

Abstract
To create potentially superior aids to smoking cessation and/or antidepressants and to elucidate bupropion’s possible mechanisms of action(s), several analogues based on its active hydroxymetabolite (2S,3S)-4a were synthesized and tested for their abilities to inhibit monoamine uptake and nAChR subtype activities in vitro and acute effects of nicotine in vivo. The 3′,4′-dichlorophenyl [(±)-4n], naphthyl (4r), and 3-chlorophenyl or 3-propyl analogues 4s and 4t, respectively, had higher inhibitory potency and/or absolute selectivity than (2S,3S)-4a for inhibition of DA, NE, or 5HT uptake. The 3′-fluorophenyl, 3′-bromophenyl, and 4-biphenyl analogues 4c, 4d, and 4l, respectively, had higher potency for antagonism of α4β2-nAChR than (2S,3S)-4a. Several analogues also had higher potency than (2S,3S)-4a as antagonists of nicotine-mediated antinociception in the tail-flick assay. The results suggest that compounds acting via some combination of DA, NE, or 5HT inhibition and/or antagonism of α4β2-nAChR can potentially be new pharmacotherapeutics for treatment of nicotine dependence.
Keywords: Nicotine, bupropion, hydroxybupropion, structure activity relationship, dopamine uptake, norepinephrine uptake, nAChR antagonism, antinociception, locomotor activity, hypothermia
Introduction
Tobacco use is the leading preventable cause of disease, disability, and death in the United States (US). According to the Centers for Disease Control (CDC)a 2008 Smoking and Tobacco Use—Fact Sheet,1 cigarette smoking results in more than 400,000 premature deaths in the US each year—about 1 in every 5 deaths. On average, adults who smoke die 14 years earlier than nonsmokers.2 Cigarette smoking accounts for about one-third of all cancers, including 90% of lung cancer cases. Smoking also causes lung diseases such as chronic bronchitis and emphysema and increases the risk of stroke, heart attack, vascular disease, and aneurysm.2 In spite of these documented connections between tobacco use and disease, an unacceptable number of people continue to use tobacco products. In 2008, 28.6% of the US population 12 years of age and older (70.9 million people) used a tobacco product at least once in the prior month to being interviewed. This figure includes 3.1 million young people aged 12–17 (12.4% of this age group).3
Nicotine (1) is considered to be the main psychoactive component in tobacco smoke that causes and maintains tobacco use.4 Nicotine’s pharmacological and behavioral effects result from the activation of different nicotinic acetylcholine receptor (nAChR) subtypes. The subtypes are either homo or hetero pentameric ion channels, consisting of different combinations of genetically-distinct subunits, (α1, α2–α10, β1–β4, γ, δ, ε).5,6 The predominant nAChR subtypes found in the brain are thought to be heteromeric α4β2-nAChR or homomeric α7-nAChR.7 However, appreciable amounts of α3β4*- and α6β2*-nAChRs (where the * indicates that other subunits are known or possible assembly partners with those specified) also are in brain regions implicated in reward and drug dependence.8–13
Nicotine exposure can stimulate activity of somatodendritic nAChRs to alter neuronal electrical activity and neurotransmitter release as a consequence of neuronal activation. However, by acting at nAChRs positioned on nerve terminals, nicotine also can increase neurotransmitter release as a consequence of local depolarization of the nerve terminal membrane potential and/or calcium ion mobilization in terminals. The integration of these effects are likely to contribute to nicotine’s actions, including those that are presumably involved in its reinforcement of tobacco product use, such as effects in monoaminergic reward pathways.14,15
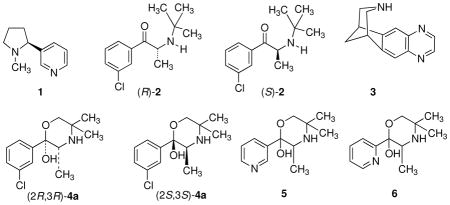
Even though nicotine dependence has a huge impact on global health, pharmacotherapies for treating tobacco use are limited. They include nicotine-replacement therapies (NRTs), bupropion (2), and varenicline (3). Since only about one-fifth of smokers are able to maintain long-term (12 months) abstinence with any of the present pharmacotherapies,16,17 new and improved drugs are needed.
Bupropion [(±)-2-tert-butylamino-3′-chloropropiophenone] is used clinically as a racemic mixture of its (R)- and (S)-isomers, (R)-bupropion [(R)-2] and (S)-bupropion [(S)-2], respectively. Bupropion is extensively metabolized with less than 1% recovered intact in urine.16 Major metabolites result from hydroxylation of the N-tert-butyl group by P450-(CYP)2B6 isoenzyme.17–19 The resulting hydroxylated metabolites cyclizes to give (2R,3R)- and (2S,3S)-hydroxybupropion [(2R,3R)-4a and (2S,3S)-4a, respectively]. Several studies suggest that (2S,3S)-hydroxybupropion [(2S,3S)-4a] contributes to the antidepressant and smoking cessation efficacy of 2.20,21 Peak plasma and cerebrospinal fluid concentrations of (2S,3S)-4a exceed those of 2 by 4- to 7-fold, and (2S,3S)-4a has a longer elimination half-life than the parent drug.22,23
In a previous study, we reported that (2S,3S)-4a was an inhibitor of both dopamine (DA) and norepinephrine (NE) uptake.21 Importantly, we found that (2S,3S)-4a was a noncompetitive functional antagonist at α4β2-nAChRs with an IC50 value of 3.3 μM, a concentration that is comparable to those needed to inhibit DA and NE uptake. In addition, (2S,3S)-4a was 3–10 times more potent than 2 after acute administration in mice in antagonizing nicotine-induced hypomobility and hypothermia and nicotine-induced analgesia in tail-flick and hot-plate tests. Compound (2S,3S)-4a also was equally potent with 2 in the mouse forced-swim test of depression. Because (2S,3S)-4a has higher potency at relevant targets compared to 2, it may be a better drug candidate for smoking cessation pharmacotherapy. Moreover, (2S,3S)-4a can serve as a lead structure to design analogues with greater potency at the relevant targets and to possess better drug-like properties.
In this study, we report the synthesis and biological evaluation of 23 hydroxybupropion analogues 4b–4v, 5 and 6 (all with the 2S,3S-stereochemistry except for 4n, 4p, 5, and 6, which are racemic). Some of the analogues have higher potencies than 2 for DA and NE uptake inhibition as well as for antagonism of the α4β2-nAChR. In addition, some of the compounds antagonize the antinociceptive, hypolocomotion, and hypothermic effects of acutely administered nicotine in mice with potencies greater than that of 2 or (2S,3S)-4a. 2-(3′-Bromophenyl)-3,5,5-trimethylmorpholin-2-ol (4d), which was 19-fold more potent in inhibition of DA uptake and 6-fold more potent as an α4β2*-nAChR antagonist than (2S,3S)-4a, has one of the more interesting in vitro properties.
Chemistry
Analogues 4a-g and 4q-t were synthesized in a fashion similar to that reported in the literature for optically active 4a starting with an aryl ketone (Scheme 1).24 The commercially unavailable propiophenones (8) were synthesized by Grignard additions to commercially available aryl nitriles (7). (Z)-tert-Butyldimethylsilylenol ether formation from these propiophenones, 8a–k, using t-buthyldimethylsilyl triflate in methylene chloride, gave high yields of the (Z)-enol ethers 9a–k. The key transformation in this sequence is a chiral Sharpless hydroxylation reaction of these enol ethers, which when using AD-mix-®, provided the (R)- α-hydroxy ketones 10a–k. The products of these reactions were not checked for optical purity but were found to be optically active, so optical induction was successful at some level. Due to possible epimerization throughout the process, it was decided to aminate the ketone before establishing optical purity. Initial efforts to reproduce the literature preparation of (2S,3S)-4a by converting (R)-1-(3-chlorophenyl)-2-hydroxypropan-1-one (10a) to the desired product [(2S,3S)-4a] using the literature conditions with 2-amino-2-methyl-1-propanol and 2,6-lutidine failed, or were low yielding. The presence of 2,6-lutidine overwhelmed silica gel chromatography making purification difficult. A modified approach was developed using proton sponge and provided (2S,3S)-4a in good yields. This modified procedure was used to synthesize compounds 4a–g and 4q–t.
Scheme 1.

a
aReagents: (a) RCH2MgBr; (b) TBSOTf, CH2Cl2, Et3N; (c) AD-mix-β, t-BuOH/H2O; (d) Tf2O, proton sponge; 2-amino-2-methyl-1-propanol, CH3CN.
Analogues 4n, 4p, 5, and 6 were synthesized as racemic mixtures by following the standard synthesis of bupropion analogues,25 except substituting 2-amino-2-methyl-1-propanol for t-butylamine, shown in Scheme 2. As illustrated in Scheme 2, the appropriate propiophenones (8l–o) were first synthesized by the addition of ethylmagnesium bromide to the nitriles (7), or in the case of the 3-pyridyl analogue was synthesized by lithium halogen exchange starting with 3-bromopyridine (11) and adding proprionitrile. Simple bromination to form the alpha-bromo ketones (12a–d) followed by amination with 2-amino-2-methyl-1-propanol provided the desired analogues in good yield. It should be noted that the optically active syntheses of 5 and 6 were attempted using the approach in Scheme 1, but the Sharpless reaction failed to provide the desired product.
Scheme 2.

a
aReagents: (a) EtMgBr; (b) nBuLi, CH3CH2CN; (c) Br2; (d) 2-amino-2-methyl-1-propanol, CH3CN.
The synthesis of 4h–m and 4o was accomplished by a novel convergent synthetic approach for the preparation of 2-substitued morpholinols as outlined in Scheme 3. This new approach utilized a nucleophilic addition of Grignard reagents to (3S)-3,5,5-trimethylmorpholin-2-one (15). Treatment of methyl (R)-(+)-lactate (13) with trifluoromethanesulfonic anhydride and 2,6-lutidine at 0 °C gave methyl (2R)-2-{[(trifluoromethyl)sulfonyl]oxy}propionate (14) in 77% yield. The alkylation of 2-amino-2-methyl-1-propanol with triflate 14 at −40 °C for 2 h and overnight at room temperature and subsequent cyclization afforded 15 in 63% yield. To test the approach, reaction of lactone 15 with 3-chlorophenylmagnesium bromide resulted in the formation of (2S,3S)-trimethyl-2-(3′-chlorophenyl)morpholin-2-ol [(2S,3S)-4a] in 32% yield (98% ee), 16% overall from (R)-(+)-lactate (13). The addition of the appropriate arylmagnesium bromide to 15 provided 4h–m and 4o.
Scheme 3.

a
aReagents: (a) Tf2O, 2,6-lutidine; (b) 2-amino-2-methyl-1-propanol, CH2Cl2, −40 °C to RT; (c) arylmagnesium bromide.
The C-3 stereocenter of these compounds was derived from the lactate, not created by a synthetic transformation such as the Sharpless hydroxylation used in Scheme 1. This center was then leveraged to create the second C-2 stereocenter. The resulting stereochemistry at C-2 was a result of either facial selectivity during the Grignard addition anti to the C-3 methyl group and/or a thermodynamic equilibrium of the final product to the S,S-configuration since the resulting product can ring open and close. The ring opened form loses its C-2 stereochemistry, forming a ketone. This route was more convergent than the Sharpless hydroxylation route, and was more reliable, requiring far less analytical work. All of the compounds could have been synthesized by the new route, but this was deemed inefficient since those compounds were already available for pharmacological testing.
N-Methylated compounds 4u and 4v were synthesized from their non-alkylated analogues (4a and 4i respectively) by reaction with methyl iodide in the presence of potassium carbonate (Scheme 4).
Scheme 4.

a
aReagents: (a) CH3I, K2CO3.
In Vitro Assays
The (2S,3S)-4a analogues 4b–4v and 5 and 6 were evaluated for their ability to block reuptake of [3H]dopamine ([3H]DA), [3H]serotonin ([3H]5HT), and [3H]norepinephrine ([3H]NE), respectively, into HEK293 cells stably expressing human DA transporters [(h)DAT], 5HT transporters [(h)SERT], or NE transporters [h(NET)] using methods similar to those previously reported.21,26 The results are given in Table 1.
Table 1.
Inhibition of monoamine uptake and nAChR function for hydroxybupropion analogs
 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compdc | R1 | R2 | X | Y | Z | monoamine uptake inhibitiona |
nAChR inhibitionb |
|||||
| IC50 (nM) | IC50 (μM) | |||||||||||
| [3H]DA | [3H]NE | [3H]5HT | α3β4* | α4β2 | α4β4 | α1*β1 | ||||||
| 660 ± 178 | 1850 ± 300 | IA | 1.8 (1.15) | 12 (1.15) | 12 (1.07) | 7.9 (1.12) | ||||||
| (2R,3R)-4a | CH3 | H | Cl | H | H | IA | 9900 ± 1400 | IA | 6.5 (1.20) | 31 (1.12) | 41 (1.07) | 7.6 (1.12) |
| (2S,3S)-4a | CH3 | H | Cl | H | H | 630 ± 50 | 241 ± 60 | IA | 11 (1.48) | 3.3 (1.07) | 30 (1.10) | 28 (1.45) |
| 4b | CH3 | H | H | H | H | 1065 ± 30 | 550 ± 90 | IA | 8.9 (1.23) | 6.4 (1.23) | 92 (1.29) | IA |
| 4c | CH3 | H | F | H | H | 1380 ± 360 | 740 ± 150 | IA | 15 (1.12) | 1.3 (1.17) | IA | IA |
| 4d | CH3 | H | Br | H | H | 3340 ± 680 | 920 ± 300 | IA | 3.2 (1.12) | 0.55 (1.23) | 30 (1.07) | 18 (1.07) |
| 4e | CH3 | H | CH3 | H | H | 2600 ± 400 | 1130 ± 20 | IA | 8.6 (1.12) | 6.0 (1.20) | 64 (1.20) | 33 (1.07) |
| 4f | CH3 | H | CH3O | H | H | 16,000 ± 2000 | 3000 ± 900 | IA | 11 (1.07) | 10 (1.26) | IA | 49 (1.07) |
| 4g | CH3 | H | NO2 | H | H | 12,000 ± 4000 | 1210 ± 340 | IA | 14 (1.10) | 4.8 (1.26) | 80 (1.10) | 96 (1.10) |
| 4h | CH3 | H | H | F | H | 4200 ± 700 | 3800 ± 600 | IA | 20 (1.15) | 12 (1.10) | IA | 69 (1.17) |
| 4i | CH3 | H | H | Cl | H | 285 ± 70 | 830 ± 90 | 4600 ± 900 | 5.1 (1.07) | 9.2 (1.17) | 33 (0.05) | 19 (1.15) |
| 4j | CH3 | H | H | CH3 | H | 832 ± 260 | 1680 ± 330 | IA | 8.6 (1.07) | 12 (1.12) | 62 (1.10) | 20 (1.15) |
| 4k | CH3 | H | H | CH3O | H | IA | IA | IA | 11 (1.10) | 27 (1.23) | 72 (1.12) | 25 (1.10) |
| 4l | CH3 | H | H | C6H5 | H | IA | 10,300 ± 1500 | IA | 1.3 (1.12) | 1.8 (1.12) | 8.1 (1.07) | 5.9 (1.15) |
| 4m | CH3 | H | F | F | H | 2140 ± 180 | 740 ± 110 | IA | 11.9 (1.15) | 12 (1.07) | IA | 36 (1.2) |
| 4nc | CH3 | H | Cl | Cl | H | 70 ± 20 | 114 ± 30 | 360 ± 40 | 2.6 (1.10) | 20 (1.07) | 14 (1.17) | 7.2 (1.12) |
| 4o | CH3 | H | F | H | F | 1020 ± 190 | 151 ± 43 | IA | 11 (1.15) | 6.3 (1.51) | 62 (1.07) | 23 (1.12) |
| 4pc | CH3 | H | Cl | H | Cl | 8250 ± 720 | 2440 ± 730 | IA | 3.9 (1.07) | 11 (1.05) | 18 (1.17) | 7.2 (1.07) |
| 4q | CH3 | H | 1-napthyl | — | 10,000 ± 4000 | 411 ± 53 | 1565 ± 215 | 5.2 (1.15) | 9.0 (1.07) | 13 (1.10) | 6.6 (1.10) | |
| 4r | CH3 | H | 2-napthyl | 453 ± 4 | 1570 ± 430 | 334 ± 42 | 2.0 (1.05) | 6.5 (1.07) | 11 (1.10) | 11 (1.12) | ||
| 4s | C2H5 | H | Cl | H | H | 204 ± 23 | 43.4 ± 72 | 2500 ± 540 | 4.3 (1.12) | 2.9 (1.10) | 16 (1.05) | 14 (1.10) |
| 4t | C3H7 | H | Cl | H | H | 30 ± 4 | 31 ± 10 | 4130 ± 770 | 4.8 (1.10) | 7.5 (1.05) | 18 (1.07) | 10 (1.07) |
| 4u | CH3 | CH3 | Cl | H | H | 3400 ± 600 | 415 ± 9 | IA | 6.5 (1.05) | 7.1 (1.07) | 43 (1.20) | 57 (1.05) |
| 4v | CH3 | CH3 | H | Cl | H | 2870 ± 820 | 527 ± 104 | 6480 ± 1280 | 4.6 (1.15) | 42 (1.17) | 91 (1.12) | 43 (1.05) |
| 5c | IA | IA | IA | IA | IA | IA | IA | |||||
| 6c | IA | 7950 ± 1800 | IA | IA | IA | IA | IA | |||||
Values for mean ± standard error of three independent experiments, each conducted with triplicate determination.
Mean micromolar IC50values (to two significant digits) for bupropion and the indicated analogs from three independent experiments for inhibition of functional responses to an EC80–EC90 concentration of carbamylcholine mediated by nAChR subtypes composed of the indicated subunits (where * indicates that additional subunits are or may be additional assembly partners with the subunits specified; see Methods and Materials). Numbers in parentheses indicate S.E.M. as a multiplication/division factor of the mean micromolar IC50 values shown [i.e., the value 1.8 (1.15) reflects a mean IC50 value of 1.8 μM with an S.E.M. range of 1.8 × 1.15 μM to 1.8/1.15 μM or 1.6–2.1 μM]. The value 11 (1.48) reflects a mean IC50 value of 11 μM with an S.E.M. range of 11 × 1.40 μM to 11/1.48 μM or 7.4–16 μM. IA: IC50 >100 μM.
Compounds 4b–4m, and 4o–4v are all (2S,3S)-isomers. Compounds 4n, 4p, 5, and 6 are racemic materials.
Compound (2S,3S)-4a and analogues 4b–4v and 5 and 6 also were evaluated for their ability to antagonize functional responses of α3β4*-, α4β2-, α4β4-, and α1*-nAChR using previously reported methods21 modified as described in Methods and Materials. Results are given in Table 1 and in Figures 1–2.
Figure 1.

Specific 86Rb+ efflux (ordinate; percentage of control) was determined for functional, human muscle-type α1β1γδ-nAChR (●), ganglionic α3β4*-nAChR (○), α4β2-nAChR (▲) or α4β4-nAChR (▽) naturally or heterologously expressed in human cell lines in the presence of a receptor subtype-specific, EC80-EC90 concentration of the full agonist, carbamylcholine, either alone or in the presence of the indicated concentrations (abscissa, log molar) of (2S,3S)-hydroxybupropion [(2S,3S)-4a] or its analogues (compounds 4d, 4c, and 4g) as indicated. Mean micromolar IC50 values and SEM as a multiplication/division factor of the mean micromolar IC50 value are provided in Table 1.
Figure 2.

Specific 86Rb+ efflux (ordinate; percentage of control) was determined for functional, human muscle-type α1β1γδ-nAChR (●), ganglionic α3β4*-nAChR (○), α4β2-nAChR (▲) or α4β4-nAChR (▽) naturally or heterologously expressed in human cell lines in the presence of a receptor subtype-specific, EC80–EC90 concentration of the full agonist, carbamylcholine, either alone or in the presence of the indicated concentrations (abscissa, log molar) of (2R,3R)-hydroxybupropion [(2R,3R)-4a] or analogues (compounds 4s, 4w, and 4t) as indicated. Mean micromolar IC50 values and SEM as a multiplication/division factor of the mean micromolar IC50 value are provided in Table 1.
In Vivo Assays
Compound (2S,3S)-4a analogues 4b–4v and 5 and 6 also were evaluated for their ability to antagonize behavioral responses to acute nicotine administration as previously described.21 Results are given in Table 2.
Table 2.
Pharmacological evaluation of hydroxybupropion analogs as non-competitive nicotinic antagonistsa
| Compdb | AD50 (mg/kg) |
|||
|---|---|---|---|---|
| Tail-flickc | Hot-platec | Locomotionc | Hypothermiac | |
| 2 | 1.2 (1–1.8) | 15 (6–19) | 4.9 (0.9–46) | 9.2 (4–23) |
| (2S,3S)-4a | 0.2 (0.1–1.1) | 1.0 (0.5–4.5) | 0.9 (0.38–3.7) | 1.5 (0.95–2.6) |
| (2R,3R)-4a | 2.5 (1.7–3.5) | 10.3 (8.9–15) | IA | IA |
| 4b | IA | IA | IA | IA |
| 4c | 0.012 (0.002–0.16) | 8.6 (0.7–10.5) | IA | 4.4 (1.3–14.5) |
| 4d | 0.16 (0.05–0.6) | 4.3 (1.8–9.8) | 2.6 (0.7–10.1) | 1.7 (0.5–6.8) |
| 4e | 0.054 (0.04–0.066) | 7.6 (2–29) | IA | 2.3 (0.4–11) |
| 4f | 5.85 (3.8–8.8) | IA | IA | IA |
| 4g | 4.9 (1.6–15) | IA | IA | 4.7 (3.5–6.2) |
| 4h | 0.013 (0.005–0.03) | IA | IA | 5.67 (2.5–12.8) |
| 4i | 0.019 (0.063–0.1) | IA | IA | IA |
| 4j | 0.004 (0.002–0.012) | IA | IA | IA |
| 4k | 0.019 (0.06–0.064) | IA | IA | IA |
| 4l | 0.021 (0.005–0.1) | IA | IA | IA |
| 4m | 0.006 (0.004–0.01) | IA | IA | IA |
| 4n | 8.8 (4.3–18) | IA | IA | IA |
| 4o | 0.0056 (0.004–0.009) | IA | IA | IA |
| 4p | IA | IA | 1.9 | IA |
| 4q | 0.034 (0.001–0.1) | IA | IA | IA |
| 4r | 0.04 (0.001–0.6) | IA | IA | IA |
| 4s | 0.004 (0.001–0.03) | 3.7 (0.8–17) | 10.3 (1.4–75) | 7 (4.5–10.8) |
| 4t | 0.004 (0.001–0.03) | IA | 4.7 | IA |
| 4u | 0.016 (0.004–0.06) | IA | IA | IA |
| 4v | 0.32 (0.04–2.5) | IA | IA | IA |
| 5 | n/a | n/a | n/a | n/a |
| 6 | n/a | n/a | n/a | n/a |
Results were expressed as AD50 (mg/kg) ± confidence limits (CL) or % effect at the highest dose tested. Dose-response curves were determined using a minimum of four different doses of test compound, and at least eight mice were used per dose group.
See footnote c in Table 1.
n/a: not assayed; IA: AD50 > 15 mg/kg; NFT = no further testing.
Results
Effects on Monoamine Uptake
Compound 2 inhibits DA reuptake (IC50 = 660 nM), which would increase synaptic levels of DA and presumed reward. Compound (2S,3S)-4a (IC50 = 630 nM), but not (2R,3R)-4a (IC50 > 10 μM), is as effective as 2 in inhibiting DA uptake inhibition (Table 1). Compound 2 also inhibits NE reuptake (IC50 = 1850 nM), which increases synaptic levels of NE. Interestingly, (2S,3S)-4a (IC50 = 241 nM), but not (2R,3R)-4a (IC50 = 9900 nM), is 7.7-times more potent than 2 in inhibiting NE uptake (Table 1). Neither 2 nor its hydroxymetabolites are active (IC50 > 10 μM; Table 1) as inhibitors of 5HT uptake.
Among the new hydroxybupropion analogues tested, the propyl extended chain form 4t and the (±)-3′,4′-dichlorophenyl derivative (±)-4n of (2S,3S)-4a with IC50 values of 30 and 70 nM, respectively, the ethyl extended chain form 4s (IC50 = 204 nM) and the 4-chlorophenyl analogue 4i (IC50 = 285 nM) have higher potency than (2S,3S)-4a (IC50 = 630 nM) as inhibitors of DA uptake.
In terms of activity for NE uptake inhibition, the ethyl and propyl extended chain forms, 4s and 4t (IC50 values of 43 nM and 31 nM, respectively), and the (±)-3′-,4′-dichlorophenyl derivative (±)-4n (IC50 = 114 nM), are more potent than (2S,3S)-4a (IC50 = 241 nM).
The only analogues that had sub-micromolar IC50 values for inhibition of 5HT uptake were the (±)-3′,4′-dichlorophenyl analogue (±)-4n (IC50 = 360 nM) and the 2-napthyl analogue 4r (IC50 = 334 nM). (2S,3S)-4a, (2R,3R)-4a and 16 of the analogues were inactive at the SERT. The remaining analogues had IC50 values greater than 1560 nM.
Compound 2 has ~3-fold selectivity for inhibition at DA over NE uptake and is inactive at inhibition of 5HT uptake. Neither (2R,3R)-4a nor (2S,3S)-4a shows activity for 5HT uptake. Compound (2R,3R)-4a’s poor activity overall precludes comments about its transporter selectivity. On the other hand, (2S,3S)-4a has the opposite DA/NE inhibition of uptake selectivity relative to 2, exhibiting ~3-fold selectivity for inhibition of uptake of NE over DA. Analogues 4b–4g, 4l–4m, 4o–4q, 4s, and 4u–4v share with (2S,3S)-4a selectivity for inhibition of NE over DA uptake inhibition. Relative to (2S,3S)-4a, 4s and 4o have the best combination of higher potency in NE uptake inhibition and selectivity for NE over DA uptake inhibition. The propyl-extended chain analogue, 4t, has much higher potency than (2S,3S)- or (2R,3R)-4a at each of the monoamine transporter targets, but it is essentially equipotent for NE and DA uptake inhibition (IC50 = 31 and 30 nM). The only analogues tested with selectivity for inhibition of DA over NE uptake [discounting the small preference for DA over NE and 5HT inhibition shown by (±)-3′,4′-dichlorophenyl analogue (±)-4n] are the 4′-chlorophenyl analogue 4i or 4′-methylphenyl analogues 4j and 2-napthyl analogue (4r) (3–5-fold). Only the 2-napthyl analogue 4r shows slight selectivity for the SERT (IC50 = 334 nM) over NET (IC50 = 1570 nM). However, by contrast to 4r, the structurally related 1-napthyl analogue (4q) has 24-fold selectivity for inhibition of NE over DA uptake. Thus, alkyl extension as well as phenyl substitution can impact inhibitory potency and selectivity of the hydroxybupropion analogues for monoamine transporters.
In Vitro Profiles for Drug Action at nAChR
Effects of hydroxybupropion analogues 4b–4v and 5 and 6 on function of diverse, human nAChR subtypes naturally or heterologously expressed by human cell lines were assessed using 86Rb+ efflux assays that are specific only for nAChR function in the cells used. None of the analogues has activity as an agonist at α1*-, α3β4*-, α4β2-, or α4β4-nAChR, because 86Rb+ efflux in the presence of these ligands alone at concentrations from ~5 nM to 100 μM (data not shown here) was indistinguishable from responses in cells exposed only to efflux buffer.
86Rb+ efflux assays also were used to assess whether ligands had activity as antagonists at human nAChR. Representative concentration-response curves for selected ligands (2S,3S)-4a, 4d, 4c, and 4g (Fig. 1) and (2R,3R)-4a, 4s, 4u, and 4t (Fig. 2) illustrate nAChR in vitro inhibitory profiles (see also Table 1). As we have shown previously,21 (2S,3S)-4a is a full antagonist at all of the nAChR subtypes tested and is selective (p<0.05) for α4β2- over α3β4*-, α4β4- and α1*-nAChR (upper left panel, Fig. 1). Compounds 4c, 4d and 4g also block all nAChR function at an adequately high concentration, 4c and 4d have higher potency (p<0.05) than (2S,3S)-4a at α4β2-nAChR, and all three compounds are more selective than (2S,3S)-4a for α4β2-nAChR over the other nAChR subtypes (Fig. 1). By contrast, (2R,3R)-4a, which also is a full antagonist at all of the nAChR subtypes tested, is selective (p<0.05) for α3β4*- and α1*-nAChR over α4β2- and α4β4-nAChR (upper left panel, Fig. 2). Compounds 4s and 4u have very similar potency as inhibitors of α4β2- and α3β4*-nAChR function and are selective for those over α1*- and α4β4-nAChR (fig. 2). Compound 4t is another full antagonist at all nAChR subtypes but with diminished selectivity for α3β4*-nAChR (Fig. 2). Note, more generally, that all of the ligands tested were full antagonists in that they blocked all nAChR function at the highest concentration tested or trended to do so if there still was sub-maximal block at 100 μM. Not shown here are other studies demonstrating that antagonism in all cases was mediated non-competitively, in that agonist concentration-ion flux response curves in the presence of ~IC50 concentrations of analogues showed diminished efficacy of agonist relative to response curves obtained in the absence of analogues and that agonist apparent EC50 values were unaffected by the presence of analogs.
As was previously noted,21 (2S,3S)-4a has an altered nAChR functional inhibitory profile relative to 2, showing higher potency at α4β2-nAChR (IC50 = 3.3 μM compared to 12 μM for 2) and higher selectivity for α4β2- over other nAChR (~3-fold) (Table 1).
Relative to (2R,3R)-4a and (2S,3S)-4a (IC50 values of 6.5 and 11 μM, respectively), the 3′-deschlorophenyl- analogue 4b, and the 3′-methylphenyl and 4′-methylphenyl- analogues 4e and 4j have comparable inhibitory potencies at α3β4*-nAChR (IC50 = 8.5–8.9 μM; Table 1). By contrast, there is slightly higher potency at α3β4*-nAChR for the 3′-bromophenyl 4d, 3′-chlorophenyl-4-(N-methyl) 4u, ethyl and propyl chain extended analogues 4s and 4t, 4′-chloro 4i, 3′,5′-dichlorophenyl 4p, 1-naphthyl 4q, and 4′-chloro-4-(N-methyl) 4v analogues (IC50 = 3.2–6.5 μM; p<0.05 for IC50 value differences between (2R,3R)-4a and 4d or 4p). Also having higher antagonist potencies (p<0.05) than (2R,3R)-4a at α3β4*-nAChR are the (±)-3′,4′-dichlorophenyl analogue, (±)-4n and the 2-naphthyl analogue 4r (IC50 values of 2.6 and 2.0 μM, respectively (Table 1). For these analogues, IC50 values for inhibition of α1*-nAChR function are higher (>33 μM for 4b, 4e, 4u, and 4v) or are in the range (5.9–19 μM for 4d, 4i–4j, 4l, (±)-4n, 4p–4t) of IC50 values for inhibition of α1*-nAChR by (2S,3S)-4a or (2R,3R)-4a (28 or 7.6 μM, respectively).
In absolute terms, the 3′-bromophenyl- (4d), 3′-fluorophenyl- (4c), biphenyl 4l, and ethyl extended chain analogue 4s have higher inhibitory potency at α4β2-nAChR than (2S,3S)-4a [IC50 values of 0.55, 1.3, 1.8 (all p<0.05) and 2.9 relative to 3.3 μM, respectively].
Of the analogues tested, 4v has the largest selectivity for α3β4*-nAChR over the other nAChR subtypes (Table 1). (2R,3R)-4a is barely selective for α3β4*-nAChR over α1*-nAChR, but about 5-fold selective for α3β4*- over α4β2-nAChR (Table 1). Of all the analogues tested, 4b–4e, 4g–4h, 4o and 4s have reasonable selectivity for α4β2-nAChR over other nAChR subtypes. Compounds 4f, 4m and 4u are about equipotent at α4β2- and α3β4*-nAChR. From another perspective, only the racemic 3′,4′-dicholoro (±)-4n (~8-fold) and 4′-chloro-4-(N-methyl) 4v (~9-fold) have higher selectivity for α3β4*- over α4β2-nAChR than (2R,3R)-4a (~5-fold) or 2 (~6.7-fold). The 3′-fluorophenyl 4c (~12-fold) and 3′-bromophenyl 4d (~6-fold) analogues have selectivity for α4β2-nAChR over other nAChR subtypes better than or comparable to that for (2S,3S)-4a (~3-fold).
Multiple Target Actions of Hydroxybupropion Analogues
We compared inhibitory potencies across transporters and nAChR relative to (2S,3S)-4a, which has ~3-fold selectivity for inhibition of NE over DA uptake inhibition and ~14-fold selectivity for inhibition of NE uptake inhibition over α4β2-nAChR function. Interestingly, there is a change to absolute selectivity for inhibition of α4β2-nAChR over DA uptake inhibition (~6-fold) and over NE uptake inhibition (~1.7-fold), and there is ~3.6-fold selectivity for inhibition of NE over DA uptake for 3′-bromophenyl 4d. There is an even more striking increase in selectivity for inhibition of α4β2-nAChR function over transporter inhibition for biphenyl analogue 4l (>5-fold over DA uptake inhibition and 6-fold over NE uptake inhibition). Selectivity for inhibition of NE uptake inhibition over α4β2-nAChR function also is reduced relative to that for (2S,3S)-4a for analogues 4c (~1.7-fold), 4h (~3-fold), 4f (~3.3-fold), 4g and 4r (~4-fold), 4e (~5.3-fold), and 4b (~12-fold), and for analogue 4p (~4.5-fold). Conversely, there is an increase in selectivity for inhibition of NE uptake over α4β2-nAChR function for 4t (~240-fold), (±)-4n (~175-fold), 4v (80-fold), 4s (~67-fold), and 4o (42-fold), although selectivity for inhibition of NE uptake over inhibition of α3β4*- instead of α4β2-nAChR function is less for 4t, (±)-4n, and 4v (~154-, 23-, and 9-fold, respectively; recall that 4t has comparable activity for DA and NE uptake inhibition). Although its selectivity for inhibition of DA over NE uptake is marginal, (±)-4n has ~37-fold selectivity for inhibition of DA uptake over α3β4*-nAChR and >285-fold selectivity for inhibition of DA uptake over α4β2-nAChR. The biphenyl analog 4l has 7.9-fold selectivity for inhibition of α3β4*-nAChR over inhibition of NE uptake, surpassing selectivity seen for (2R,3R)-4a (~1.5-fold).
In Vivo Effects of Analogues
Compound 2 blocks nicotine-induced increases in locomotor activity with an AD50 value of 4.9 mg/kg, while (2S,3S)-4a has a lower AD50 value of 0.9 mg/kg in the same assay, but none of the analogues is better than (2S,3S)-4a, although 4d and 4p (2.6 and 1.9 mg/kg AD50, respectively) have slightly higher potency than 2 (Table 2).
The ability of (2S,3S)-4a to block the nicotine-induced decrease in body temperature is better than that for 2 [AD50 = 1.5 and 9.2 mg/kg, respectively (Table 2)]. Compound 4d (AD50 = 1.7 mg/kg) also rivals (2S,3S)-4a in this assay, and analogues 4c, 4e, 4g, 4h, and 4s have intermediate potencies (2.3–7 mg/kg AD50 values).
In the hot-plate assay, 2 blocks nicotine-induced, supraspinally-mediated analgesia with an AD50 value of 15 mg/kg. Compound (2S,3S)-4a is 15-fold more potent (AD50 value of 1 mg/kg), but blockade of nicotine-induced hot-plate analgesia is no better for any of the new analogues tested. However, 4c, 4d, 4e, and 4s (3.7–8.6 mg/kg AD50 values) are more potent than 2 (Table 2).
Nicotine-induced analgesia in the tail flick assay, which assesses spinal processes involved in nicotine antinociception,27 is blocked by 2 with an AD50 of 1.2 mg/kg. Compound (2S,3S)-4a with an AD50 = 0.2 mg/kg (Table 2) has a 6-fold increase in effectiveness in this assay. Fifteen of the new analogues have even higher potency. AD50 values (in mg/kg) are 0.004 for 4j, 4s, and 4t; ~0.006 for 4m and 4o; 0.012–0.013 for 4c and 4h; 0.016 for 4u; 0.019 for 4i and 4k; 0.021 for 4l; between 0.034 and 0.054 for 4q, 4r, and 4e; and 0.16 for 4d (Table 2).
At the doses tested, none of the compounds showed sedative, convulsive or other obvious toxic effects in mice.
Thus, in four assays of the ability of analogues to block acute actions of nicotine, 3′-bromophenyl analogue 4d was more effective than 2 in each and rivaled effects of (2S,3S)-4a in tail-flick and hypothermia assessments. The 3′-fluorophenyl analogue 4c, 3′-methylphenyl analogue 4e, and ethyl extended chain analogue 4s exceeded 2’s potency in three assays and had higher potency than (2S,3S)-4a in the tail-flick assay. Compounds 4c, 4d, and 4s also are remarkable for their higher inhibitory effectiveness at α4β2-nAChR than (2S,3S)-4a. Eleven of the other ligands had better effectiveness than 2 in one of the acute assays, with potency in the tail-flick assay correlating with IC50 values <10 μM for inhibition of α4β2-nAChR function for 4c–4e, 4l, 4o, and 4q–4u, but not for 4h–4k, 4m and 4v, which were potent in the tail-flick assay but not as α4β2-nAChR antagonists, or for 4b, 4f and 4g, which lacked effectiveness relative to (2S,3S)-4a in the tail-flick assay but have sub-10 μM IC50 values at α4β2-nAChR. Lacking potency in tail-flick or inhibition of α4β2-nAChR function, but having activity as antagonists of α3β4*-nAChR, are 4n and 4p. Increased in vitro inhibition of DA and NE uptake did not necessarily correlate with increased in vivo inhibitory potency in nicotine-sensitive assays if there also was no increase in effectiveness at α4β2-nAChR or selectivity for that target (e.g., 4n). On the other hand, the ligands with the highest inhibitory potency for NE uptake inhibition (and highest or higher effectiveness than (2S,3S)-4a at DA uptake inhibition) were among the most potent inhibitors of nicotine-induced analgesia in the tail-flick assay (4s and 4t). Improvement over (2S,3S)-4a in the ability to block nicotine-induced analgesia in the tail-flick assay as seen for 4j, 4m, and 4o has no obvious basis as judged from in vitro assays.
Discussion
New analogues of the hydroxybupropion isomer (2S,3S)-4a were generated and assessed for their ability to affect monoamine uptake, the function of four different nAChR subtypes, and the acute effects of nicotine on nociception, locomotor activity, and hypothermia.
We succeeded in generating analogues with reasonably higher inhibitory potency than 2 or either of its hydroxymetabolite isomers (2R,3R)-4a and (2S,3S)-4a for DA uptake inhibition (4i, 4n,4s, and 4t), NE uptake inhibition [(±)-4n,4s, and 4t], or 5HT uptake inhibition [(±)-4n and 4r], or for functional inhibition of α3β4*-nAChR (4l) or α4β2-nAChR (4c, 4d, 4l and 4s). Selectivity for inhibition of DA uptake over nAChR or NE uptake inhibition better than that of 2 was achieved with retention of reasonable potency at DAT for 4i. As predicted based on our previous studies of hydroxymetabolites, many of the compounds evaluated have some selectivity for inhibition of NE over DA uptake. This was improved relative to (2S,3S)-4a for 4f–4g, 4s, 4v, 4o, 4u and 4q, but only 4s and 4o have improved potency at NE uptake inhibition. Compounds (±)-4n, 4t, and 4s are more selective than (2S,3S)-4a for inhibition of NE uptake inhibition over nAChR function. Only (±)-4n and 4v have increased selectivity for α3β4*-nAChR over other nAChR subtypes relative to 2 or (2R,3R)-4a. Compounds 4l and 4k have improved selectivity for α3β4*-nAChR functional inhibition over DA and NE uptake inhibition relative to that of (2R,3R)-4a. Compound 4d has absolute selectivity for inhibition of α4β2-nAChR over inhibition of α3β4*-nAChR as well as over NE, 5HT or DA uptake inhibition, and 4l’s similar potency at α4β2- and α3β4*-nAChR also means that it is selective for inhibition of α4β2-nAChR over monoamine transporters. Selectivity for α4β2-nAChR over α3β4*- and other nAChR subtypes was increased for 4c and 4d relative to that for 2 and (2S,3S)-4a. Selectivity for inhibition of α4β2-nAChR over NE uptake inhibition was increased relative to that of (2S,3S)-4a for 4b, 4c, 4e, 4f, 4g, 4p, and 4r, with 4d actually being absolutely selective for inhibition of α4β2-nAChR function over other nAChR subtypes and over inhibition of DA and NE uptake.
From a chemical structure perspective, changes in the 3′-chlorophenyl group in (2S,3S)-4a to 3′-bromophenyl 4d or 3′-fluorophenyl 4c afforded ligands with improved affinity and selectivity for α4β2-nAChRs. Selectivity for monoamine transporter uptake over α4β2-nAChRs functional block was decreased while selectivity for α4β2-nAChRs over other nAChR subtypes was preserved by other changes in the phenyl substitution (nitro 4g, methyl 4e) or lack of chloro group (4b) but not for 3′-methoxyphenyl substitution (4f). The dichlorophenyl analogues (±)-4n and 4p have increased selectivity for α3β4*-nAChR over other nAChR subtypes. In addition, the (±)-3′,4′-dichlorophenyl analogue [(±)-4n] also has a marked increase in affinity for inhibition of DA uptake. Replacement of the 3-methyl group on the morpholinol ring with an ethyl or propyl group (4s and 4t) affords ligands with notable improvements in selectivity for inhibition of NE uptake over nAChR and increases in potencies for inhibition of DA and NE uptake. Changing the 3-chlorophenyl ring to a pyridine ring leads to ligands 5 and 6, which are without activity. Naphthyl analogues 4q and 4r have modestly altered activities compared to (2S,3S)-4a. Interestingly, moving the 3-substituent of the phenyl group of (2S,3S)-4a and 4e to the 4′ position affords ligands (4i–4j) that are selective for DA over NE uptake and nAChR functional inhibition and for inhibition of α3β4*- over α4β2-nAChR. However, the biphenyl analogue (4l) has almost no activity at monoamine transporters yet is very potent as an inhibitor of α4β2 and α3β4*-nAChR. Moving the 3′-substituent of the phenyl group of the N-methyl analogue 4u to the 4′ position (4v) has little effect on inhibition or DA or NE uptake but increases selectivity for functional blockage of α3β4*- over α4β2-nAChRs from negligible to ~9-fold.
In silico predictions that all of the compounds synthesized would have drug-like character and activity in the central nervous system were consistent with the results of behavioral studies. We obtained several analogues with higher potency than (2S,3S)-4a or 2 as antagonists of nicotine-mediated antinociception in the tail-flick assay. The ethyl and propyl extended chain ligands 4s and 4t with high affinity for inhibition of DA and NE uptake, the 3′-fluoro and 3′-bromo phenyl substituted analogues 4c and 4d with high affinity and good selectivity for α4β2-nAChR, and to a lesser extent the 3′-methylphenyl analogue 4e also with good activity at α4β2-nAChR, also had better potency in the tail-flick assay than 2 or (2S,3S)-4a. The naphthyl analogues 4q and 4r with selectivity for inhibition of NE over DA uptake and for inhibition of DA over NE uptake, respectively, but having comparable effects on nAChR function, both had ~5-fold higher activity in the tail-flick assay than (2S,3S)-4a. However, the other in vivo assays mostly did not reveal striking inhibition by analogues of acute nicotine action. Moreover, the tail-flick assay could reflect CNS actions of ligands at a higher level than the presumed spinal level of nicotine-mediated antinociception in the test.
The current studies dovetail with those based on modifications to the bupropion template28 to provide insights into further target-directed drug development. The new analogues also illuminate roles of specific molecular targets in nicotine-mediated behavioral effects. Extensions of the R1 methyl group in (2S,3S)-4a to an ethyl or propyl group (4s and 4t) preferentially improve activities for inhibition of DA and NE uptake. The naphthyl analogues (4q and 4r) afford ligands with better potency for 5HT uptake inhibition, and changing the 3′-chlorophenyl moiety of (2S,3S)-4a to a 3′-fluoro or 3′-bromophenyl group leads to ligands with better activity at α4β2-nAChRs (4c and 4d).
Greater challenges come in correlating the in vivo effects to each other and to the in vitro fingerprints. Formation of metabolites, differences in pharmacokinetics or difficulties in brain penetration following systemic administration could confound interpretations. However, even for assays that are done soon after delivery, the chemical modifications that selectively increase in vitro activity at transporters or at α4β2-nAChR afford ligands with potential as nicotine antagonists in vivo. That is, increased activity at either α4β2-nAChR or for inhibition of DA or NE uptake (or for one ligand, at 5HT uptake inhibition) correlates well with improvement in ligand antagonist potency in the tail-flick assay. Thus, the behavioral results suggest that effects of nicotine dependence and/or depression could be countered by ligands acting at DAT, NET, or α4β2-nAChR or any combination of the three.
The current findings provide part of a preclinical foundation for development of compounds related to (2S,3S)-4a. Recently, Silverstone et al.29 reported that the racemic hydroxybupropion induced convulsions in mice after i.p. administration. However, the potency of (2S,3S)-4a in our mouse testing is close to 50-fold lower than the racemic hydroxy bupropion metabolite active doses reported by Silverstone et al.29 Furthermore, we found no evidence for seizure or convulsive activities at any of the doses tested in our study. Thus, we will continue exploring use of hydroxybupropion analogues as potentially superior aids to smoking cessation.
Experimental Section
Nuclear magnetic resonance (1H NMR and 13C NMR) spectra were recorded on a 300 MHz (Bruker AVANCE 300) unless otherwise noted. Chemical shift data for the proton resonances were reported in parts per million (δ) relative to internal (CH3)4Si (δ 0.0). Optical rotations were measured on an AutoPol III polarimeter, purchased from Rudolf Research. Elemental analyses were performed by Atlantic Microlab, Norcross, GA. Purity of compounds (>95%) was established by elemental analyses. Analytical thin-layer chromatography (TLC) was carried out on plates precoated with silica gel GHLF (250 μM thickness). TLC visualization was accomplished with a UV lamp or in an iodine chamber or ninhydrin staining. All moisture-sensitive reactions were performed under a positive pressure of nitrogen maintained by a direct line from a nitrogen source. Anhydrous solvents were purchased from Aldrich Chemical Co. 3-Methoxypropiophenone (8f), 3,4-dichloropropiophenone (8l), 3,5-dichloropropiophenone (8m), 1-(pyridin-2-yl)propan-1-one (8n) and 1-(pyridin-3-yl)propan-1-one (8o), were synthesized, but are now commercially available. 3-Chlorobutyrophenone (8j) and 3-chloropentaphenone (8k) were described in an earlier paper on bupropion analogs.30
Synthesis of (2S,3S)-4a using optically active Sharpless hydroxylation chemistry
Step 1: (Z)-tert-Butyl-[1-(3-chlorophenyl)prop-1-enyloxy]dimethylsilane (9a)
In a 250-mL flask 3′-chloropropiophenone (8a, 10 g, 0.059 mol) was dissolved in 100 mL in CH2Cl2 and cooled with an ice water bath. Et3N (13 mL, 95 mmol) was added to the solution, followed by slow addition of TBDMSOTf (15 mL, 65 mmol). After stirring overnight at room temperature, the reaction mixture was diluted with CH2Cl2 and washed with NaHCO3. The organic layer was separated, dried (Na2SO4) and concentrated. The oily residue was purified by column chromatography on neutral alumina using hexanes (a few drops of Et3N were added) as the eluent to give 16.4 g (98%) of title product as a colorless oil: 1H NMR (CDCl3) δ 7.46–7.44 (m, 1H), 7.35–7.32 (m, 1H), 7.21–7.20 (m, 2H), 5.23 (q, 1H, J = 6.9 Hz), 1.73 (d, 3H, J = 6.9 Hz), 0.99 (s, 9H), −0.03 (s, 6H). C15H23ClOSi.
Step 2: (R)-1-(3-Chlorophenyl)-2-hydroxypropan-1-one ((R)-10a)
A mixture of AD-mix-β (50.6 g) and CH3SO2NH2 (3.5 g, 0.037 mol) in tert-butyl alcohol-water (120 mL/120 mL) was cooled at 0 °C and treated with 9a (10 g, 0.036 mol). The reaction mixture was stirred for 16 h at 0 °C. Sodium sulfite (36 g) was added and the mixture was stirred for another hour. The mixture was filtered through a Celite pad and washed with ether. The filtrate was transferred to a separation funnel and the lower dark colored layer was discarded. The upper yellowish phase was separated, dried (Na2SO4), filtered, and concentrated. The crude product was purified by column chromatography on silica gel using hexanes-EtOAc (10:1 to 3:1) as the eluent to give 5.8 g (87%) of title product as light greenish oil: [α]20D +64.2° (c 1.2, CHCl3); 1H NMR (CDCl3) δ 7.91 (s, 1H), 7.80 (d, 1H, J = 7.8 Hz), 7.62–7.57 (m, 1H), 7.46 (t, 1H, J = 7.8 Hz), 5.15–5.08 (m, 1H), 3.68–3.65 (m, 1H), 1.45 (d, 3H, J = 7.1 Hz). C9H9ClO2. Characterization data is similar with that reported in the literature.24
Step 3: (2S,3S)-2-(3-Chlorophenyl)-3,5,5-trimethylmorpholin-2-ol [(2S,3S)-4a] Hemi-D-tartrate
A sample of (R)-10a (2.47 g, 0.0134 mol) was added to a 250 mL flask and dissolved in CH2Cl2 (40 mL). Proton sponge (3.5 g, 0.016 mol) was added to the reaction flask, and the reaction mixture was cooled to −50 °C. Triflic anhydride (2.47 mL, 14.7 mmol) was slowly added to the reaction flask, the temperature was allowed to rise to 0 °C and stirred for an additional hour. The resulting orange slurry was transferred by syringe to another flask containing a solution of 2-amino-2-methyl-1-propanol (2.6 g, 0.029 mol) in CH3CN (40 mL) at −10 °C. After stirring for 4 h at 0 °C, the precipitate was removed by filtration and the filtrate was concentrated. The residue was extracted with ether and solid was removed by filtration and discarded. The filtrate was concentrated to an oil. Purification of the residue by chromatography on silica gel using EtOAc with 1% NH4OH as the eluent gave 1.39 g (41%) of (2S,3S)-4a as a white solid. Characterization data is similar with data reported in the literature.24
The product freebase (1.2 g, 0.0047 mol) was dissolved in 20 mL of MeOH and treated with a solution of D-tartaric acid (350 mg, 2.30 mmol) in MeOH (3 mL). After stirring for 5 min at room temperature, the reaction mixture was concentrated, the sample dissolved in CH2Cl2 (30 mL) and MeOH was added until the solution was clear. Next, ether was added slowly until it became cloudy or small crystals started to form. After keeping the mixture at 0 °C for 1 h, the white solid was collected by filtration and recrystallized to give 0.7 g of (2S,3S)-4a•0.5D-tartrate as a white solid (ee 98.4%): mp 128–131 °C; [α]23D +13.7° (c 0.76, CH3OH); Anal. (C15H21ClNO5•0.5H2O). C, H, N.
(2R,3R)-2-(3-Chlorophenyl)-3,5,5-trimethylmorpholin-2-ol ((2R,3R)-4a) Hemi-L-tartrate
Following the procedure described for (2S,3S)-4a, a sample of (S)-10a (4.5 g, 0.024 mol), was dissolved in CH2Cl2 (75 mL) and treated with Proton sponge (6.3 g) and cooled to −50 °C. Next, triflic anhydride (4.5 mL, 32 mmol) was added slowly, and the reaction mixture was stirred at 0 °C for an additional hour. The resulting orange slurry was transferred by syringe to another flask containing a solution of 2-amino-2-methyl-1-propanol, (4.7 g, 0.052 mol) in CH3CN. After purification, 3.3 g (78%) of the (2R,3R)-4a was isolated and converted to 2.7 g of the Hemi-L-tartrate salt (>99%ee): mp 128–131 °C; [α]23D −13.0° (c 0.79, CH3OH). Characterization data is similar with data reported in the literature.24
(2S,3S)-2-Phenyl-3,5,5-trimethylmorpholin-2-ol (4b) Hemi-D-tartrate
Compound 4b was synthesized by a procedure similar to that described for (2S,3S)-4a using (R)-1-phenyl-2-hydroxypropan-1-one (10b, 3.49 g, 0.0233 mol), Proton sponge (5.9 g), triflic anhydride (4.2 mL, 26 mmol), and 2-amino-2-methyl-1-propanol (4.6 g, 0.052 mol) in CH2Cl2 (50 mL). After purification by chromatography on silica gel, 3.52 g (68%) of the free base 4b was isolated, and converted to 1.19 g of the hemi-D-tartrate salt, which had >99%ee: mp 112–113 °C; [α]20D +15.8° (c 1.1, CH3OH); 1H NMR (methanol-d4) δ 7.61–7.59 (m, 2H), 7.44–7.36 (m, 3H), 4.32 (s, 1H), 4.24 (d, 1H, J = 12.3 Hz), 3.58–3.48 (m, 2H), 1.64 (s, 3H), 1.39 (s, 3H), 1.09 (d, 3H, J = 6.6 Hz); 13C NMR (methanol-d4) δ 179.1, 142.4, 130.3, 129.6 (2C), 127.9 (2C), 97.3, 75.4, 67.4, 55.47, 55.27, 24.1, 21.3, 14.3; LCMS (ESI) m/z 222.4 (M-tartrate)+; Anal. (C15H22NO5•0.5H2O). C, H, N.
(2S,3S)-2-(3-Fluorophenyl)-3,5,5-trimethylmorpholin-2-ol (4c) Hemi-D-tartrate
Compound 4c was synthesized by a procedure similar to that described for (2S,3S)-4a using (R)-1-(3-fluorophenyl)-2-hydroxypropan-1-one (10c, 3.94 g, 0.024 mol), Proton sponge (6.0 g), triflic anhydride (4.6 mL, 25.8 mmol), and 2-amino-2-methyl-1-propanol (4.6 g, 0.052 mol) in acetonitrile (50 mL). After purification, 2.2 g (39%) of the free base 4c was isolated and converted to the hemi-D-tartrate salt, which had >99%ee: mp 131–132 °C; [α]20D +20.4° (c 1.0, CH3OH); 1H NMR (methanol-d4) δ 7.45–7.41 (m, 2H), 7.35–7.31 (m, 1H), 7.11–7.09 (m, 1H), 4.33 (s, 1H), 4.26 (d, 1H, J = 12.0 Hz), 3.56–3.45 (m, 2H), 1.59 (s, 3H), 1.34 (s, 3H), 1.06 (d, 3H, J = 6.6 Hz); 13C NMR (methanol-d4) δ 178.1, 165.4, 162.1, 144.8, 130.8, 123.2, 116.3 (d), 114.3 (d), 96.2, 74.5, 67.0, 54.3, 23.6, 20.8, 13.7; LCMS (ESI) m/z 240.0 [(M – tartrate)+, M = C15H21FNO5]; Anal. (C15H21FNO5•0.5H2O). C, H, N.
(2S,3S)-2-(3-Bromophenyl)-3,5,5-trimethylmorpholin-2-ol (4d) Hemi-D-tartrate
Compound 4d was synthesized by a procedure similar to that described for (2S,3S)-4a using (R)-1-(3-bromophenyl)-2-hydroxypropan-1-one (10d, 4.0 g, 0.018 mol), Proton sponge (4.5 g, 0.021 mol), triflic anhydride (3.2 mL, 192 mmol), and 2-amino-2-methyl-1-propanol (3.4 g, 0.038 mol) in acetonitrile (50 mL). After purification, 2.04 g (39%) of the free base 4d was isolated and converted to 1.6 g of the hemi-D-tartrate salt, which had >99%ee: mp 129–130 °C; [α]20D +9.6° (c 1.0, CH3OH); 1H NMR (methanol-d4) δ 7.77–7.76 (m, 1H), 7.63–7.53 (m, 2H), 7.38–7.32 (m, 1H), 4.37 (s, 1H), 4.14 (d, 1H, J = 12.0 Hz), 3.58–3.39 (m, 2H), 1.57 (s, 3H), 1.32 (s, 3H), 1.07–1.02 (m, 3H); 13C NMR (methanol-d4) δ 178.8, 145.1, 133.3, 131.1, 126.9, 123.6, 96.7, 75.2, 67.7, 55.0, 24.4, 21.5, 14.4; LCMS (ESI) m/z 300.6 [(M – tartrate)+, M = C15H21BrNO5]; Anal. (C15H21BrNO5•0.25H2O): C, H, N.
(2S,3S)-2-(m-Tolyl)-3,5,5-trimethylmorpholin-2-ol (4e) Hemi-D-tartrate
Compound 4e was synthesized by a procedure similar to that described for (2S,3S)-4a using (R)-1-(3-methylphenyl)-2-hydroxypropan-1-one (10e, 4.2 g, 0.026 mol), Proton sponge (6.5 g, 0.030 mol), triflic anhydride (4.60 mL, 282 mmol), and 2-amino-2-methyl-1-propanol (5.0 g, 0.056 mol) in acetonitrile (55 mL). After purification, 4.0 g (66%) of the free base 4e was isolated and converted to 3.6 g of the hemi-D-tartrate salt, which had 94%ee: mp 104–105 °C; [α]20D +11.9° (c 0.85, CH3OH); 1H NMR (methanol-d4) δ 7.43–7.38 (m, 2H), 7.28 (t, 1H, J = 7.8 Hz), 7.21–7.18 (m, 1H), 4.33 (s, 1H), 4.20 (d, 1H, J = 12.0 Hz), 3.52–3.45 (m, 2H), 2.38 (s, 3H), 1.61 (s, 3H), 1.36 (s, 3H), 1.06 (d, 3H, J = 6.6 Hz); 13C NMR (methanol-d4) δ 179.0, 142.3, 139.4, 130.9, 129.5, 128.4, 125.0, 97.2, 75.3, 67.4, 55.2, 24.2, 21.9, 21.4, 14.4; LCMS (ESI) m/z 236.2 [(M – tartrate)+, M = C16H24NO5]; Anal. (C16H24NO5•0.75H2O: C, H, N.
(2S,3S)-2-(3-Methoxyphenyl)-3,5,5-trimethylmorpholin-2-ol (4f) Hemi-D-tartrate
Compound 4f was synthesized by a procedure similar to that described for (2S,3S)-4a using (R)-1-(3-methoxyphenyl)-2-hydroxypropan-1-one (10f, 4.2 g, 0.023 mol), Proton sponge (5.9 g, 0.028 mol), triflic anhydride (4.2 mL, 257 mmol), and 2-amino-2-methyl-1-propanol (4.5 g, 0.051 mol) in CH2Cl2 (50 mL). After purification, 4.16 g (71%) of the free base 4f was isolated and converted to 1.24 g the hemi-D-tartrate salt, which had 91%ee: mp 99–100 °C; [α]20D +7.9° (c 1.1, CH3OH); 1H NMR (methanol-d4) δ 7.32 (t, 1H, J = 7.8 Hz), 7.19–7.14 (m, 2H), 6.96–6.92 (m, 1H), 4.33 (s, 1H), 4.18 (d, 1H, J = 12.3 Hz), 3.81 (s, 3H), 3.52 (d, 1H, J = 12.3 Hz), 3.48–3.45 (m, 1H), 1.59 (s, 3H), 1.34 (s, 3H), 1.06 (d, 3H, J = 6.6 Hz). 13C NMR (methanol-d4) δ 178.8, 161.4, 144.1, 130.7, 120.1, 115.4, 113.8, 97.1, 75.2, 67.6, 56.1, 55.2, 24.4, 21.5, 14.5; LCMS (ESI) mlz 252.3 [(M – tartrate)+, M = C16H24NO6]; Anal. (C16H24NO6•0.5H2O). C, H, N.
(2S,3S)-2-(3-Nitrophenyl)-3,5,5-trimethylmorpholin-2-ol (4g) Hemi-D-tartrate
Compound 4g was synthesized by a procedure similar to that described for (2S,3S)-4a using (R)-1-(3-nitrophenyl)-2-hydroxypropan-1-one (10g, 4.0 g, 0.021 mol), Proton sponge (5.2 g, 0.0246 mol), triflic anhydride (3.7 mL, 0.023 mol), and 2-amino-2-methyl-1-propanol (4.0 g, 0.045 mol) in acetonitrile (45 mL). After purification, 1.0 g (18%) of the free base 4g was isolated and converted to the hemi-D-tartrate salt, which had 94%ee: mp 192–193 °C; [α]20D +6.5° (c 1.0, CH3OH); 1H NMR (methanol-d4) δ 8.47–8.25 (m, 1H), 8.31–8.26 (m, 1H), 8.05–8.01 (m, 1H), 7.73–7.66 (m, 1H), 4.34 (s, 1H), 4.18 (d, 1H, J = 12.1 Hz), 3.59 (d, 1H, J = 6.6 Hz), 3.50 (q, 1H, J = 6.6 Hz), 1.60 (s, 3H), 1.35 (s, 3H), 1.07 (d, 3H, J = 6.6 Hz); 13C NMR (methanol-d4) δ 178.3, 149.9, 145.3, 134.4, 131.0, 125.0, 123.0, 96.8, 75.0, 68.1, 54.8, 24.7, 21.7, 14.6; LCMS (ESI) mlz 267.3 [(M – tartrate)+, M = C15H21N2O7]; Anal. (C15H21N2O7•0.25H2O). C, H, N.
(2S,3S)-2-(4-Fluorophenyl)-3,5,5-trimethylmorpholin-2-ol (4h) Hemi-fumarate
A solution of 15 (166 mg, 1.16 mmol) in dry THF (1.2 mL, 1M) under an N2 atmosphere was cooled to −78 °C and treated with 4-fluorophenylmagnesium bromide (1.3 equiv., 1.5 mmol, 1.9 mL, 0.8 M solution in THF). The reaction mixture was stirred at −78 °C for 3 h. A saturated aqueous solution of NH4Cl was added to the reaction vessel, and the mixture was allowed to warm to room temperature. EtOAc (5 mL) was added to the reaction vessel and the organic layer was separated. The aqueous phase was extracted with EtOAc (three times). The combined organic extracts were washed (water, brine), dried (Na2SO4) and concentrated. The residue was purified by column chromatography on silica gel using CH2Cl2 to CH2Cl2-MeOH (90:10) as the eluent to afford 80 mg of 4h as a white solid: [α]22D +31.2° (c 0.5, CHC13); 1H NMR (CDCl3) δ 7.60–7.55 (m, 2H), δ 7.08–7.00 (m, 2H), 3.83 (d, 1H, J = 11.3 Hz), 3.40 (d, 1H, J = 11.3 Hz), 3.17 (q, 1H, J = 6.4 Hz), 1.38 (s, 3H), 1.08 (s, 3H), 0.78 (d, 3H, J = 6.4 Hz); 13C NMR (CDCl3) δ 128.0, 127.9, 114.87, 114.58, 103.2, 98.4, 96.8, 69.5, 53.5, 27.3, 22.8, 16.4; LCMS (ESI) m/z 240.0 [(M + H)+, M = C13H18FNO2].
A sample of 4h (56.0 mg, 0.234 mmol) in ether (2 mL) was treated with a solution of fumaric acid (30.0 mg, 0.258 mmol) in MeOH (0.6 mL). The mixture was stirred at room temperature overnight. Filtration and washing of the filter cake with ether, followed by recrystallization of the solid from MeOH-ether gave 45 mg (54%) of 4h•0.5 fumarate: mp 178–182 °C; [α]22D +29° (c 0.6, MeOH); 1H NMR (methanol-d4) δ 7.65–7.59 (m, 2H), 7.15–7.08 (m, 2H), 6.66 (s, 1H), 4.15 (d, 1H, J = 12.2 Hz), 3.52 (d, 1H, J = 12.2 Hz), 3.41–3.33 (m, 1H), 1.56 (s, 3H), 1.32 (s, 3H), 1.03 (d, 3H, J = 6.6 Hz); 13C NMR (methanol-d4) δ 136.7, 129.75, 129.64, 115.92, 115.63, 67.5, 54.8, 54.1, 24.3, 21.3, 14.3; LCMS (ESI) m/z 240.3 [(M–fumaric)+, M = C13H18FNO2•0.5C4H4O4]; Anal. (C15H20FNO4•H2O), C, H, N.
(2S,3S)-2-(4-Chlorophenyl)-3,5,5-trimethylmorpholin-2-ol (4i) Hemi-Fumarate
A solution of 15 (357 mg, 2.50 mmol) in dry THF (2.5 mL, 1M) under an N2 atmosphere was cooled to −78 °C and treated with 4-chlorophenylmagnesium bromide (2 equiv., 5.00 mmol, 5.00 mL, 1M solution in ether). The reaction mixture was stirred at −78 °C for 2 h. A saturated aqueous solution of NH4Cl was added to the reaction vessel, and the mixture was allowed to warm to room temperature. EtOAc was added and the organic layer was separated and the aqueous phase was extracted with EtOAc (trice). The combined organic extracts were washed (water, brine), dried (Na2SO4) and concentrated. The residue was purified by column chromatography on silica gel using CH2Cl2 to CH2Cl2-MeOH (90:10) as the eluent to afford 180 mg (29%) of 4i as a white solid: [α]23D +33° (c 0.4, CHC13): 1H NMR (CDCl3) δ 7.54 (d, 2H, J = 8.5 Hz), 7.32 (d, 2H, J = 8.5 Hz), 3.83 (d, 1H, J = 11.3 Hz), 3.40 (d, 1H, J = 11.3 Hz), 3.18 (q, 1H, J = 6.5 Hz), 1.38 (s, 3H), 1.08 (s, 3H), 0.78 (d, 3H, J = 6.5 Hz); 13C NMR (CDCl3) δ 128.5, 128.1, 127.6, 127.2, 101.6, 95.9, 69.5, 53.4, 49.7, 27.3, 22.8, 16.4; LCMS (ESI) m/z 256.3 [(M + H)+, M = C13H18ClNO2].
A sample of 4i (161 mg, 0.629 mmol) in ether (3 mL) was treated with a solution of fumaric acid (73.0 mg, 0.629 mmol) in MeOH (1.2 mL). The mixture was stirred at room temperature overnight. Filtration and washing of the filter cake with ether, followed by recrystallization from MeOH-ether gave 123 mg (53%) of 4i•0.5 fumarate as a white solid; mp 187–190 °C; [α]22D +22° (c 0.75, MeOH); 1H NMR (methanol-d4) δ 7.58 (d, 2H, J = 8.6 Hz), 7.41 (d, 1H, J = 8.6 Hz), 6.67 (s, 1H), 4.11 (d, 1H, J = 12.0 Hz), 3.51 (d, 1H, J = 12.0 Hz), 3.38–3.36 (m, 1H), 1.53 (s, 3H), 1.28 (s, 3H), 1.00 (d, 3H, J = 6.6 Hz); 13C NMR (methanol-d4) δ 172.9, 141.1, 136.7, 135.8, 129.3, 96.6, 67.4, 54.68, 54.37, 24.1, 21.2, 14.1; LCMS (ESI) m/z 256.6 [(M – fumaric)+, M = C13H18ClNO2•0.5C4H4O4]; Anal. (C15H20ClNO4•0.5H2O). C, H, N.
(2S,3S)-3,5,5-Trimethyl-2-(4-methylphenyl)morpholin-2-ol (4j) Hemi-Fumarate
A solution of morpholin-2-one 15 (270 mg, 1.88 mmol) in anhydrous THF (1.9 mL) was cooled to −78 °C and treated with p-tolylmagnesium bromide (1.2 equiv., 2.26 mmol, 2.26 mL, 1 M solution in THF) under an N2 atmosphere. After stirring the reaction mixture at −78 °C for 1.5 h, saturated aqueous solution of NH4Cl (30 mL) was added to the reaction vessel, and the mixture was allowed to warm to room temperature. Ether (30 mL) was added to the reaction flask, the organic layer was separated. The aqueous phase was extracted with ether (twice). The combined organic extracts were washed (water, brine), dried (Na2SO4) and concentrated. The residue was purified by column chromatography on silica gel using CH2Cl2 to CH2Cl2-MeOH (90:10) as the eluent to give 271 mg (41%) of 4j as a yellow foam: [α]22D +21.2° (c 0.9, CHCl3); 1H NMR (CDCl3) δ 7.47 (d, 2H, J = 8.2 Hz), 7.16 (d, 2H, J = 8.0 Hz), 3.85 (d, 1H, J = 11.3 Hz), 3.40 (d, 1H, J = 11.2 Hz), 3.20–3.07 (m, 1H), 2.35 (s, 3H), 1.39 (s, 3H), 1.08 (s, 3H), 0.81 (d, 3H, J = 6.5 Hz); 13C NMR (CDCl3) δ 129.6, 128.60, 128.49, 125.9, 103.2, 96.2, 69.4, 53.9, 53.4, 49.6, 27.3, 22.8, 16.5; LCMS (ESI) m/z 236.3 [(M + H)+, M = C14H21NO2].
A sample of 4j (240 mg, 0.683 mmol) in ether (3 mL) was treated with a solution of fumaric acid (87.0 mg, 0.751 mmol) in MeOH (2 mL) and stirred at room temperature overnight. Ether was added to the reaction mixture. The solid was recrystallized from MeOH-ether to give 140 mg (67%) of 4j•0.5 fumarate as a white solid: mp 178–182 °C; [α]22D +19° (c 0.6, MeOH); 1H NMR (methanol-d4) δ 7.47 (d, 2H, J = 8.2 Hz), 7.21 (d, 2H, J = 8.0 Hz), 6.65 (s, 1H), 4.18 (d, 1H, J = 12.2 Hz), 3.51 (d, 1H, J = 12.2 Hz), 3.46 (q, 1H, J = 6.6 Hz), 2.35 (s, 3H), 1.58 (s, 3H), 1.34 (s, 3H), 1.04 (d, 1H, J = 6.6 Hz); 13C NMR (methanol-d4) δ 139.9, 139.2, 136.9, 129.8, 127.4, 96.8, 67.2, 54.9, 54.6, 24.1, 21.1, 14.1; LCMS (ESI) m/z 236.2 [(M – fumaric)+ C14H21NO2•0.5C4H4O4]; Anal. (C16H23NO4•0.25H2O). C, H, N.
(2S,3S)-2-(4-Methoxyphenyl)-3,5,5-trimethylmorpholin-2-ol (4k) Hydrochloride
A solution of 15 (434 mg, 3.03 mmol) in anhydrous THF (3 mL, 1M) was cooled to −78 °C and treated with 4-methoxyphenylmagnesium bromide (1.3 equiv., 3.93 mmol, 3.9 mL of 1 M solution in THF). The reaction mixture was stirred at −78 °C and under an N2 atmosphere for 1.5 h. A saturated aqueous solution of NH4Cl (30 mL) was added to the reaction vessel, and the mixture was allowed to warm to room temperature. The mixture was extracted with ether (three times). The combined ether extracts were washed (water, brine), dried (Na2SO4) and concentrated. The residue was purified by column chromatography on silica gel using CH2Cl2 to CH2Cl2-MeOH-NH4OH (90:9:1) as the eluent to give 380 mg of a yellow solid.
The sample (379 mg, 0.783 mmol) in ether (5 mL) was treated with HCl (1.00 mmol, 0.250 mL, 4 M solution in dioxane). The mixture was stirred at room temperature overnight. Ether was added to the reaction mixture. The suspension was sonicated, centrifuged, and decanted three times to afford a solid pellet. The solid material was recrystallized from MeOH-ether to give 215 mg (95%) of 4k•HCl as a pale yellow solid: mp 170–172 °C; [α]20D +19.6° (c 1.0, MeOH). 1H NMR (methanol-d4) δ 7.52 (d, 2H, J = 8.9 Hz), 6.95 (d, 2H, J = 8.9 Hz), 4.22 (d, 1H, J = 12.4 Hz), 3.81 (s, 3H), 3.58–3.45 (m, 2H), 1.62 (s, 3H), 1.39 (s, 3H), 1.10 (d, 3H, J = 6.6 Hz); 13C NMR (methanol-d4) δ 161.8, 133.5, 132.8, 128.8, 114.6, 96.7, 66.6, 55.80, 55.7, 55.2, 23.4, 20.8, 13.7; LCMS (ESI) m/z 286.4 (M − H)+, M = C14H21NO3•HCl]; Anal. (C14H22ClNO3). C, H, N.
(2S,3S)-2-Biphenyl-4-yl-3,5,5-trimethylmorpholin-2-ol (4l) Hemi-Fumarate
A solution of 15 (394 mg, 2.75 mmol) in anhydrous THF (2.75 mL, 1M) was cooled to −78 °C and treated with 4-biphenylmagnesium bromide (1.2 equiv., 3.30 mmol, 6.60 mL, 0.5 M solution in THF) under an N2 atmosphere. After stirring at −78 °C for 1.5 h, the reaction mixture was treated with saturated aqueous solution of NH4Cl (40 mL) and EtOAc was added (30 mL). The organic layer was separated. The aqueous phase was extracted with EtOAc trice. The combined organic extracts were washed (water, brine), dried (Na2SO4) and concentrated. Purification of the residue by column chromatography on silica gel using CH2Cl2 to CH2Cl2-MeOH (90:10) as the eluent afforded 300 mg (37%) of 4l as a white solid: [α]23D +15.2° (c 0.3, CHC13); 1H NMR (CDCl3) δ 7.70–7.33 (m, 9H), 3.88 (d, 1H, J = 11.3 Hz), 3.44 (d, 1H, J = 11.3 Hz), 3.29–3.25 (m, 1H), 1.42 (s, 3H), 1.10 (s, 3H), 0.86 (d, 3H, J = 6.5 Hz); 13C NMR (CDCl3) δ 140.9, 129.0, 128.97, 128.74, 128.4, 127.54, 127.30, 127.13, 126.7, 126.5, 96.2, 69.5, 53.4, 51.4, 49.6, 27.4, 22.8, 16.6; LCMS (ESI) m/z 298.4, 280.3 [(M + H)+, M = C19H23NO2].
A sample of 4l (379 mg, 1.27 mmol) in CH2Cl2 (4 mL) was treated with a solution of fumaric acid (148 mg, 1.27 mmol) in MeOH (4 mL). The mixture was stirred at room temperature overnight. Filtration and washing of the filter cake with ether, followed by recrystallization from MeOH-ether gave 130 mg (25%) of 4l•0.5fumarate as a white solid: mp 197–202 °C; [α]22D +18° (c 1.6, MeOH); 1H NMR (methanol-d4) δ 7.70–7.63 (m, 6H), 7.49–7.35 (m, 3H), 6.69 (s, 1H), 4.20 (d, 1H, J = 12.1 Hz), 3.55 (d, 1H, J = 12.1 Hz), 3.47 (q, 1H, J = 6.7 Hz), 1.60 (s, 3H), 1.34 (s, 3H), 1.08 (d, 3H, J = 6.6 Hz); 13C NMR (methanol-d4) δ 142.9, 141.8, 141.4, 136.7, 129.9, 128.6, 128.04, 128.02, 127.7, 96.9, 67.5, 54.8, 54.1, 30.7, 24.4, 21.3, 14.4; LCMS (ESI) m/z 298.6, 280.3 [(M – fumaric)+, M = C19H23NO2•0.5C4H4O4]; Anal (C21H25NO4•0.75H2O). C, H, N.
(2S,3S)-2-(3,4-Difluorophenyl)-3,5,5-trimethylmorpholin-2-ol (4m) Hemi-Fumarate
A solution of morpholin-2-one 15 (410 mg, 2.86 mmol) in anhydrous THF (2.9 mL, 1M) was cooled to −78 °C and treated with 3,4-difluorophenylmagnesium bromide (1.2 equiv., 3.43 mmol, 6.90 mL, 0.5 M solution in THF) under an N2 atmosphere. After stirring the reaction mixture at −78 °C under an inert atmosphere for 1.5 h, saturated aqueous solution of NH4Cl was added to the reaction vessel. The reaction mixture was allowed to warm to room temperature and extracted with ether (three times). The combined organic extracts were washed (water, brine), dried (Na2SO4) and concentrated. Purification of the residue by column chromatography on silica gel using CH2Cl2 to CH2Cl2-MeOH-NH4OH (90:9:1) as the eluent gave 190 mg (25%) of 4m as a yellow solid: [α]22D +18.2° (c 1.0, CHCl3); 1H NMR (CDCl3) δ 7.45–7.39 (m, 1H), 7.34–7.32 (m, 1H), 7.18–7.10 (m, 1H), 3.82 (d, 1H, J = 11.3 Hz), 3.40 (d, 1H, J = 11.3 Hz), 3.18 (q, 1H, J = 6.5 Hz), 1.38 (s, 3H), 1.08 (s, 3H), 0.78 (d, 3H, J = 6.5 Hz); 13C NMR (CDCl3) δ 122.40, 122.31 116.74, 116.51, 115.8, 115.6, 96.5, 69.6, 53.4, 49.8, 27.3, 22.8, 16.5; LCMS (ESI) m/z 258.6, [(M + H)+, M = C13H17F2NO2].
A sample of 4m (180 mg, 0.699 mmol) in ether (3 mL) was treated with a solution of fumaric acid (80.0 mg, 0.689 mmol) in MeOH (2.5 mL). The mixture was stirred at room temperature overnight. Ether was added to the reaction mixture. The suspension was sonicated, centrifuged, and decanted to afford a solid pellet that was recrystallization from MeOH-ether. The suspension was sonicated, centrifuged, and decanted three times to afford 119 mg (53%) of 4m•0.5fumarate as a white solid: mp 187–189 °C; [α]22D +29.6° (c 0.5, MeOH); 1H NMR (methanol-d4) δ 7.52–7.41 (m, 2H), 7.38–7.22 (m, 1H), 6.65 (s, 1H), 4.34 (s, 1H), 4.15 (d, 1H, J = 12.2 Hz), 3.54 (d, 1H, J = 12.2 Hz), 3.46 (q, 1H, J = 6.6 Hz), 1.57 (s, 3H), 1.33 (s, 3H), 1.06 (d, 3H, J = 6.6 Hz); 13C NMR (methanol-d4) δ 136.7, 124.4, 118.1, 117.8, 117.1, 116.9, 98.2, 67.5, 54.58, 54.36, 24.1, 21.2, 14.1; LCMS (ESI) m/z 258.6, [(M – fumaric)+ M = C13H17F2NO2•0.5C4H4O4]; Anal. (C15H19F2NO4•0.5H2O). C, H, N.
2-(3,4-Dichlorophenyl)-3,5,5-trimethylmorpholin-2-ol [(±)-4n] Hemi-Fumarate
To a solution of 3′,4′-dichloropropiophenone (8l, 5.02 g, 0.247 mol) in CH2Cl2 (100 mL) was added ten drops of bromine. After stirring at room temperature under nitrogen for several min, the characteristic red color of bromine disappeared indicating initiation of the reaction. The remainder of the bromine (1.27 mL, 24.7 mmol) was added dropwise and the reaction solution was allowed to stir at room temperature under nitrogen atmosphere for 1.75 h. Analysis by TLC (silica, 2:1 hexane:CH2Cl2) indicated consumption of starting material. The reaction solution was quenched and brought to a pH of 9 with a saturated aqueous solution of NaHCO3 and concentrated NH4OH. The solution was extracted with CH2Cl2, dried (Na2SO4), filtered, concentrated, and dried to give 7.14 g (100%) of 2-bromo-(3′,4′-dichlorophenyl)propan-1-one as a white solid. Characterization data is similar with data reported in the literature.31
2-Bromo-(3′,4′-dichlorophenyl)propan-1-one (6.97 g, 0.025 mol) in a minimal amount of CH2Cl2 was transferred to a sealable reaction tube. Most of the CH2Cl2 was removed via positive nitrogen flow. 2-Amino-2-methyl-1-propanol (23.6 mL, 247 mmol) was added in one portion, and the tube was sealed and placed in an oil bath heated to 75 °C. After stirring at 75 °C overnight, analysis by TLC (silica, 9:1:20 ether-Et3N-hexane) showed only a trace amount of starting material remaining and the reaction was allowed to cool to room temperature. The reaction mixture was quenched and brought to a pH of 10 with a saturated aqueous solution of NaHCO3 and the product was extracted with CH2Cl2. The organic layer was separated, dried (Na2SO4), filtered, concentrated, and dried to give 11.29 g of a yellow oil. The residue was purified by column chromatography on silica gel using ether-Et3N-hexane (9:1:50) as eluent to give 3.50 g (49%) of 4n as a white solid: 1H NMR (250 MHz, DMSO-d6) δ 7.70–7.67 (m, 1H), 7.47–7.39 (m, 2H), 3.81 (d, 1H), 3.40 (d, 1H), 3.22–3.14 (m, 1H), 1.38 (s, 3H), 1.08 (s, 3H), 0.78 (d, 3H).
A solution of (±)-4n (3.34 g, 0.012 mol) in methanol was treated with fumaric acid (1.34 g, 0.012 mol). The mixture was allowed to stir for 15 min and a white solid precipitated out of solution, which was collected by vacuum filtration to afford 2.23 g (53%) of 4n•0.5 fumarate as a white solid: mp 188–189 °C; 1H NMR (250 MHz, DMSO-d6) δ 7.68–7.62 (m, 2H), 7.52–7.48 (m, 1H), 3.77 (d, 1H), 3.36 (d, 1H), 3.13–3.07 (q, 1H), 1.33 (s, 3H), 1.07 (s, 3H), 0.76 (d, 3H); Anal. (C15H19Cl2NO4). C, H, N.
(2S,3S)-2-(3,5-Difluorophenyl)-3,5,5-trimethylmorpholin-2-ol (4o) Hydrochloride
A solution of 15 (448 mg, 3.13 mmol) in anhydrous THF (3 mL, 1M) was cooled to −78 °C and treated with 3,5-dipfluorophenylmagnesium bromide (1.2 equiv., 3.75 mmol, 7.5 mL, 0.5 M solution in THF). The reaction mixture was stirred at −78 °C and under an N2 atmosphere for 1.5 h. Saturated aqueous solution of NH4Cl was added to the reaction vessel, and the mixture was allowed to warm to room temperature. The reaction mixture was extracted with ether (three times). The combined ether extracts were washed (water, brine), dried (Na2SO4) and concentrated. Purification of the residue by column chromatography on silica gel using CH2Cl2 to CH2Cl2-MeOH-NH4OH (90:9:1) as the eluent gave 105 mg (13%) of 4o as a yellow solid: [α]22D +19.5° (c 0.8, CHCl3); 1H NMR (CDCl3) δ 7.16–7.10 (m, 2H), 6.82–6.67 (m, 1H), 3.81 (d, 1H, J = 12.0 Hz), 3.40 (d, 1H, J = 12.0 Hz), 3.18 (q, 1H, J = 6.0 Hz), 1.39 (s, 3H), 1.12 (s, 3H), 0.83 (d, 3H, J = 6.5 Hz); 13C NMR (CDCl3) δ 109.63, 109.40, 103.9, 103.5, 103.2, 95.8, 69.2, 53.3, 50.1, 49.6, 26.9, 22.5, 15.2; LCMS (ESI) m/z 258.8 [(M + H)+, M = C13H17F2NO2].
A sample of 4o (71.0 mg, 0.275 mmol) in CH2Cl2 (1.5 mL) was treated with HCl (0.386 mmol, 0.100 mL solution 4M in dioxane). The mixture was stirred at room temperature overnight and ether was added to the reaction mixture. The suspension was sonicated, centrifuged, and decanted to afford a solid pellet; this procedure was repeated three times. The solid material was recrystallized from methanol-ether. The suspension was sonicated, centrifuged, decanted and dried to give 60 mg (74%) of 4o•HCl as a pale yellow solid: mp 199–204 °C; [α]22D +21.5° (c 1.0, MeOH); 1H NMR (methanol-d4) δ 7.23–7.18 (m, 2H), 7.03–6.92 (m, 1H), 4.19 (d, 1H, J = 12.0 Hz), 3.65–3.54 (m, 2H), 1.62 (s, 3H), 1.39 (s, 3H), 1.12 (d, 3H, J = 6.0 Hz); 13C NMR (methanol-d4) δ 166.1, 162.8, 111.1, 110.9, 105.6, 105,2, 104.9, 66.9, 55.6, 54.5, 23.5, 20.9, 13.7; LCMS (ESI) m/z 258.5 [(M – HCl)+, M = C13H17F2NO2•HCl]; Anal. (C13H18ClF2NO2). C, H, N.
2-(3,5-Dichlorophenyl)-3,5,5-trimethylmorpholin-2-ol [(±)-4p] Hemi-Fumarate
To a stirred solution of 3,5-dichloropropiophenone (8m, 3.81 g, 0.0188 mol) in CH2Cl2 (28 mL) was added bromine (0.986 mL, 19.1 mmol) dropwise. The bromine was immediately consumed upon addition of each drop until the end of addition when it had a consistent brownish color, indicative of excess bromine. The reaction was immediately quenched with saturated aqueous NaHCO3, extracted three times with CH2Cl2. The organic layer was separated, combined and concentrated under reduced pressure, without drying to afford a yellow oil. The oil was dissolved in anhydrous diethyl ether (50 mL) and 2-amino-2-methyl-1-propanol (6.7 g, 0.075 mol) was added. The reaction mixture was stirred overnight and quenched with saturated aqueous NaHCO3. The aqueous layer was extracted three times with CH2Cl2, and the organic layers concentrated to afford another yellow oil. Purification by column chromatography on silica gel afforded 2.51 g (46%) of the title compound. The hemi-fumarate salt was prepared by dissolving the free base in methanol and adding 1.0 g of fumaric acid to form the title compound: mp 188–190 °C; 1H NMR (DMSO-d6) δ 7.58– 7.57 (m, 1H), 7.47 (m, 2H), 6.67 (s, 0.5H), 4.09 (d, 1H, J = 11.9 Hz), 3.53 (d, 1H, J = 12.0 Hz), 3.41–3.37 (m, 1H), 1.53 (s, 3H), 1.28 (s, 3H), 1.02 (d, 3H, 6.6 Hz); 13C NMR (DMSO-d6) δ 134.9, 133.6, 127.7, 125.2, 94.7, 67.2, 52.5, 50.1, 25.0, 21.6, 14.6; Anal. (C15H19Cl2NO4•0.25H2O). C, H, N.
(2S,3S)-2-(Naphthalen-1-yl)-3,5,5-trimethylmorpholin-2-ol (4q) Hemi-D-tartrate
Compound 4q was synthesized by a procedure similar to that described for (2S,3S)-4a employing (R)-2-hydroxy-1-(naphthalen-1-yl)propan-1-one (10h, 1.15 g, 0.058 mol), Proton sponge (1.5 g, 0.070 mol), triflic anhydride (1.1 mL, 64 mmol), and 2-amino-2-methyl-1-propanol (1.1 g, 0.012 mol) in CH3CN (45 mL). After purification, 1.35 g (86%) of the free base 4q was isolated and converted to the D-tartrate salt, which was recrystallized from H2O-MeOH-Et2O solvent system: mp 115–116 °C; [α]20D −9.5° (c 0.74, CH3OH); 1H NMR (methanol-d4) δ 8.89 (d, 1H, J = 8.4 Hz), 7.97–7.89 (m, 3H), 7.54–7.50 (m, 3H), 4.33 (s, 1H), 3.70 (d, 1H, J = 12.2 Hz), 3.48 (q, 1H, J = 7.0 Hx), 1.77 (s, 3H), 1.41 (s, 3H), 0.96 (d, 3H, J = 6.6 Hz); 13C NMR (methanol-d4) δ 136.5, 132.3, 131.9, 130.5, 130.2, 128.1, 127.4, 127.0, 126.1, 99.0, 75.1, 68.1, 67.3, 53.9, 24.8, 22.9, 15.8, 15.2; LCMS (ESI) m/z 272.3 [(M-tartrate)+, M = C19H24NO5]; Anal. (C19H24NO5•0.75H2O). C, H, N.
(2S,3S)-2-(Naphthalen-2-yl)-3,5,5-trimethylmorpholin-2-ol (4r) Hemi-D-tartrate
Compound 4r was synthesized by a procedure similar to that described for (2S,3S)-4a using (R)-2-hydroxy-1-(naphthalen-2-yl)propan-1-one (10i, 2.5 g, 0.013 mol), Proton sponge (3.24 g, 0.0151 mol), triflic anhydride (2.4 mL, 137 mmol), and 2-amino-2-methyl-1-propanol (2.4 g, 0.027 mol) in acetonitrile (40 mL). After purification, 2.2 g (65%) of the free base 4r was isolated and converted to the hemi-D-tartrate salt: mp 179–180 °C; [α]20D −1.5° (c 0.55, CH3OH); 1H NMR (methanol-d4) δ 8.13–8.12 (m, 1H), 7.95–7.88 (m, 3H), 7.75–7.71 (m, 1H), 7.57–7.50 (m, 2H), 4.33 (s, 1H), 4.25 (d, 1H, J = 11.9 Hz), 3.66–3.57 (m, 2H), 1.65 (s, 3H), 1.37 (s, 3H), 1.09 (d, 3H, J = 6.6 Hz); 13C NMR (methanol-d4) δ 178.7, 139.9, 135.3, 134.6, 129.8, 129.33, 129.00, 128.0, 127.8, 127.4, 125.4, 97.4, 75.2, 67.7, 55.1, 24.5, 21.6, 14.6; LCMS (ESI) m/z 272.5 [(M – tartrate)+, M = C19H24NO5]; Anal. (C19H24NO5). C, H, N.
(2S,3S)-2-(3-Chlorophenyl)-3-ethyl-5,5-dimethylmorpholin-2-ol (4s) Hemi-D-tartrate
Compound 4s was synthesized by a procedure similar to that described for (2S,3S)-4a using (R)-1-(3-chlorophenyl)-2-hydroxybutan-1-one (10j, 1.13 g, 0.0568 mol), Proton sponge (1.43 g, 0.0667 mol), triflic anhydride (1.0 g, 0.063 mol), and 2-amino-2-methyl-1-propanol (1.09 g, 0.0122 mol) in CH2Cl2 (13 mL). After purification, the free base 4r was converted to 0.230 g of its D-tartrate salt: mp 160–161 °C; [α]20D +5.6° (c 0.8, CH3OH); 1H NMR (methanol-d4) δ 7.49–7.46 (m, 1H), 7.41–7.35 (m, 3H), 4.37–4.34 (m, 1H), 4.30–4.24 (m, 1H), 3.81–3.62 (m, 2H), 1.57 (s, 3H), 1.47–1.36 (m, 2H), 1.33 (s, 3H), 0.81–0.71 (m, 3H); 13C NMR (methanol-d4) δ 178.5, 145.2, 135.6, 131.2, 130.3, 128.2, 126.5, 97.1, 75.0, 67.8, 61.1, 55.0; LCMS (ESI) m/z 270.4 [(M – tartrate)+, M = C16H23ClNO5); Anal. (C16H23ClNO5•0.25H2O). C, H, N.
(2S,3S)-2-(3-Chlorophenyl)-5,5-dimethyl-3-propyl-morpholin-2-ol (4t) Hemi-D-tartrate
Compound 4t was synthesized by a procedure similar to that described for (2S,3S)-4a using (R)-1-(3-chlorophenyl)-2-hydroxypent-1-one (10k, 1.5 g, 0.0704 mol), Proton sponge (1.8 g, 0.0840 mol), triflic anhydride (1.29 g, 0.081 mol), and 2-amino-2-methyl-1-propanol (1.42 g, 0.0159 mol) in CH2Cl2 (7 mL). After purification, 790 mg (40%) of the free base 4t was isolated and converted to the hemi-D-tartrate salt, which had 99%ee: mp 151–152 °C, [α]20D −10.1° (c 0.77, CH3OH); 1H NMR (methanol-d4) δ 7.50–7.47 (m, 1H), 7.43–7.36 (m, 3H), 4.38 (s, 1H), 4.32 (d, 1H, J = 10.0 Hz), 3.81–3.66 (m, 1H), 1.58 (s, 3H), 1.49–1.38 (m, 2H), 1.35 (s, 3H), 1.34–1.27 (m, 1H), 1.02–0.92 (m, 1H), 0.77 (t, 3H, J = 7.0 Hz); 13C NMR (methanol-d4) δ 176.1, 142.9, 133.3, 128.9, 128.0, 125.9, 124.2, 94.8, 72.7, 65.4, 57.0, 52.7, 29.9, 22.0, 19.3, 18.2, 12.1; Anal. (C17H25ClNO5). C, H, N.
(2S,3S)-2-(3-Chlorophenyl)-3,4,5,5-tetramethylmorpholin-2-ol (4u) Di-p-tolyl-L-tartrate
A sample of (2S,3S)-2-(3-Chlorophenyl)-3,5,5-trimethylmorpholin-2-ol (4i, 107 mg, 0.42 mmol) in DMF (2.0 mL) was treated with K2CO3 (174 mg, 1.26 mmol). After stirring the reaction mixture at room temperature under an inert atmosphere for 1.5 h, CH3I (19.0 μL, 0.3 mmol) was added to the reaction flask and the reaction mixture was stirred at 70 °C for 24 h. The reaction mixture was cooled to 0 °C water added, followed by extraction with ether (thee times). The combined organic extracts were washed (water, brine), dried (Na2SO4) and concentrated to a pale yellow oil. Purification of the residue by column chromatography gave a 72 mg (63%) of 4u as a white solid. The compound 4u was converted to the corresponding di-p-tolyl-L-tartrate salt: mp 128–129 °C; [α]20D −50.0° (c 0.83, CH3OH); 1H NMR (DMSO-d6) δ 7.85 (d, 4H, J = 7.8 Hz), 7.50–7.40 (m, 4H), 7.32 (d, 4H, J = 7.8 Hz), 5.6(s, 2H), 4.07 (d, 1H, J = 12.4 Hz), 3.50–3.40 (m, 1H), 2.56 (s, 3H), 2.36 (s, 6H), 1.40 (s, 3H), 1.21 (s, 3H), 0.90 (d, 3H, J = 6.2 Hz); 13C NMR (DMSO-d6) δ 168.2, 164.9, 143.9, 132.7, 129.9, 129.3 (d), 128.6, 126.67, 126.31, 125.3, 96.4, 72.2, 66.2, 60.4, 59.2, 33.3, 21.1, 15.9, 11.5; Anal. (C34H38ClNO10•2H2O). C, H, N.
(2S,3S)-2-(4-Chlorophenyl)-3,4,5,5-tetramethylmorpholin-2-ol (4v) Fumarate
A sample of (4i) (144 mg, 0.563 mmol) in anhydrous THF (1.9 mL) was treated with K2CO3 (4 folds, 311 mg, 2.25 mmol). After stirring the reaction mixture at room temperature under inert atmosphere for 1 h, CH3I (1.3 equiv., 46.0 μL, 0.731 mmol) was added to the reaction flask. The reaction mixture was stirred at room temperature for 24 h, cooled to 0 °C and water added, followed by extraction with ether (thee times). The combined organic extracts were washed (water, brine), dried (Na2SO4) and concentrated to a pale yellow oil. Purification of the residue by column chromatography on silica gel and CH2Cl2-MeOH-NH4OH (90:9:1) as the eluent gave 93.0 mg (61%) of a white solid: mp = 75–78 °C; [α]22D +28.0° (c 1.0, CHCl3); 1H NMR δ 7.58–7.53 (m, 2H), 7.34–7.31 (m, 2H), 4.52–4.49 (br, 1H), 3.90 (d, 1H, J = 11.6 Hz), 3.32 (d, 1H, J = 11.6 Hz), 2.85 (q, 1H, J = 6.5 Hz), 2.20 (s, 3H), 1.19 (s, 3H), 1.07 (s, 3H), 0.76 (d, 3H, J = 6.5 Hz ); 13C NMR δ 133.9, 128.59, 128.36, 128.05, 127.7, 97.5, 70.4, 59.3, 53.8, 32.3, 25.5, 14.4, 13.1; MS (ESI) m/z 270.4 [(M + H)+ M = C14H20ClNO2].
A sample of 4v (90 mg, 0.33 mmol) in ether (1.5 mL) was treated with a solution of fumaric acid (38 mg, 0.33 mmol) in MeOH (1 mL). The mixture was stirred at room temperature overnight. Ether was added to the reaction mixture. The suspension was sonicated, centrifuged, and decanted to afford a solid pellet; this procedure was repeated three times. Recrystallization from MeOH/ether afforded the title product as white solid 101 mg (77%): mp = 167–169 °C; [α]22D +44.3° (c 1.0, MeOH); 1H NMR (methanol-d4) δ 7.60 (d, 2H, J = 8.6 Hz), 7.43 (d, 2H, J = 8.6 Hz), 6.68 (s, 2H), 4.35 (d, 1H, J = 12.8 Hz), 3.64–3.54 (m, 2H), 2.79 (s, 3H), 1.60 (s, 3H), 1.41 (s, 3H), 1.14 (d, 3H, J = 6.5 Hz); 13C NMR (methanol-d4) δ 171.4, 140.9, 136.3, 129.4, 98.1, 67.6, 63.4, 62.5, 34.3, 21.8, 20.8, 16.9, 12.3; MS (ESI) m/z 270.4 [(M – fumaric)+, M = C14H20ClNO2•C4H4O4]; Anal. (C18H24ClNO6•0.5H2O) C, H, N.
3,5,5-Trimethyl-2-(pyridin-3-yl)morpholin-2-ol (5) D-Tartrate
A sample of 1-(pyridin-3-yl)propan-1-one (8o, 685 mg, 5.00 mmol) was dissolved in CCl4 (20 mL). Bromine (0.26 mL, 5 mmol) was added to the reaction flask, and the reaction mixture was gently refluxed for 1 h. The solvent was decanted. The deep red solid at the bottom of flask was washed with ether, dried, suspended in CH3CN (40 mL), and treated with 2-amino-2-methyl-1-propanol (0.89 g, 0.01 mol). The reaction mixture was stirred for 8 h. The precipitate was removed by filtration and the filtrate was extracted with EtOAc. The organic layer was washed with aqueous NaHCO3, dried (Na2SO4) and concentrated to a deep red oil residue. The oily residue was purified by column chromatography on silica gel and CH2Cl2-MeOH (20:1 to 5:1) with 1% NH4OH) to give 56 mg free base that was converted to 5•tartrate as a yellow solid: mp 115–116 °C; 1H NMR (CDCl3) δ 8.85–8.83 (m, 1H), 8.54 (dd, 1H, J = 4.7, 1.7 Hz), 7.91 (tt, 1H, J = 8.0, 2.0 Hz), 7.31–7.25 (m, 1H), 3.85–3.82 (m, 1H), 3.42 (d, 1H, J = 11.2 Hz), 3.21 (q, 1H, J = 6.5 Hz), 1.40 (s, 3H), 1.10 (s, 3H), 0.81 (d, 3H, J = 6.5 Hz); 13C NMR (CDCl3) δ 149.4, 148.1, 137.2, 134.1, 122.8, 95.4, 69.4, 53.55, 49.8, 27.4, 22.8, 16.4; LCMS (ESI) m/z 223.3 [(M – tartrate)+, M = C16H24N2O8]; Anal. (C16H24N2O8•0.75H2O). C, H, N.
3,5,5-Trimethyl-2-(pyridin-2-yl)morpholin-2-ol (6) D-Tartrate
1-(Pyridin-2-yl)propan-1-one (8n, 1.71 g, 0.013 mol) was dissolved in CCl4 (50 mL). Bromine (0.66 mL, 12.7 mmol) was added and gently refluxed for 1 h. The solvent was decanted. The orange solid at the bottom of flask was washed with ether, vacuum dried, and then suspend in CH3CN (40 mL), followed by the addition of 2-amino-2-methyl-1-propanol (2.30 g, 0.0254 mol). The reaction mixture was stirred for 24 h. The precipitate was separated by filtration and was extracted with EtOAc. The organic layer was washed with aqueous NaHCO3 and dried (Na2SO4) and concentrated to give a orange oil. The residue was purified by column chromatography on silica gel using, CH2Cl2-MeOH (50:1 to 20:1) with 1% NH4OH) to give 0.94 g (33%) of free base that was converted to the corresponding tartrate salt: 1H NMR (CDCl3) δ 8.55–8.51 (m, 1H), 7.84–7.59 (m, 1H), 7.63–7.57 (m, 1H), 7.34–7.26 (m, 1H), 6.00 (s, 1H), 3.97–3.91 (m, 1H), 3.47–3.37 (m, 2H), 1.42 (s, 3H), 1.10 (s, 3H), 0.74 (d, 3H, J = 6.5 Hz); 13C NMR (CDCl3) δ 158.6, 147.2, 137.5, 123.6, 120.5, 94.2, 70.0, 52.1, 48.9, 27.1, 22.9, 16.2; LCMS (ESI) m/z 223.4 [(M – tartrate)+, M = C16H24N2O8]; Anal. (C16H24N2O8•0.5H2O). C, H, N.
1-Naphthalen-1-ylpropan-1-one (8q)
A sample of 1-cyanonaphthylene (7a, 5.0 g, 0.0327 mol) in dry diethyl ether (200 mL) was treated with ethyl magnesium bromide (49.2 mol, 16.4 mL 3 M solution in ether) under nitrogen atmosphere at room temperature. The solution was stirred overnight at 25 °C and then cooled to 0 °C. The reaction was quenched slowly with 1 N aqueous HCl and allowed to warm to room temperature over 1 h with stirring. The aqueous layer was extracted with ether. The combined organic layers were washed with aqueous NaHCO3, water and brine, and dried (Na2SO4). The organic layers were concentrated and the resulting yellow oil was purified by column chromatography on silica gel using hexane-EtOAc (20:1) to (10:1) as the eluent afforded 3.55 g (60%) of the title product: 1H NMR (CDCl3) δ 8.59–8.52 (m, 1H), 8.00–7.94 (m, 1H), 7.90–7.81 (m, 2H), 7.62–7.45 (m, 3H), 3.08 (q, 2H, J = 7.3 Hz), 1.29 (t, 3H, J = 7.3 Hz). C13H12O.
1-Naphthalen-2-ylpropan-1-one (8r)
Compound 8r was synthesized by a procedure similar to that described for a procedure similar to 8q using identical amounts, except commercially available 2-cyanonaphthylene (7b) was used in place of 1-cyanonaphthylene. Purification by column chromatography on silica gel using hexane-EtOAc (20:1) to (10:1) afforded 4 g (66%) of the title product as a white solid: 1H NMR (CDCl3) δ 8.48 (s, 1H), 8.05 (d, 1H, J = 7.5 Hz), 8.00–7.94 (m, 1H), 7.93–7.85 (m, 2H), 7.64–7.51 (m, 2H). 3.15 (q, 2H, J = 7.2 Hz), 1.29 (t, 3H, J = 7.2 Hz); 13C NMR (CDCl3) δ 129.9, 128.8 (d), 128.2, 127.1, 124.3, 32.3, 8.8. C13H12O.
(Z)-tert-Butyl(1-phenylprop-1-enyloxy)dimethylsilane (9b)
Compound 9b was synthesized by a procedure similar to 9a using commercially available propiophenone (8b, 5.0 g, 0.037 mol), TBDMSOTf (9.4 mL, 41.0 mmol), and of Et3N (8.3 mL) in CH2Cl2 (50 mL). After purification, 7.32 g (79%) of 9b was isolated as colorless oil: 1H NMR (CDCl3) δ 7.50–7.40 (m, 2H), 7.35–7.22 (m, 3H), 5.23 (q, 1H, J = 6.9 Hz), 1.77 (d, 3H, J = 6.9 Hz), 1.03 (s, 9H), −0.04 (s, 6H); 13C NMR (CDCI3) δ 150.4, 140.0, 128.0, 127.4, 125.9, 105.9, 25.9, 18.5, 11.9, −3.9; C15H24OSi.
(Z)-tert-Butyl(1-(3-fluorophenyl)prop-1-enyloxy)dimethylsilane (9c)
Compound 9c was synthesized by a procedure similar to that described for 9a using commercially available 3-fluoropropiophenone (8c, 5.0 g, 0.033 mol), TBDMSOTf (8.3 mL, 36.1 mmol), and Et3N (7.3 mL) in CH2Cl2 (50 mL). After purification, 8.1 g (93%) of 9c was isolated as colorless oil: 1H NMR (CDCl3) δ 7.38–7.10 (m, 2H), 7.00–6.87 (m, 1H), 5.26 (q, 1H, J = 6.9 Hz), 1.75 (d, 3H, J = 6.9 Hz), 1.00 (s, 9H), −0.02 (s, 6H); 13C NMR (CDCl3) δ 164.5, 161.2, 149.2, 142.4, 129.4, 121.4, 114.3, 114.0, 112.8, 112.5, 107.1, 105.9, 26.0, 18.5, 11.9, −3.9. C15H23FOSi.
(Z)-tert-Butyl(1-(3-bromophenyl)prop-1-enyloxy)dimethylsilane (9d)
Compound 9d was synthesized by a procedure similar to that described for 9a using commercially available 3-bromopropiophenone (8d, 1.0 g, 0.0047 mol), TBDMSOTf (1.4 mL, 5.1 mmol), and Et3N (1.1 mL) in CH2Cl2 (10 mL). After workup, 1.15 g (75%) of crude 9d was isolated as colorless oil with a Z-E ratio of 94:6: 1H NMR (CDCl3) δ 7.64–7.58 (m, 1H), 7.41–7.34 (m, 2H), 7.17 (t, 1H, J = 8.04 Hz), 5.25 (q, 1H, J = 6.9 Hz), 1.75 (d, 3H, J = 6.9 Hz), 1.02 (s, 9H), −0.03 (s, 6H); 13C NMR (CDCl3) δ 148.8, 141.9, 130.2, 129.5, 128.7, 124.1, 122.1, 25.8, 18.3, 11.8, −4.0. C15H23BrOSi.
(Z)-tert-Butyl(1-(M-tolyl)prop-1-enyloxy)dimethylsilane (9e)
Compound 9e was synthesized by a procedure similar to that described for 9a using commercially available 3-methylpropiophenone (8e, 5.0 g, 0.033 mol), TBDMSOTf (8.5 mL, 36.1 mmol), and Et3N (7.5 mL) in CH2Cl2 (50 mL). After purification by column chromatography on silica gel, 7.45 g (84%) of 9e was isolated as colorless oil: 1H NMR (CDCl3) δ 7.31–7.24 (m, 2H), 7.23–7.17 (m, 1H), 7.10–7.05 (m, 1H), 5.22 (q, 1H, J = 6.9 Hz), 2.37 (s, 3H), 1.76 (d, 3H, J = 6.9 Hz), 1.03 (s, 9H), −0.03 (s, 6H); 13C NMR (CDCl3) δ 150.7, 140.1, 137.7, 128.4, 127.9, 126.8, 123.2, 105.9, 26.3, 21.8, 18.7, 12.1, −3.6. C16H26OSi.
(Z)-tert-Butyl-(1-(3-methoxyphenyl)prop-1-enyloxy)dimethylsilane (9f)
Compound 9f was synthesized by a procedure similar to that described for 9a using 3-methoxypropiophenone (8f, 6.00 g, 0.0366 mol), TBDMSOTf (9.2 mL, 40.2 mmol), and Et3N (8.1 mL) in CH2Cl2 (60 mL). After purification on alumina, 8.14 g (80%) of 9f was isolated as colorless oil: 1H NMR (CDCl3) δ 7.24–7.16 (m, 1H), 7.07–6.98 (m, 2H), 6.84–6.76 (m, 1H), 5.23 (q, 1H, J = 6.9 Hz), 3.81 (s, 3H), 1.75 (d, 3H, J = 6.9 Hz), 1.01 (s, 9H), −0.01 (s, 6H); 13C NMR (CDCl3) δ 159.4, 150.1, 141.5, 129.0, 118.4, 113.3, 111.2, 106.1, 55.3, 26.0, 18.5, 11.9, −3.9. C16H26O2Si.
(Z)-tert-Butyl-(1-(3-nitrophenyl)prop-1-enyloxy)dimethylsilane (9g)
Compound 9g was synthesized by a procedure similar to that described for 9a using commercially available 3-nitropropiophenone (8g, 5.0 g, 0.028 mol), TBDMSOTf (7.1 mL, 30.7 mmol), and Et3N (6.2 mL) in CH2Cl2 (50 mL). After purification on alumina, 8.0 g (98%) of 9g was isolated as colorless oil: 1H NMR (CDCl3) δ 8.34–8.30 (m, 1H), 8.12–8.06 (m, 1H), 7.80–7.75 (m, 1H), 7.52–7.42 (m, 1H), 5.40 (q, 1H, J = 6.9 Hz), 1.78 (d, 3H, J = 6.9 Hz), 1.02 (s, 9H), −0.02 (s, 6H); 13C NMR (CDCI3) δ 148.8, 141.6, 131.3, 129.0, 122.1, 120.5, 108.6, 106.8, 25.9, 18.4, 12.0, −3.8. C15H23NO3Si.
(Z)-tert-Butyldimethyl-(1-(naphthalen-1-yl)prop-1-enyloxy)silane (9q)
Compound 9q was synthesized by a procedure similar to that described for 9a using 1-naphthalen-1-ylpropan-1-one (8h, 3.5 g, 0.019 mol), TBDMSOTf (4.8 mL, 0.021 mol), and Et3N (4.3 mL) in CH2Cl2 (26 mL). After purification, 4.58 g (81%) of title product was isolated as colorless oil: 1H NMR (CDCl3) δ 8.27–8.24 (m, 1H), 7.83–7.75 (m, 2H), 7.48–7.38 (m, 4H), 5.04 (q, 1H, J = 6.0 Hz), 1.83 (d, 3H, J = 6.0 Hz), 0.87 (s, 9H), −0.29 (s, 6H); 13C NMR (CDCl3) δ 150.0, 138.3, 133.6, 131.6, 128.1, 128.0, 126.8, 126.2, 125.7, 125.1, 108.4, 26.3, 25.7, 18.2, −4.6. C19H26OSi.
(Z)-tert-Butyldimethyl-(1-(naphthalen-2-yl)prop-1-enyloxy)silane (9r)
Compound 9r was synthesized by a procedure similar to that described for 9a using 1-naphthalen-2-ylpropan-1-one (8i, 4.0 g, 0.0217 mol, TBDMSOTf (5.5 mL, 23.9 mmol), and Et3N (4.9 mL) in CH2Cl2 (26 mL). After purification, 4.7 g (73%) of the title product was isolated as colorless oil: 1H NMR (CDCl3) δ 7.86–7.74 (m, 1H), 7.86–7.74 (m, 3H), 7.59 (dd, 1H, J = 8.6, 1.7 Hz), 7.51–7.41 (m, 2H), 5.38 (q, 1H, J = 6.9 Hz), 1.81 (d, 3H, J = 6.9 Hz), 1.04 (s, 9H), 0.02 (s, 6H); 13C NMR (CDCl3) δ 150.5, 137.5, 133.5, 133.2, 128.5, 127.9, 127.8, 126.4, 126.0, 107.0, 105.9, 26.3, 26.1, 18.8, −3.6. C19H26OSi.
(Z)-tert-Butyl(1-(3-chlorophenyl)but-1-enyloxy)dimethylsilane (9s)
Compound 9s was synthesized by a procedure similar to that described for 9a using 3-chlorobutyrophenone (8j, 3.1 g, 0.0169 mol), TBDMSOTf (4.3 mL, 18.6 mmol), and Et3N (3.8 mL) in CH2Cl2 (30 mL). After purification, 3.7 g (74%) of the title product was isolated as colorless oil: 1H NMR (CDCl3) δ 7.48–7.44 (m, 1H), 7.38–7.32 (m, 1H), 7.31–7.29 (m, 2H), 5.16 (t, 1H, J = 7.1 Hz), 2.30–2.19 (m, 2H), 1.09– 1.03 (m, 3H), 1.01 (s, 9H), −0.04 (s, 6H); 13C NMR (CDCl3) δ 147.5, 141.7, 133.9, 129.2, 127.3, 125.9, 123.8, 114.9, 25.8, 19.5, 14.1, 0.0, −4.1. C16H25ClOSi.
(Z)-tert-Butyl(1-(3-chlorophenyl)pent-1-enyloxy)dimethylsilane (9t)
Compound 9t was synthesized by a procedure similar to that described for 9a using 3-chloropentaphenone (8k, 2.7 g, 0.014 mol), TBDMSOTf (3.5 mL, 15 mmol), and Et3N (3.1 mL) in CH2Cl2 (25 mL). After purification, 4.18 g (96%) of the title product was isolated as colorless oil: 1H NMR (CDCl3) δ 7.47–7.44 (m, 1H), 7.38–7.32 (m, 1H), 7.30–7.28 (m, 1H), 7.25–7.22 (m, 1H), 5.18 (t, 1H, J = 7.2 Hz), 2.20 (q, 2H, J = 7.5 Hz), 1.54–1.38 (m, 2H), 1.02 (s, 9H), 1.00–0.90 (m, 3H), −0.05 (s, 6H); 13C NMR (CDCl3) δ 148.0, 141.8, 133.9, 129.2, 127.6, 125.9, 123.9, 113.0, 28.3, 25.8, 22.8, 14.0, 0.0, −4.01. C17H27ClOSi.
(S)-1-(3-Chlorophenyl)-2-hydroxypropan-1-one [(S)-10a]
Compound (S)-10a was synthesized by a procedure similar to that described for (R)-10a using (Z)-tert-butyl(1-(3-chlorophenyl)prop-1-enyloxy)dimethylsilane (9a, 8.8 g, 0.031 mol), AD-mix-α (43.5 g), and CH3SO2NH2 (3a, 3 g, 0.032 mol) in tert-butyl alcohol-water (120 mL:120 mL). The reaction mixture was quenched with sodium sulfite (31.1 g). After purification, 4.51 g (78%) of the (S)-10a was isolated. Characterization data is similar with data reported in the literature.24
(R)-1-Phenyl-2-hydroxypropan-1-one (10b)
Compound 10b was synthesized by a procedure similar to that described for (R)-10a using (Z)-tert-butyl(1-phenprop-1-enyloxy)dimethylsilane (9b, 7.2 g, 0.029 mol), AD-mix-β 40.7 g), and CH3SO2NH2 (2.8 g, 0.0294 mol) in tert-butyl alcohol-water (110 mL:110 mL). The reaction was quenched with sodium sulfite (29.1 g). After purification, 2.49 g (80%) of the title product was isolated: [α]20D +84.9° (c 1.6, CHCl3); 1H NMR (CDCI3) δ 7.96–7.90 (m, 2H), 7.66–7.58 (m, 1H), 7.54–7.47 (m, 2H), 5.23–5.12 (m, 1H), 3.84 (d, 1H, J = 6.3 Hz), 1.45 (d, 3H, J = 7.05 Hz); 13C NMR (CDCI3) δ 202.4, 134.0, 133.4, 128.9, 128.7, 69.3, 22.3. C9H10O2.
(R)-1-(3-Fluorophenyl)-2-hydroxypropan-1-one (10c)
Compound 10c was synthesized by a procedure similar to that described for (R)-10a using (Z)-tert-butyl(1-(3-fluorophenyl)prop-1-enyloxy)dimethylsilane, (9c, 8.0 g, 0.030 mol), AD-mix-β 42.1 g), and CH3SO2NH2 (2.90 g, 0.0301 mol) in tert-butyl alcohol-water (120 mL:120 mL). The reaction was quenched with sodium sulfite (30.1 g). After purification, 4.4 g (87%) of the desired product 10c was isolated: [α]20D +58.1° (c 3.2, CHCl3); 1H NMR (CDCl3) δ 7.74–7.59 (m, 2H), 7.54–7.45 (m, 1H), 7.37–7.28 (m, 1H), 5.19– 5.07 (m, 1H), 1.76 (d, 1H, J = 6.3 Hz), 1.46 (d, 3H, J = 6.9 Hz); 13C NMR (CDCl3) δ 201.6, 164.9, 131.0, (d), 124.7, 121.3 (d), 115.8 (d), 69.9, 22.4. C9H9FO2.
(R)-1-(3-Bromophenyl)-2-hydroxypropan-1-one (10d)
Compound 10d was synthesized by a procedure similar to that described for (R)-10a using (Z)-tert-butyl(1-(3-bromophenyl)prop-1-enyloxy)dimethylsilane, (9d, 1.15 g, 0.0035 mol), AD-mix-β 4.9 g), and CH3SO2NH2 (334 mg, 3.5 mmol) in tert-butyl alcohol-water (17.5 mL:17.5 mL). The reaction was quenched with sodium sulfite (3.5 g). After purification by column chromatography on silica gel, 0.700 g (88%) of the desired product was isolated: [α]20D +61.1° (c 1.3, CHCl3); 1H NMR (CDCl3) δ 8.09–8.04 (m, 1H), 7.87–7.81 (m, 1H), 7.78–7.72 (m, 1H), 7.39 (t, 1H, J = 8.0 Hz), 5.18–5.05 (m, 1H), 3.66 (d, 1H, J = 6.4 Hz), 1.45 (d, 3H, J = 7.2 Hz); 13C NMR (CDCl3) δ 201.2, 136.8, 135.2, 131.6, 130.4, 127.1, 123.3, 69.5, 22.1. C9H9BrO2.
(R)-1-(M-Tolyl)-2-hydroxypropan-1-one (10e)
Compound 10e was synthesized by a procedure similar to that described for (R)-10a using (Z)-tert-butyl(1-(3-methylphenyl)prop-1-enyloxy)dimethylsilane, (9e, 7.4 g, 0.028 mol), AD-mix-β 39.5 g), and CH3SO2NH2 (2.73 g, 0.029 mol) in tert-butyl alcohol-water (110 mL:110 mL). The reaction was quenched with sodium sulfite (28.3 g). After purification, 4.2 g (85%) of the desired 10e was isolated: [α]20D +83.9° (c 2.0, CHCl3); 1H NMR (CDCl3) δ 7.78–7.67 (m, 2H), 7.46–7.33 (m, 2H), 5.20–5.09 (m, 1H), 3.86 (d, 1H, J = 6.3 Hz), 2.42 (s, 3H), 1.44 (d, 3H, J = 7.0 Hz); 13C NMR (CDCl3) δ 202.6, 138.8, 134.8, 133.5, 129.1, 128.7, 125.9, 69.4, 22.3, 21.4. C10H12O2.
(R)-1-(3-Methoxyphenyl)-2-hydroxypropan-1-one (10f)
Compound 10f was synthesized by a procedure similar to that described for (R)-10a using (Z)-tert-butyl(1-(3-methoxyphenyl)prop-1-enyloxy)dimethylsilane, (9f, 8.1 g, 0.029 mol), AD-mix-β 40.7 g), and CH3SO2NH2 (2.8 g, 0.0294 mol) in tert-butyl alcohol-water (110 mL:110 mL). The reaction was quenched with sodium sulfite (29.1 g). After purification, 4.2 g (80%) of the desired 10f was isolated: [α]20D +71.1° (c 1.1, CHCl3); 1H NMR (CDCl3) δ 7.51–7.45 (m, 2H), 7.44–7.37 (m, 1H), 7.19–7.13 (m, 1H), 5.19–5.09 (m, 1H), 3.87 (s, 3H), 3.76 (d, 1H, J = 6.5 Hz), 1.45 (d, 3H, J = 7.1 Hz); 13C NMR (CDCl3) δ 202.3, 160.0, 134.7, 129.9, 121.1, 120.3, 113.1, 69.4, 55.5, 22.4. C10H12O3.
(R)-1-(3-Nitrophenyl)-2-hydroxypropan-1-one (10g)
Compound 10g was synthesized by a procedure similar to that described for (R)-10a using (Z)-tert-butyl(1-(3-nitrophenyl)prop-1-enyloxy)dimethylsilane, (9g, 8.0 g, 0.027 mol), of AD-mix-β (38 g), and CH3SO2NH2 (2.64 g, 0.0277 mol) in tert-butyl alcohol-water (110 mL:110 mL). The reaction was quenched with sodium sulfite (27.4 g). After purification by column chromatography on silica gel, 4.0 g (75%) of the desired product was isolated: [α]20D +63.8° (c 1.3, CHCl3); 1H NMR (CDCI3) δ 8.80–8.74 (m, 1H), 8.52–8.44 (m, 1H), 8.31–8.24 (m, 1H), 7.83–7.68 (m, 1H), 5.27–5.13 (m, 1H), 3.59 (d, 1H, J = 6.5 Hz), 1.49 (d, 3H, J = 7.08 Hz); 13C NMR (CDCI3) δ 200.4, 148.6, 134.9, 134.1, 130.2, 128.1, 121.6, 69.8, 21.9; C9H9NO4.
(R)-2-Hydroxy-1-(naphthalen-1-yl)propan-1-one (10h)
Compound 10h was synthesized by a procedure similar to that described for (R)-10a using (Z)-tert-butyl(1-(naphthalen-1-yl)prop-1-enyloxy)dimethylsilane, (9h, 4.58 g, 0.015 mol), AD-mix-β 21.4 g), and CH3SO2NH2 (1.5 g, 0.0158 mol) in tert-butyl alcohol-water (60 mL:60 mL). The reaction was quenched with sodium sulfite (15.3 g). After purification by column chromatography on silica gel, 1.15 g (38%) of the title product was isolated plus 2.1 g of starting olefin was recoverd raising the effective yield to 70%: [α]20D +140.2° (c 3.2, CHCl3); 1H NMR (CDCl3) δ 8.51–8.46 (m, 1H), 8.04 (d, 1H, J = 8.3 Hz), 7.93–7.87 (m, 1H), 7.80–7.75 (m, 1H), 7.66–7.48 (m, 3H), 5.30–5.17 (m, 1H), 3.96 (d, 1H, J = 5.8 Hz), 1.36 (d, 3H, J = 7.1 Hz); 13C NMR (CDCl3) δ 205.7, 134.1, 133.5, 132.5, 130.7, 128.7, 128.4, 127.7, 126.9, 125.5, 124.4, 71.2, 21.3; LCMS (ESI) m/z 201.2 [(M + H)+, M = C13H12O2].
(R)-2-Hydroxy-1-(naphthalen-2-yl)propan-1-one (10i)
Compound 10i was synthesized by a procedure similar to that described for (R)-10a using (Z)-tert-butyl(1-(naphthalen-2-yl)prop-1-enyloxy)dimethylsilane, (9i, 4.7 g, 0.016 mol), AD-mix-β 22.0 g), and CH3SO2NH2 (1.55 g, 0.016 mol) in tert-butyl alcohol-water (60 mL:60 mL). The reaction was quenched with sodium sulfite (15.7 g). After purification by column chromatography on silica gel, 2.5 g (80%) of the desired product was isolated: [α]20D +115° (c 0.7, CHCl3); 1H NMR (CDCl3) δ 8.44 (s, 1H), 8.01–7.86–8.00 (m, 4H), 7.69–7.54 (m, 2H), 5.37–5.27 (m, 1H), 3.86 (d, 1H, J = 6.5 Hz), 1.52 (d, 3H, J = 7.0 Hz); 13C NMR (CDCl3) δ 202.3, 136.0, 132.4, 130.7, 130.5, 129.7, 129.0, 128.8, 127.9, 127.1, 124.0, 69.4, 22.5. C13H12O2.
(R)-1-(3-Chlorophenyl)-2-hydroxybutan-1-one (10j)
Compound 10j was synthesized by a procedure similar to that described for (R)-10a using (Z)-tert-butyl(1-(3-chlorophenyl)but-1-enyloxy)dimethylsilane (9j, 3.7 g, 0.013 mol), AD-mix-β 17.5 g), and CH3SO2NH2 (1.2 g, 0.0126 mol) in tert-butyl alcohol-water (45 mL:45 mL). The reaction was quenched with sodium sulfite (12.5 g). After purification by column chromatography on silica gel, 2.2 g (79%) of the title product was isolated: [α]20D +31.4° (c 1.0, CHCl3); 1H NMR (CDCl3) δ 7.91–7.88 (m, 1H), 7.81–7.75 (m, 1H), 7.62–7.56 (m, 1H), 7.45 (t, 1H, J = 7.8 Hz), 5.06–4.98 (m, 1H), 3.60 (d, 1H, J = 6.5 Hz), 2.04–1.87 (m, 1H), 1.70–1.51 (m, 1H), 0.94 (t, 3H, J = 7.4 Hz); 13C NMR (CDCl3) δ 201.4, 135.8, 135.7, 134.2, 130.6, 128.9, 126.9, 74.5, 29.1, 9.2. C10H11ClO2.
(R)-1-(3-Chlorophenyl)-2-hydroxypentan-1-one (10k)
Compound 10k was synthesized by a procedure similar to that described for (R)-10a using (Z)-tert-butyl(1-(3-chlorophenyl)pent-1-enyloxy)dimethylsilane (9k, 4.1 g, 0.013 mol), AD-mix-β (18.5 g), and CH3SO2NH2 (1.3 g, 0.014 mol) in tert-butyl alcohol-water (50 mL:50 mL). The reaction was quenched with sodium sulfite (13.2 g). After purification, 2.4 g (77%) of the desired product was isolated: [α]20D +33.3° (c 1.1, CHCl3); 1H NMR (CDCl3) δ 7.91–7.87 (m, 1H), 7.80–7.75 (m, 1H), 7.63–7.17 (m, 1H), 7.45 (t, 1H, J = 7.6 Hz), 5.08–5.00 (m, 1H), 3.58 (d, 1H, J = 6.5 Hz), 1.89–1.75 (m, 1H), 1.61–1.35 (m, 3H), 0.93 (t, 3H, J = 7.2 Hz); 13C NMR (CDCl3) δ 201.1, 135.4, 133.8, 130.2, 128.6, 126.5, 73.2, 37.8, 18.2, 13.8. C11H13ClO2.
Methyl (2R)-2-[(trifluoromethyl)sulfonyl]oxypropionate) (14)
Following a reported procedure27 with modification, a solution of methyl-(R)-lactate (13) (5.20 g, 0.05 mol) in anhydrous CH2Cl2 (200 mL, 0.25 M) was cooled to 0 °C and was treated with trifluromethane sulfonic anhydride (8.8 mL, 52.5 mmol) and 2,6-lutidine (6.10 mL, 52.5 mmol) under an N2 atmosphere. After stirring for 20 min at 0 °C, the reaction mixture was concentrated to a pink oil residue. Column chromatography on silica gel using CH2Cl2 as the eluent afforded 9.15 g (77%) of 14 as a light pink oil with characterization data as previously reported:32 [α]25D +40.5° (c 1.0, CHC13); 1H NMR (CDCl3) δ 5.27 (q, 1H, J = 6.9 Hz), 3.85 (s, 3H), 1.71 (d, 3H, J = 6.9 Hz); 13C NMR (CDCl3) δ 167.8, 79.9, 53.3, 18.0; LCMS (ESI) m/z 240.1 [(M + 4H)+, M = C5H7F3O5S].
(3S)-3,5,5-Trimethylmorpholin-2-one (15)
A solution of triflate 14 (5.00 g, 0.021 mol) in anhydrous CH2Cl2 (80 mL) under an N2 atmosphere, was cooled to −40 °C and was treated with a solution of 2-amino-2-methyl-1-propanol (2.5 folds, 4.68 g, 0.0525 mol) in anhydrous CH2Cl2 (10 mL). After stirring for 2 h at −40 °C, the reaction mixture was warmed slowly to 0 °C then to room temperature and stirred overnight. The reaction mixture was treated with saturated aqueous NaHCO3 solution (100 mL). The organic phase was washed (water, brine), separated, and dried (Na2SO4). The aqueous layer was extracted with EtOAc (twice). The organic layer was separated, washed (water, brine) and dried (Na2SO4). The organic extracts were combined and concentrated to give a yellow oil. Column chromatography on silica gel using hexanes-EtOAc (1:2) to EtOAc gave 1.88 g (63%) of 15 as a light yellow oil: [α]23D −75° (c 1.0, CHC13); 1H NMR (CDCl3) δ 4.11 (s, 2H), 3.71 (q, 1H, J = 6.8), 1.39 (d, 3H, J = 6.8 Hz), 1.26 (s, 3H), 1.18 (s, 3H); 13C NMR (CDCl3) δ 229.2, 77.6, 49.5, 49.3, 27.2, 24.2, 18.4; LCMS (APCI) m/z 144.3 [(M + H)+, M = C7H13NO2].
Note: 1H NMR data is similar with the data reported in the literature for the racemic compound.33
Cell lines and culture
Human embryonic kidney (HEK-293) cells stably expressing human DAT, NET or SERT were maintained as previously described.26
Use was made of several human cell lines that naturally or heterologously express specific, functional, human nAChR subtypes.34 Cells of the TE671/RD line naturally expresses muscle-type nAChR (α1β1γδ- or α1*-nAChR), and SH-SY5Y neuroblastoma cells naturally expresses autonomic α3β4*-nAChRs (containing α3, β4, probably α5, and sometimes β2 subunits). Different clones of SH-EP1 epithelial cell lines have been engineered to heterologously express either α4β2-nAChR, which are thought to be the most abundant, high affinity nicotine-binding nAChR in mammalian brain, or α4β4-nAChR, another possible brain nAChR subtype (SH-EP1-hα4β2 or α4β4 cells, respectively).35,36 These cells were maintained as low passage number (1–26 from our frozen stocks) cultures to ensure stable expression of native or heterologously-expressed nAChR as previously described.37 Cells were passaged once weekly by splitting just-confluent cultures 1/300 (TE671/RD), 1/5 (SH-SY5Y), or 1/20 (transfected SH-EP1) in serum-supplemented medium to maintain log-phase growth.
Transporter Assays
The abilities of 2 and its analogues to inhibit uptake of [3H]dopamine ([3H]DA), [3H]serotonin ([3H]5-HT), or [3H]norepinephrine ([3H]NE) by the respective, human transporters were evaluated using the appropriate HEK-293 cell line as previously reported.26
nAChR Functional Assays
Cells were harvested at confluence from 100-mm plates by mild trypsinization (Irvine Scientific, Santa Ana, CA) and trituration or (for SH-SY5Y cells) by trituration alone before being suspended in complete medium and evenly seeded at a density of 1.25–2 confluent 100-mm plates per 24-well plate (Falcon; ~100–125 μg of total cell protein per well in a 500 μL volume). After cells had adhered (generally overnight, but no sooner than 4 h later), the medium was removed and replaced with 250 μL per well of complete medium supplemented with ~350,000 cpm of 86Rb+ (PerkinElmer Life and Analytical Sciences, Boston, MA) and counted at 40% efficiency using Cerenkov counting (TriCarb 1900 liquid scintillation analyzer, 59% efficiency; PerkinElmer Life Sciences). After at least 4 h and typically overnight, 86Rb+ efflux was measured using the “flip-plate” technique.37 Briefly, after aspiration of the bulk of 86Rb+ loading medium from each well of the “cell plate,” each well containing cells was rinsed 3× with 2 mL of fresh 86Rb+ efflux buffer (130 mM NaCl, 5.4 mM KCl, 2 mM CaCl2, 5 mM glucose, 50 mM HEPES, pH 7.4) to remove extracellular 86Rb+. Following removal of residual rinse buffer by aspiration, the flip-plate technique was used again to simultaneously introduce 1.5 mL of fresh efflux buffer containing drugs of choice at indicated final concentrations from a 24-well “efflux/drug plate” into the wells of the cell plate. After a 5 min incubation, the solution was “flipped” back into the efflux/drug plate, and any remaining buffer in the cell plate was removed by aspiration. Cells remaining in the cell plate were lysed and suspended by addition of 1.5 mL of 0.1 M NaOH, 0.1% sodium dodecyl sulfate to each well. Suspensions in each well were then subjected to Cerenkov counting (Wallac Micobeta Trilux 1450; 25% efficiency) after placement of inserts (Wallac 1450–109) into each well to minimize cross-talk between wells.
For quality control and normalization purposes, the sum of 86Rb+ in cell plates and efflux/drug plates was defined to confirm material balance (i.e., that the sum of 86Rb+ released into the efflux/drug plates and 86Rb+ remaining in the cell plate were the same for each well). This assured that 86Rb+ efflux was the same whether measured in absolute terms or as a percentage of loaded 86Rb+. Similarly, the sum of 86Rb+ in cell plates and efflux/drug plates also determined the efficiency of 86Rb+ loading (the percentage of applied 86Rb+ actually loaded into cells).
Control, total 86Rb+ efflux was assessed in the presence of only a fully efficacious concentration of carbamylcholine (1 mM for SH-EP1-hα4β2, SH-EP1-hα4β4 cells or TE671/RD cells; 3 mM for SH-SY5Y cells). Control, non-specific 86Rb+ efflux was measured either in the presence of the fully efficacious concentration of carbamylcholine plus 100 μM mecamylamine, which gave full block of agonist-induced and spontaneous nAChR-mediated ion flux, or in the presence of efflux buffer alone. Either determination of non-specific efflux was equivalent. Specific efflux was then taken as the difference in control samples between total and non-specific 86Rb+ efflux. Any intrinsic agonist activity of test drugs was ascertained using samples containing test drug only at different concentrations and was normalized, after subtraction of non-specific efflux, to specific efflux in test drug-free, control samples. Antagonism of carbamylcholine-evoked 86Rb+ efflux was assessed in samples containing the full agonist at a concentration where it stimulates 80–90% of maximal function (i.e., its EC80–EC90 value) when exposed alone to a given nAChR subtype (i.e, 460 μM for TE671/RD cells, 2 mM for SH-SY5Y cells; 200 μM for SH-EP1-hα4β2 or -α4β4 cells) and test drugs at the concentrations shown. After subtraction of non-specific efflux, results were normalized to specific ion flux in control samples. For studies of mechanism of antagonism, concentration-response curves were obtained using samples containing the full agonist, carbamylcholine, at the indicated concentrations alone or in the presence of a concentration of the test ligand close to its IC50 value for inhibition of nAChR function.
Ion flux assay results were fit using Prism (GraphPad) to the Hill equation, F = Fmax/(1 + (X/Z)n), where F is the test sample specific ion flux as a percentage of control, Fmax is specific ion flux in the absence of test drug (i.e., for control samples), X is the test ligand concentration, Z is the EC50 (n>0 for agonists) or IC50 (n<0 for antagonists), and n is the Hill coefficient. All concentration-ion flux response curves were simple and fit well allowing maximum and minimum ion flux values to be determined by curve fitting, but in cases where antagonists had weak functional potency, minimum ion flux was set at 0% of control. Note that because agonist concentrations used for test ligand antagonism assessments were EC80–EC90 values, not all of the data, even at the lowest concentrations of test antagonist, approaches 100% of specific efflux as separately determined in sister samples exposed to fully efficacious concentrations of agonist.
Behavior
Mice were tested for nicotine-induced antinociception, hypothermia and hypomotility. The complex set of nicotinic effects makes it difficult to measure a single representative “nicotinic” acute response in animals. We opted instead in our study for a battery of tests where these different effects of the drug are measured. Factors such as agonist potency, time-course, site of action and nicotinic receptor subtypes mediating these effects, were taken into consideration. For obvious reasons, these nicotinic effects measured are centrally-mediated. Although little is known about the different nicotinic receptor subtypes activated for some of these different responses, the antinociceptive effects of nicotine were among the best described. Recent report showed that neuronal α4β2 nAChRs subtypes are involved in nicotine-induced antinociception in the tail-flick and hot-plate tests. However, the primary sites of action are different for the two tests (the tail-flick assay involves a spinal reflex but in contrast, supraspinal sites are more likely to be involved in the hot-plate test). Moreover, spinal sites seems to involve both α4β2 and non-α4β2 receptors, whereas supraspinal sites are more likely to involve α4β2 neuronal subtypes as major component.38 We therefore decided to measure nicotinic responses in both pain tests. All animal experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and Institutional Animal Care and Use Committee guidelines.
Animals
Male Institute of Cancer Research (ICR) mice (weighing 20–25 g) obtained from Harlan (Indianapolis, IN) were used throughout the study. Animals were housed in an Association for Assessment and Accreditation of Laboratory Animal Care-approved facility, were placed in groups of six, and had free access to food and water. Studies were approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
Tail-Flick Test
Antinociception for pain mediated at the spinal level was assessed by the tail-flick method of D’Amour and Smith.39 In brief, mice were lightly restrained while a radiant heat source was shone onto the upper portion of the tail. To minimize tissue damage, a maximum latency of 10 s was imposed. Latency to remove the tail from the heat source was recorded for each animal. A control response (2–4 s) was determined for each mouse before treatment, and a test latency was determined after drug administration (nicotine as an analgesic 5 min after subcutaneous administration at 2.5 mg/kg; nicotine administration 15 min after exposure to saline of bupropion analogue to assess the latter drug’s ability to block nicotine-mediated antinociception). Antinociceptive response was calculated as the percentage of maximum possible effect (%MPE), where %MPE = [(test control)/(10 control)] × 100.
Hot-Plate Test
Mice were placed into a 10-cm wide glass cylinder on a hot plate (Thermojust Apparatus) maintained at 55 °C for assessment of pain responses mediated at supraspinal levels. To minimize tissue damage, a maximum exposure to the hot plate 40 s was imposed. Measures of control latencies (time until the animal jumped or licked its paws; typically 8–12 s) were done twice for stimuli applied at least 10 min apart for each mouse. Antinociceptive responses after test drug administrations were determined and calculated as the %MPE, where %MPE = [(test latency in s – control latency in s)/(40 s – control latency in s) × 100]. Groups of 8 to 12 animals were used for each drug condition. Antagonism studies were carried in mice pretreated with either saline or bupropion metabolites 15 min before nicotine administration. The animals were then tested 5 min after administration of a subcutaneous dose of 2.5 mg/kg nicotine.
Locomotor Activity
Mice were placed into individual Omnitech photocell activity cages (28 × 16.5 cm; Omnitech Electronics, Columbus, OH) 5 min after subcutaneous administration of either 0.9% saline or nicotine (1.5 mg/kg). Interruptions of the photocell beams (two banks of eight cells each) were then recorded for the next 10 min. Data were expressed as the number of photocell interruptions. Antagonism studies were carried out by pretreating the mice with either saline or bupropion metabolites 15 min before nicotine administration.
Body Temperature
Rectal temperature was measured by a thermistor probe (inserted 24 mm) and digital thermometer (YSI Inc., Yellow Springs, OH). Readings were taken just before and 30 min after subcutaneous injection of either saline or 2.5 mg/kg nicotine. The difference in rectal temperature before and after treatment was calculated for each mouse. The ambient temperature of the laboratory varied from 21 to 24 °C from day to day. Antagonism studies were carried out by pretreating the mice with either saline or bupropion metabolites 15 min before nicotine administration. The animals were then tested 30 min after administration of a subcutaneous dose of 2.5 mg/kg nicotine.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health National Cooperative Drug Discovery Group grant U19 DA019377. Other effort was supported by grants (to RJL) from the National Institutes of Health (DA015389) and the Barrow Neurological Foundation.
Footnotes
Abbreviations: CDC, Centers for Disease Control; DA, dopamine; 5HT, serotonin; NE, norepinephrine; SR, sustained release; HEK, human embryonic kidney; DAT, dopamine transporter; SERT, serotonin transporter; NET, norepinephrine transporter; nAChR, nicotine acetylcholine receptor; VTA, ventral tegmental area; NRT, nicotine replacement therapy; MPE, maximum possible effect.
Supporting Information Available: Elemental analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Centers for Disease Control and Prevention. Smoking and Tobacco Use-Fact Sheet: Health Effects of Cigarette Smoking (updated January 2008) http://www.cdc.gov/tobacco/data_statistics/fact_sheets/health_effects/effects_cig_smoking/
- 2.US Department of Health and Human Services. The Health Consequences of Smoking: what it means to you. NIH; 2004. [Google Scholar]
- 3.SAMHSA. (Substance Abuse and Mental Health Services Administration) Results from the 2008 National Survey on Drug Use and Health. Department of Health and Human Services; Washington, DC: 2009. [Google Scholar]
- 4.Lerman C, LeSage MG, Perkins KA, O’Malley SS, Siegel SJ, Benowitz NL, Corrigall WA. Translational research in medication development for nicotine dependence. Nat Rev Drug Discov. 2007;6:746–762. doi: 10.1038/nrd2361. [DOI] [PubMed] [Google Scholar]
- 5.Le Novere N, Corringer PJ, Changeux JP. The diversity of subunit composition in nAChRs: evolutionary origins, physiologic and pharmacologic consequences. J Neurobiol. 2002;53:447–456. doi: 10.1002/neu.10153. [DOI] [PubMed] [Google Scholar]
- 6.Gotti C, Moretti M, Gaimarri A, Zanardi A, Clementi F, Zoli M. Heterogeneity and complexity of native brain nicotinic receptors. Biochem Pharmacol. 2007;74:1102–1111. doi: 10.1016/j.bcp.2007.05.023. [DOI] [PubMed] [Google Scholar]
- 7.Marks MJ, Collins AC. Characterization of nicotine binding in mouse brain and comparison with the binding of α-bungarotoxin and quinuclidinyl benzilate. Mol Pharmacol. 1982;22:554–564. [PubMed] [Google Scholar]
- 8.Rahman S, Lopez-Hernandez GY, Corrigall WA, Papke RL. Neuronal nicotinic receptors as brain targets for pharmacotherapy of drug addiction. CNS Neurol Disord Drug Targets. 2008;7:422–441. doi: 10.2174/187152708786927831. [DOI] [PubMed] [Google Scholar]
- 9.Adams DJ, Nutter TJ. Calcium permeability and modulation of nicotinic acetylcholine receptor-channels in rat parasympathetic neurons. J Physiol Paris. 1992;86:67–76. doi: 10.1016/s0928-4257(05)80009-9. [DOI] [PubMed] [Google Scholar]
- 10.Le Novere N, Zoli M, Changeux JP. Neuronal nicotinic receptor alpha 6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. Eur J Neurosci. 1996;8:2428–2439. doi: 10.1111/j.1460-9568.1996.tb01206.x. [DOI] [PubMed] [Google Scholar]
- 11.Goldner FM, Dineley KT, Patrick JW. Immunohistochemical localization of the nicotinic acetylcholine receptor subunit alpha6 to dopaminergic neurons in the substantia nigra and ventral tegmental area. Neuroreport. 1997;8:2739–2742. doi: 10.1097/00001756-199708180-00019. [DOI] [PubMed] [Google Scholar]
- 12.Exley R, Clements MA, Hartung H, McIntosh JM, Cragg SJ. Alpha6-containing nicotinic acetylcholine receptors dominate the nicotine control of dopamine neurotransmission in nucleus accumbens. Neuropsychopharmacology. 2008;33:2158–2166. doi: 10.1038/sj.npp.1301617. [DOI] [PubMed] [Google Scholar]
- 13.Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov. 2009;8:733–750. doi: 10.1038/nrd2927. [DOI] [PubMed] [Google Scholar]
- 14.Corrigall WA, Franklin KB, Coen KM, Clarke PB. The mesolimbic dopaminergic system is implicated in the reinforcing effects of nicotine. Psychopharmacology. 1992;107:285–289. doi: 10.1007/BF02245149. [DOI] [PubMed] [Google Scholar]
- 15.Brody AL, Olmstead RE, London ED, Farahi J, Meyer JH, Grossman P, Lee GS, Huang J, Hahn EL, Mandelkern MA. Smoking-induced ventral striatum dopamine release. Am J Psychiatry. 2004;161:1211–1218. doi: 10.1176/appi.ajp.161.7.1211. [DOI] [PubMed] [Google Scholar]
- 16.Johnston AJ, Ascher J, Leadbetter R, Schmith VD, Patel DK, Durcan M, Bentley B. Pharmacokinetic optimisation of sustained-release bupropion for smoking cessation. Drugs. 2002;(Suppl 2):11–24. doi: 10.2165/00003495-200262002-00002. [DOI] [PubMed] [Google Scholar]
- 17.Hesse LM, Venkatakrishnan K, Court MH, von Moltke LL, Duan SX, Shader RI, Greenblatt DJ. CYP2B6 mediates the in vitro hydroxylation of bupropion: potential drug interactions with other antidepressants. Drug Metab Dispos. 2000;28:1176–1183. [PubMed] [Google Scholar]
- 18.Faucette SR, Hawke RL, Lecluyse EL, Shord SS, Yan B, Laethem RM, Lindley CM. Validation of bupropion hydroxylation as a selective marker of human cytochrome P450 2B6 catalytic activity. Drug Metab Dispos. 2000;28:1222–1230. [PubMed] [Google Scholar]
- 19.Faucette SR, Hawke RL, Shord SS, Lecluyse EL, Lindley CM. Evaluation of the contribution of cytochrome P450 3A4 to human liver microsomal bupropion hydroxylation. Drug Metab Dispos. 2001;29:1123–1129. [PubMed] [Google Scholar]
- 20.Bondarev ML, Bondareva TS, Young R, Glennon RA. Behavioral and biochemical investigations of bupropion metabolites. Eur J Pharmacol. 2003;474:85–93. doi: 10.1016/s0014-2999(03)02010-7. [DOI] [PubMed] [Google Scholar]
- 21.Damaj MI, Carroll FI, Eaton JB, Navarro HA, Blough BE, Mirza S, Lukas RJ, Martin BR. Enantioselective effects of hydroxy metabolites of bupropion on behavior and on function of monoamine transporters and nicotinic receptors. Mol Pharmacol. 2004;66:675–682. doi: 10.1124/mol.104.001313. [DOI] [PubMed] [Google Scholar]
- 22.Jefferson JW, Pradko JF, Muir KT. Bupropion for major depressive disorder: Pharmacokinetic and formulation considerations. Clin Ther. 2005;27:1685–1695. doi: 10.1016/j.clinthera.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 23.Kharasch ED, Mitchell D, Coles R. Stereoselective bupropion hydroxylation as an in vivo phenotypic probe for cytochrome P4502B6 (CYP2B6) activity. J Clin Pharmacol. 2008;48:464–474. doi: 10.1177/0091270008314254. [DOI] [PubMed] [Google Scholar]
- 24.Fang QK, Han Z, Grover P, Kessler D, Senanayake CH, Wald SA. Rapid access to enantiopure bupropion and its major metabolite by stereospecific nucleophilic substitution on an α-ketotriflate. Tetrahedron: Asymmetry. 2000;11:3659–3663. [Google Scholar]
- 25.Kelley JL, Musso DL, Boswell GE, Soroko FE, Cooper BR. (2S,3S,5R)-2-(3,5-difluorophenyl)-3,5-dimethyl-2-morpholinol: a novel antidepressant agent and selective inhibitor of norepinephrine uptake. J Med Chem. 1996;39:347–349. doi: 10.1021/jm950630p. [DOI] [PubMed] [Google Scholar]
- 26.Eshleman AJ, Carmolli M, Cumbay M, Martens CR, Neve KA, Janowsky A. Characteristics of drug interactions with recombinant biogenic amine transporters expressed in the same cell type. J Pharmacol Exp Ther. 1999;289:877–885. [PubMed] [Google Scholar]
- 27.Damaj MI, Fei-Yin M, Dukat M, Glassco W, Glennon RA, Martin BR. Antinociceptive responses to nicotinic acertylcholine receptors ligands after systemic amd inthathecal administration in mice. J Pharmacol Exp Ther. 1998;284:1058–1065. [PubMed] [Google Scholar]
- 28.Carroll FI, Blough BE, Mascarella SW, Navarro HA, Eaton JB, Lukas RJ, Damaj MI. Synthesis and Biological Evaluation of Bupropion Analogues as Potential Pharmacotherapies for Smoking Cessation. J Med Chem. 2010;53:2204–2214. doi: 10.1021/jm9017465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silverstone PH, Williams R, McMahon L, Fleming R, Fogarty S. Effect of increasing intraperitoneal infusion rates on bupropion hydrochloride-induced seizures in mice. Ann Gen Psychiatry. 2008;7:27. doi: 10.1186/1744-859X-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carroll FI, Blough B, Abraham P, Mills AC, Holleman JA, Wolckenhauer SA, Decker AM, Landavazo A, McElroy KT, Navarro HA, Gatch MB, Foster MJ. Synthesis and Biological Evaluation of Bupropion Analogues as Potential Pharmacotherapies for Cocaine Addiction. J Med Chem. 2009;52:6768–6781. doi: 10.1021/jm901189z. [DOI] [PubMed] [Google Scholar]
- 31.Anderson WK, Jones AN. Synthesis and evaluation of furan, thiophene, and azole bis[(carbamoyloxy)methyl] derivatives as potential antineoplastic agents. J Med Chem. 1984;27:1559–1565. doi: 10.1021/jm00378a006. [DOI] [PubMed] [Google Scholar]
- 32.Hoffman RV, Tao J. A simple, stereoselective synthesis of ketomethylene dipeptide isosteres. Tetrahedron. 1997;53:7119–7125. [Google Scholar]
- 33.Koch TH, Olesen JA, DeNiro J. Unusually weak carbon-carbon single bond. J Am Chem Soc. 1975;97:7285–7288. [Google Scholar]
- 34.Lukas RJ, Fryer JD, Eaton JB, Gentry CL. Some methods for studies of nicotinic acetylcholine receptor pharmacology. In: Levin ED, editor. Nicotinic Receptors and the Nervous System. CRC Press; Boca Raton: 2002. pp. 3–27. [Google Scholar]
- 35.Eaton JB, Peng JH, Schroeder KM, George AA, Fryer JD, Krishnan C, Buhlman L, Kuo YP, Steinlein O, Lukas RJ. Characterization of human alpha 4 beta 2-nicotinic acetylcholine receptors stably and heterologously expressed in native nicotinic receptor-null SH-EP1 human epithelial cells. Mol Pharmacol. 2003;64:1283–1294. doi: 10.1124/mol.64.6.1283. [DOI] [PubMed] [Google Scholar]
- 36.Gentry CL, Lukas RJ. Local anesthetics noncompetitively inhibit function of four distinct nicotinic acetylcholine receptor subtypes. J Pharmacol Exp Ther. 2001;299:1038–1048. [PubMed] [Google Scholar]
- 37.Lukas RJ, Fryer JD, Eaton JBLGC. Some methods for studies of nicotinic acetylcholine receptor pharmacology. In: Levine ED, editor. Nicotinic receptors and the Nervous System. CRC Press; Boca Raton: 2002. pp. 3–27. [Google Scholar]
- 38.Marubio LM, del Mar Arroyo-Jimenez M, Cordero-Erausquin M, Lena C, Le Novere N, de Kerchove d’Exaerde A, Huchet M, Damaj MI, Changeux JP. Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature. 1999;398:805–810. doi: 10.1038/19756. [DOI] [PubMed] [Google Scholar]
- 39.D’Amour FE, Smith DL. A method for determining loss of pain sensation. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.