33640. doi: 10.1074/jbc.M806026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller BG, Hassell AM, Wolfenden R, Milburn MV, Short SA. Anatomy of a proficient enzyme: The structure of orotidine 5′-monophosphate decarboxylase in the presence and absence of a potential transition state analog. Proc Natl Acad Sci U S A. 2000;97:2011–2016. doi: 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawson CL, Zhang RG, Schevitz RW, Otwinowski Z, Joachimiak A, et al. Flexibility of the DNA-binding domains of Trp repressor. Proteins. 1988;3:18–31. doi: 10.1002/prot.340030103. [DOI] [PubMed] [Google Scholar]
- 31.Park SY, Yokoyama T, Shibayama N, Shiro Y, Tame JRH. 1.25 angstrom resolution crystal structures of human haemoglobin in the oxy, deoxy and carbonmonoxy forms. J Mol Biol. 2006;360:690–701. doi: 10.1016/j.jmb.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 32.Morera S, Lariviere L, Kurzeck J, Aschke-Sonnenborn U, Freemont PS, et al. High resolution crystal structures of T4 phage beta-glucosyltransferase: Induced fit and effect of substrate and metal binding. J Mol Biol. 2001;311:569–577. doi: 10.1006/jmbi.2001.4905. [DOI] [PubMed] [Google Scholar]
- 33.Swarbrick JD, Bashtannyk T, Maksel D, Zhang XR, Blackburn GM, et al. The three-dimensional structure of the Nudix enzyme diadenosine tetraphosphate hydrolase from Lupinus angustifolius L. J Mol Biol. 2000;302:1165–1177. doi: 10.1006/jmbi.2000.4085. [DOI] [PubMed] [Google Scholar]
- 34.Fletcher JI, Swarbrick JD, Maksel D, Gayler KR, Gooley PR. The structure of Ap4A hydrolase complexed with ATP-MgFx reveals the basis of substrate binding. Structure. 2002;10:205–213. doi: 10.1016/s0969-2126(02)00696-2. [DOI] [PubMed] [Google Scholar]
- 35.Ridgway D, Broderick G, Lopez-Campistrous A, Ru'aini M, Winter P, et al. Coarse-grained molecular simulation of diffusion and reaction kinetics in a crowded virtual cytoplasm. Biophys J. 2008;94:3748–3759. doi: 10.1529/biophysj.107.116053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolf-Watz M, Thai V, Henzler-Wildman K, Hadjipavlou G, Eisenmesser EZ, et al. Linkage between dynamics and catalysis in a thermophilic-mesophilic enzyme pair. Nat Struct Mol Biol. 2004;11:945–949. doi: 10.1038/nsmb821. [DOI] [PubMed] [Google Scholar]
- 37.Maragakis P, Karplus M. Large amplitude conformational change in proteins explored with a plastic network model: Adenylate kinase. J Mol Biol. 2005;352:807–822. doi: 10.1016/j.jmb.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 38.Arora K, Brooks CL. Large-scale allosteric conformational transitions of adenylate kinase appear to involve a population-shift mechanism. Proc Natl Acad Sci U S A. 2007;104:18496–18501. doi: 10.1073/pnas.0706443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henzler-Wildman KA, Thai V, Lei M, Ott M, Wolf-Watz M, et al. Intrinsic motions along an enzymatic reaction trajectory. Nature. 2007;450:838–844. doi: 10.1038/nature06410. [DOI] [PubMed] [Google Scholar]
- 40.Wood BM, Chan KK, Amyes TL, Richard JP, Gerlt JA. Mechanism of the orotidine 5′-monophosphate decarboxylase-catalyzed reaction: effect of solvent viscosity on kinetic constants. Biochemistry. 2009;48:5510–5517. doi: 10.1021/bi9006226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westphal V, Darby NJ, Winther JR. Functional properties of the two redox-active sites in yeast protein disulphide isomerase in vitro and in vivo. J Mol Biol. 1999;286:1229–1239. doi: 10.1006/jmbi.1999.2560. [DOI] [PubMed] [Google Scholar]
- 42.Horvath SM, Malenfant A, Rossi F, Rossi-Bernardi L. The oxygen affinity of concentrated human hemoglobin solutions and human blood. Am J Hematol. 1977;2:343–354. doi: 10.1002/ajh.2830020404. [DOI] [PubMed] [Google Scholar]
- 43.Rotter M, Aprelev A, Adachi K, Ferrone FA. Molecular crowding limits the role of fetal hemoglobin in therapy for sickle cell disease. J Mol Biol. 2005;347:1015–1023. doi: 10.1016/j.jmb.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 44.McNulty BC, Young GB, Pielak GJ. Macromolecular crowding in the Escherichia coli periplasm maintains alpha-synuclein disorder. J Mol Biol. 2006;355:893–897. doi: 10.1016/j.jmb.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 45.Shtilerman MD, Ding TT, Lansbury PT., Jr Molecular crowding accelerates fibrillization of α-synuclein: could an increase in the cytoplasmic protein concentration induce Parkinson's disease? Biochemistry. 2002;41:3855–3860. doi: 10.1021/bi0120906. [DOI] [PubMed] [Google Scholar]
- 46.Uversky VN, Cooper EM, Bower KS, Li J, Fink AL. Accelerated α-synuclein fibrillation in crowded milieu. FEBS Lett. 2002;515:99–103. doi: 10.1016/s0014-5793(02)02446-8. [DOI] [PubMed] [Google Scholar]
- 47.Schmeing TM, Ramakrishnan V. What recent ribosome structures have revealed about the mechanism of translation. Nature. 2009;461:1234–1242. doi: 10.1038/nature08403. [DOI] [PubMed] [Google Scholar]
- 48.Johansson M, Lovmar M, Ehrenberg M. Rate and accuracy of bacterial protein synthesis revisited. Curr Opin Microbiol. 2008;11:141–147. doi: 10.1016/j.mib.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 49.Liang ST, Xu YC, Dennis P, Bremer H. mRNA composition and control of bacterial gene expression. J Bacteriol. 2000;182:3037–3044. doi: 10.1128/jb.182.11.3037-3044.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Widom B. Some topics in the theory of fluids. J Chem Phys. 1963;39:2802–2812. [Google Scholar]
- 51.Qin SB, Zhou H-X. Generalized fundamental measure theory for atomistic modeling of macromolecular crowding. Phys Rev E. 2010;81:031919. doi: 10.1103/PhysRevE.81.031919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oversteegen SM, Roth R. General methods for free-volume theory. J Chem Phys. 2005;122:214502. doi: 10.1063/1.1908765. [DOI] [PubMed] [Google Scholar]
- 53.Richards FM. Areas, volumes, packing and protein structure. Annu Rev Biophys Bioeng. 1977;6:151–176. doi: 10.1146/annurev.bb.06.060177.001055. [DOI] [PubMed] [Google Scholar]
- 54.Hubbard SJ, Thornton JM. ‘NACCESS’, Computer Program, Department of Biochemistry and Molecular Biology, University College London. 1993 [Google Scholar]
- 55.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. The weighted histogram analysis method for free-energy calculations on biomolecules.1. The method. J Comp Chem. 1992;13:1011–1021. [Google Scholar]
- 56.Roux B. The calculation of the potential of mean force using computer simulations. Comp Phys Commun. 1995;91:275–282. [Google Scholar]
- 57.Porter DJT, Short SA. Yeast Orotidine-5 ′-Phosphate decarboxylase: Steady-state and pre-steady-state analysis of the kinetic mechanism of substrate decarboxylation. Biochemistry. 2000;39:11788–11800. doi: 10.1021/bi001199v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Structural differences between open and closed states of seven proteins. (A) AdK. (B) yPDI. (C) ODCase. (D) TrpR. (E) Hb. (F) BGT. (G) Ap4Aase. For each protein, the open structure is in blue and the closed structure is in red; the two structures are superimposed on regions that exhibit relatively small changes. The PDB codes for these structures are listed in Table 1.
(3.02 MB TIF)
Effects of crowding on the open-to-closed population ratios of seven proteins. The crowder radius is 30 Å.
(0.54 MB TIF)
Comparison of GFMT predictions and results obtained by the particle-insertion algorithm. (A) Crowding-induced change, Δμo, in the chemical potential of AdK in the open state. (B) Corresponding quantity in the closed state. The crowder radius is 15 Å.
(0.52 MB TIF)
Root-mean-square-deviations of conformations during simulations from the starting X-ray or NMR structures. (A) AdK. (B) yPDI. (C) ODCase. (D) TrpR. (E) Hb. (F) BGT. (G) Ap4Aase. For each protein, Cα RMSDs of the open and closed states are displayed in blue and red, respectively. For ODCase, results averaged over four independent trajectories are shown.
(0.55 MB TIF)
Geometrical parameters of seven proteins for the prediction of Δμo and Δμc by the generalized fundamental measure theory
(0.07 MB DOC)