

Abstract
Age-related changes in immune regulation are likely to account for the age-associated increase in serum autoantibody levels and in certain autoimmune disorders, like myasthenia gravis (MG). To demonstrate directly a loss of immune tolerance in older individuals, responses to the acetylcholine receptor (AChR), the autoantigen in MG, were assessed in transgenic mice expressing the Torpedo californica AChR (TAChR) α-chain as a neo-self antigen. T cells from young transgenic mice had been shown to be tolerant to p146-162, the TAChR α-chain peptide that dominated young nontransgenic T-cell responses in vitro. The immunodominance of p146-162 was not lost with age; fine specificity was preserved. Moreover, T-cell tolerance to p146-162, as well as to other epitopes of the TAChR α chain extracellular domain, was maintained in old transgenic mice. Even multiple TAChR immunizations coupled with the MG-enhancing cytokine, IL-12, did not break tolerance. Additionally, T cells exhibiting CD4 upregulation, an early activation marker, were reduced in frequency equivalently in old and young transgenic animals suggesting that immune regulation in this model was not impacted by aging. Moreover, B-cell tolerance was also maintained with age. The persistence of immune tolerance was accompanied by an increase in the proportion of Tregs; it is speculated that this may compensate for deficiencies in central tolerance that occur due to thymic involution. In summary, our study reveals, for the first time, that some immune tolerance mechanisms do survive aging; this suggests that certain late onset autoimmune disorders may be induced by a specific insult that disrupts immune homeostasis.
Keywords: T lymphocytes, T regulatory cells, immune tolerance, aging, autoimmunity
Introduction
Although immune responsiveness to foreign antigens decreases with aging, there is a concomitant increase in the level of circulating autoantibodies, suggesting a loss of immune tolerance in older individuals (1). Consistent with this finding is the increased incidence of some autoimmune disorders with age; these include rheumatoid arthritis, Sjogren's syndrome and myasthenia gravis (2-5). However, studies directly addressing the effects of age on immune tolerance are limited (6-8). In one case, T-cell tolerance to an exocrine gland autoantigen was reduced in 18 to 20 month old animals in a thymectomized mouse model for Sjogren's syndrome (7). Also, in a human study, increased reactivity to the amyloid β protein was detected in T cells from older individuals (≥50 years of age) (8). Such immune dysregulation in old age could be due to a loss of self-tolerance associated with the effects of thymic involution (9, 10) and/or age-associated changes in B-cell or T-cell development, immune activation, or cytokine regulation (10-13). While all of these age-associated changes in immune regulation have been reported (9-13) and are associated with the potential to lose tolerance, other factors may help counterbalance this effect. Specifically, CD4+ T regulatory cells (Tregs) which help to control autoimmune responses in young individuals (14) reportedly increase in frequency in old humans and mice (15-18). Moreover, suppressive functions of Tregs are generally well maintained (18). Thus, the increased proportion of Tregs in old individuals could help to promote immune tolerance in the face of other age-related alterations that favor tolerance loss.
To directly assess whether tolerance is rendered “leaky” with age, we focused on autoimmune MG, a T-cell-regulated disorder in which autoantibodies bind the acetylcholine receptor (AChR) at the neuromuscular junction and produce severe muscle weakness (19). The effect of age on tolerance to the AChR is of particular interest since 40-60% of MG patients first experience symptoms after 50 years of age (2, reviewed in 3). Late onset MG can be distinguished from forms of the disease affecting younger individuals based on HLA-linked susceptibilities (reviewed in 20) and gender predilections (3). Thus, the disease etiologies associated with the two forms of MG might be distinct and it is plausible that late-onset MG would be promoted by an age-related loss of tolerance to the AChR while the early onset form could involve other factors.
In order to assess the effects of aging on MG, we had previously used the rodent model in which C57BL/6 mice are immunized with AChR purified from an electric fish, Torpedo californica (TAChR) in CFA, with an accompanying course of the TH1-enhancing cytokine IL-12 (21). Under these conditions, approximately 50% of young mice will develop symptoms of MG after the second or third immunization. However, there are two serious limitations to this model for studies of aging and tolerance. First, immunization of old mice does not result in MG, due to the relatively low titers of antibodies produced to the immunogen, TAChR; this limitation can be overcome by immunizing the animals when they are young and then recalling this memory response when old (22). The second limitation to this rodent model is that T-cell tolerance cannot be assessed. Specifically, when C57BL/6 mice are immunized with TAChR, the T-cell response is largely restricted to an immunodominant epitope, p146-162 of the TAChR α chain. Since the homologous mouse α chain region lacks a critical lysine residue that is required for TCR binding to Torpedo p146-162, the responding TAChR-reactive T cells do not recognize “self” (mouse AChR) and would therefore not have been susceptible to tolerance (23, 24). In order to circumvent this limitation to the classical mouse MG model, we generated a transgenic mouse in which the TAChR α chain is expressed as a neo-self antigen; transgene expression shows the appropriate physiological profile of muscle > brain > thymus (21). As expected, expression of TAChR α as a “self” protein did result in T-cell tolerance (21). Moreover, when the transgenic mice were immunized with Talpha [recombinant TAChR α chain (aa 1-210)] alone to eliminate by-stander help, B-cell tolerance could also be demonstrated (21). Therefore, this transgenic model will allow us, for the first time, to examine the effects of aging on immune tolerance to this important autoantigen.
Materials and Methods
Antigens
The three different immunogens used have been previously described, as follows. The intact TAChR was purified from the electric ray, Torpedo californica, (Pacific Biomarine, Venice, CA) by affinity chromatography on cobra-toxin conjugated to Sepharose 4B as detailed previously (25). The AChR α chain peptides used were synthesized by the protein core facility of the UTHSCSA or by AnaSpec (San Jose, CA) from sequences previously described (23, 26, 27); they included: Torpedo α chain peptides, p111-126, p146-162, p182-198, and p360-378 and human α61-76 (negative control peptide). The recombinant TAChR α chain fragment (Talpha; aa 1-210) was generated, purified and refolded as previously described (21).
Mice
C57BL/6 mice were obtained from the National Institutes of Aging (NIA) colony (Harlan Sprague Dawley, Indianapolis, IN and Charles River, Stone Ridge, NY). The TAChR α transgenic animals (21) were bred and aged in our facility. All mice were housed in a specific pathogen-free animal facility and used as prescribed in protocols approved by our Institutional Animal Care and Use Committee.
Immunizations
For T-cell proliferation assays, mice were immunized subcutaneously (s.c.) at the base of the tail with 50 μl of emulsion containing 25 μg of TAChR or 3 μg of p146-162 and 25 μl of complete Freund's adjuvant (CFA, H37Ra, Difco, Detroit, MI). To attempt to break tolerance, mice were immunized s.c. at five sites (above each shoulder, above each thigh, and at the base of the tail) with a total of 100 μl of emulsion, containing 50 μg of TAChR and 50 μl of CFA. The immunizations were repeated twice at monthly intervals. IL-12 (Genetics Institute, Cambridge, MA) in 100 μl PBS with 1% normal mouse serum was given intraperitoneally where indicated; the cytokine was administered for 5 consecutive days along with the primary and secondary TAChR immunizations, as described above. The dosage was 0.5 μg of IL-12 for days one and two and 0.25 μg of IL-12 on each of the next three days for the primary immunization and 0.25 μg/day for the 5 days prior to the secondary inoculation. To assess B- and T-cell responses to the alpha chain, mice were immunized s.c. at five sites (as described above) with a total of 100 μl of emulsion containing 10 μg recombinant Talpha (aa 1-210) and 50 μl CFA or, as a negative control, with 100 μl of PBS/CFA. Secondary and tertiary immunizations were given at monthly intervals using IFA as adjuvant.
Lymph Node Stimulation and Proliferation
T-cell proliferation was assayed in vitro as described previously (21). Briefly, draining lymph nodes were removed from mice 7 days after primary TAChR immunization or 7 days after tertiary TAChR α chain immunization. Single cell suspensions were prepared by mechanical disruption in RPMI 1640 supplemented with 10% fetal bovine serum, 10 mM HEPES, 2 mM L-glutamine, 50 μM 2-mercaptoethanol, 50 μg/ml streptomycin and 50 U/ml penicillin. The lymph node cells (2.5 × 105 cells/0.2ml/well) were cultured in flat bottom 96-well plates with indicated concentrations of individual synthetic peptides derived from the TAChR α chain, the intact TAChR, or the recombinant T α-chain fragment (aa 1-210). Negative control wells received either no stimulus or a nonstimulating peptide, AChRα p61-76, as indicated in individual figures. In the wells containing the recombinant protein, 2 μg polymyxin B was added to eliminate any LPS effects. As additional controls, cells were stimulated with either anti-CD3 (antibody supernatant from clone 145-2C11) to assess general T-cell reactivity or heat-killed Mycobacterium tuberculosis (Difco Labs, H37RA), a CFA component. The plates were incubated for 96 hr, pulsed with 1μCi [3H]TdR (ICN, Irvine, CA) per well, and harvested 18 hr later. [3H]TdR uptake was measured by liquid scintillation spectrometry and the mean counts/min of duplicate wells (or triplicate wells, when cell numbers were sufficient) was calculated. Results were expressed as average counts/min ± S.E.M. with background subtracted. For responses from young and old, transgenic and nontransgenic mice, statistical differences in treatment groups were assessed by higher order ANOVA (p<0.05) and the significance of differences was determined using a Tukey multiple range test. Prior to analysis the data were squared to remove the confound of negative values and a log base ten transformation performed to satisfy the assumption of equality of variances (Minitab software).
Immunofluorescent staining and flow cytometry analysis
Lymph node cells were cultured in complete media (2.5 × 106 cells/ml/well in a 12 well plate) and stimulated for 96 hrs with 20 nM p146-162 or p61-76 as previously described (28). Cells were washed and stained extracellularly with fluorescently labeled anti-CD4 (clone RM4-5), anti-CD25 (clone PC61, eBioscience) and anti-Vβ6 (clone RR4-7, BD Pharmingen). After washing, the cells were fixed, permeabilized, stained with either anti-Foxp3 (clone FJK-16s) or an isotype-matched control antibody (IgG2a) conjugated to PerCP-cy5.5 (eBioscience, San Diego, CA). Multicolor flow cytometry analysis was performed on a Becton Dickinson FACSCalibur (UTHSCSA Core Facility). The CD4high gate was separated from CD4norm gate based upon the pattern of CD4 expression following in vitro culture with a non-stimulatory control peptide (p61-76). FACS data were analyzed using a higher order ANOVA protocol, as described above.
Measuring anti-TAChR antibody titers
Young and old mice were immunized twice (as described above) with Talpha (aa 1-210) and anti-Talpha antibody titers measured by ELISA (21-22). In short, 96-well plates (Nunc Maxisorb, Thermo Fisher Scientific, Rochester, NY) were coated with 50 μl/well of Talpha (aa1-210) (20 μg/ml in PBS) and stored overnight at 4 °C. The next day, after washing wells with 0.05% Tween 20 in PBS, the plates were blocked with 200 μl per well 1% BSA/PBS for 1 hr at room temperature and washed again. Mouse serum diluted in 1% BSA/PBS was added in five-fold dilutions from 1/25 to 1/78125 and tested in duplicate. The 1% BSA/PBS diluent also served as a background control. After incubation for 2 hr at room temperature and washing, 50 μl/well of rabbit anti-mouse IgG (whole molecule) conjugated to HRP (Sigma-Aldrich, St. Louis, MO) was added and plates were incubated for 1 hr. To develop color, 100 μl of ABTS with 0.03% hydrogen peroxide was added per well and absorbance read after 15 to 30 min using a Dynatech MRX ELISA plate reader (Dynatech Labs, Chantilly, VA). To determine titer endpoints, the measured absorbances were fitted to a sigmoidal curve (GraphPad Prism 5) and the reciprocal dilution of serum required to obtain a 0.1 O.D. above background was determined by interpolation. For statistical analysis, the assumption of variance equivalence among transgenic and nontransgenic groups was satisfied by a log base ten transformation and a one-way ANOVA (p≤0.05) was performed on the transformed data for each age group.
Results
Maintenance of the fine specificity of the T-cell response to TAChR with aging
The T-cell response to TAChR in young C57BL/6 mice is dominated by a single epitope, p146-162, of the TAChR α chain (23, 24, 26). However, it was possible that a shift had occurred such that p146-162 was no longer immunodominant. Indeed, changes in fine specificity with age had been previously reported for both B-cell and T-cell responses (29-31). Should such an alteration occur in the profile of TAChR-responding T cells, it would change the focus of our analysis of tolerance in the old TAChR α transgenic mice. Thus, to assess age-related alterations in fine specificity, nontransgenic C57BL/6 mice of different ages were immunized with intact TAChR which should elicit T-cell responses both to p146-162 and to other TAChR epitopes. The draining lymph nodes, containing T cells activated by TAChR in vivo, were harvested and the T cells were tested by in vitro stimulation with a panel of TAChR alpha chain peptides. To enhance detection of potential intramolecular spreading with age, a peptide concentration that produced responses to p146-162 toward the upper limit of the linear range for most mice was used. Resulting proliferation data are summarized graphically in Figure 1. As expected, the T-cell responses from young mice showed a dominant response to p146-162, with only minor reactivities to the other Torpedo alpha chain peptides tested, p111-126, p182-198, and p360-378. This fine specificity profile was apparently unaffected by aging since T cells from older animals (both 12 months and 20 months of age) exhibited the same enhanced response to the immunodominant epitope, p146-162 (Fig. 1). The lack of proliferation to a negative control peptide, p61-76, derived from the alpha chain of the human AChR, was also preserved with age. Thus, p146-162 remained the immunodominant epitope in the TAChR response in old mice and the effects of age on tolerance to this peptide could be addressed.
Figure 1.

Effects of age on the fine specificity of the T-cell response to TAChR. Female C57BL/6 mice 2, 12, and 20 months of age were immunized with TAChR/CFA and lymph nodes were extracted 7 days later. Single cell suspensions were stimulated with individual alpha chain peptides (p146-162, p111-126, p182-198, or p360-378) or p61-76 (a negative control). A peptide concentration toward the upper limit of the linear range for the p146-162 T-cell responses (∼142 nM) was chosen in order to enhance evidence of epitope spreading. Proliferation was measured by [3H] thymidine incorporation and expressed as the mean cpm with peptide minus the mean cpm without peptide. The experiment was repeated twice and the results summarized graphically.
Maintenance of T-cell tolerance to the alpha chain p146-162 with aging
Expression of the TAChR α transgene had been shown previously to result in tolerance of T cells specific for the immunodominant α-chain peptide, p146-162, in young mice (21). To assess whether tolerance in this model would be lost with increasing age, young (6 months) and middle-aged (12-13 months) mice were immunized with TAChR/CFA. The draining lymph node T cells were assayed for the ability to proliferate in vitro in response to the immunodominant peptide, p146-162. As expected, the T cells from the young TAChR α transgenic mice were tolerant, showing significantly lower responses to p146-162 as compared with those from the nontransgenic animal (Fig. 2A). By middle age, there was no evidence that the nontransgenic T-cell response to the p146-162 α-chain peptide was waning or that the tolerance observed in T-cell responses from transgenic mice was diminished (Fig. 2B). In addition, the T cells from the transgenic and nontransgenic mice responded well to positive control stimuli, anti-CD3 antibody and Mycobacterium tuberculosis (Mtb), and to the intact TAChR (Fig. 2B). The response to TAChR in T cells from transgenic mice was possible since, even though the transgenic mice express the TAChR α chain and would be tolerant to this polypeptide, they do not express the TAChR β, δ, or γ subunits. T cells from the transgenic animals could therefore proliferate to intact TAChR, although reactivity would potentially be lower than that of the nontransgenic responders, due to the lack of α-chain-specific T cells. Overall, the responses to positive control and p146-162 stimuli reveal that both T-cell immunity and T-cell regulation are maintained in middle-age animals. It is notable that loss of humoral immunity to the TAChR had been seen in ten month old mice (22). It could be that changes in T-cell responses to this antigen occur at a later age, lagging B cell alterations.
Figure 2.

T-cell tolerance to TAChR p146-162 in young and middle-aged mice. Young (6 months old) (Panel A), and middle-aged (12-13 month old) (Panel B) mice were immunized with TAChR/CFA. Draining lymph nodes were harvested 7 days later and T cells were stimulated with incremental concentrations of the α chain peptide, p146-162. The cells were stimulated in parallel with the control stimuli, intact TAChR (1.9 nM), Mycobacterium tuberculosis (Mtb) (20 μg/well), or anti-CD3 antibody supernatant (clone 145-2C11). [3H] thymidine incorporation was measured and background (media only) was subtracted.
Since it was possible that dysfunction of regulation would be seen with increasing age, tolerance was examined in mice of “early” old age (18-19 months of age) and in mice that were truly old (23-33 months of age). Draining lymph node T cells from TAChR immunized mice were assessed for proliferation responses, as described above. Since it was critical to differentiate tolerance from lack of reactivity due to immunosenescence, based upon in vitro reactivity to the TAChR control each old animal was judged as “responsive” >2000 cpm in vitro (indicated with gray bars) or “minimally responsive” <2000 cpm (lightly striped bars), as shown in Figure 3A. In addition, in three mice, in vivo immunization elicited a low response and few draining lymph node cells could be obtained; these animals were categorized as “senescent” (white bars in Figure 3A). Lack of immune responsiveness was seen most markedly in the 23-33 month old age group, as expected. Nevertheless, mice with T-cell responsiveness to TAChR were present in both of the old age groups, so the effects of age on tolerance in these mice could be addressed.
Figure 3.

Effects of increasing age on T-cell tolerance to TAChR p146-162. Old mice were immunized once with TAChR and draining lymph node T cells were stimulated in vitro and [3H]-thymidine incorporation measured. In vitro responses to TAChR were judged as “responsive” (>2000 cpm) or “minimally responsive” (<2000 cpm) (Panel A). The TAChR stimulus used (1.9 nM for experiments B and C or 1.2 nM for experiments A and D) was within the linear range for the responses from most old mice. In three old mice, low numbers of draining lymph node cells prohibited complete in vitro analysis; these mice were labeled “senescent” (Panel A). T-cell populations from individual “responsive” old mice were used to assess reactivity to the α-chain peptide, p146-162; T cells from young immunized mice were tested in parallel (Panel B). Proliferation to 19 nM p146-162 was graphed for all animals using filled symbols; this peptide concentration was within the linear range for responses from nontransgenic mice. The upper 95% confidence limit for the mean of young transgenic responses (dashed line) and the mean value for each experiment group (solid lines) are indicated. Several mice (numbers B61, 140, 216) had enlarged spleens (> 7 mg/g body weight); they were included in Panel A for completeness. In an additional experiment (Old/IL-12 in Panel B), a more strenuous immunization protocol was used as described in Materials in Methods. Briefly, 24 month old mice were given three TAChR/CFA immunizations and were also injected with the enhancing cytokine, IL-12. The T cell responses to p146-162 for these animals are shown with open squares (Panel B).
T cells from the old “responsive” mice (Fig. 3A) were analyzed for responses to the α chain p146-162 peptide and the results for individual animals are shown graphically (indicated by the filled symbols in Figure 3B). Proliferation readouts from young responders, tested in parallel with the old, are also shown. As expected, the T-cell populations derived from young transgenic mice showed evidence of tolerance (lower proliferation, Figure 3B). However, somewhat surprisingly, tolerance was also observed in the lymph node T-cell populations from the older (18 to 30 month) transgenic mice. That is, the T-cell responses from the old transgenic animals were for the most part lower than in age-matched nontransgenic controls. Thus, tolerance to the α chain p146-162 peptide was preserved with aging.
In an additional experiment we asked whether tolerance to the AChR in old mice could easily be broken. A more stringent immunization protocol (used to induce MG in young animals) was applied. One nontransgenic and two transgenic mice (24 months old) were given three immunizations with TAChR/CFA, one month apart, and were further stimulated with the MG enhancing cytokine, IL-12 (21). When in vitro lymph node T-cell responses to p146-162 were assessed, tolerance was well maintained in the transgenic animals (open square symbols in Figure 3B). The lack of responsiveness was not due to immunosenescence since parallel in vitro stimulation with 1.9 nM TAChR invoked a vigorous response in T cells from the transgenic mice (>50,000 cpm; data not shown). This suggests that tolerance to the immunodominant α chain peptide is maintained in old animals even under conditions designed to promote a strong immune response.
Maintenance of T-cell tolerance to other α chain epitopes with aging
Despite the maintenance of tolerance to the p146-162 peptide in old transgenic mice (Fig. 3B), it was still possible that tolerance to other alpha chain epitopes could be altered by aging. To address this possibility, young and old, transgenic and nontransgenic, mice were immunized with recombinant Talpha (aa 1-210) protein. The immunization was repeated twice one month apart, allowing for further expansion of T-cell clones directed against epitopes of the α-chain. Lymph node T cells were harvested seven days after tertiary immunization and the cells were stimulated in vitro with recombinant Talpha, the α-chain peptide, p146-162, or positive control stimuli (anti-CD3 antibody and Mtb). T cells from most of the young and old mice were responsive to the anti-CD3 stimulation and to treatment with the Mtb positive control, a component of the CFA used in the primary Talpha immunizations (data not shown). Mice that were unresponsive to Mtb (<5000 cpm) or that lacked sufficient draining lymph node cells to test for control responses were not included in this analysis. T cells from all other mice were stimulated with the recombinant TAChR α chain fragment or the immunodominant epitope and proliferation was assessed. As shown in Figure 4, T cells derived from nontransgenic mice, both young and old, were generally responsive when restimulated in vitro with either the Talpha immunogen (Panel A) or p146-162 (Panel B) following the tertiary immunization. On the other hand, T cells from the transgenic animals showed tolerance, as expected. The responses from the old transgenic mice were essentially equivalent to the low levels of proliferation seen with younger transgenic individuals; this was true whether the in vitro stimulus was the α-chain fragment or the peptide. Taken together, these data suggest that T-cell tolerance to other α-chain epitopes, as well as to p146-162, are maintained with age.
Figure 4.

Effects of age on tolerance to other TAChR α-chain epitopes. Young (5 months of age) and old (23-25 months of age) mice were immunized s.c. in multiple sites with the recombinant Talpha (aa 1-210) as described in Methods. The immunizations were repeated twice at monthly intervals. The tertiary immunization was given at staggered time points (experiments A, B, and C) and draining lymph nodes were harvested 7 days later. T cells were stimulated with incremental concentrations of Talpha (aa 1-210) or the TAChR peptide, p146-162 and [3H]-thymidine incorporation was measured. The responses to 19 nM Talpha (Panel A) or 19 nM p146-162 (Panel B) are shown graphically for individual mice. One old nontransgenic and two old transgenic mice had enlarged spleens (>7 mg/g body weight) but responded to positive controls and thus were included in the tolerance study. Two mice with insufficient draining lymph node cells or with T cells that were not responsive to the Mtb control were not included; these mice also had enlarged spleens. The upper 95% confidence limit for the mean of young transgenic responses (dashed line) and the mean values for each group (solid line) are indicated.
Reduced antigen-specific T-cell clonal expansion is maintained with age
We next investigated potential mechanisms responsible for maintaining tolerance in old mice. T-cell upregulation of CD4 is an early event following T-cell receptor activation and it has been shown by us (28) and others (32, 33) that the CD4high population contains the majority of the antigen-specific T cells following challenge. In young nontransgenic C57BL/6 mice immunized with p146-162, an expansion of CD4high cells expressing the canonical TCRBV6 gene segment is seen (10% of gated lymphocytes for the young nontransgenic mouse shown in Fig 5A). In contrast, a greatly diminished expansion of the CD4high T cells is evident in the young transgenic animals (1% in the representative plot, Fig. 5A). To ask whether this decreased frequency of activated (Vβ6+CD4high) T cells, consistent with a tolerogenic phenotype, was maintained with age, old nontransgenic and transgenic mice were immunized with p146-162 and the draining lymph node cells expanded in vitro. As illustrated in Figure 5A, the old nontransgenic mice were able to upregulate CD4 (5% of lymphocytes), while the old transgenic animals maintained a clear imprint of tolerance very similar to the pattern seen in young (1% of lymphocytes). This experiment was performed on several mice of each age and genotype; the data are summarized in Figure 5B. The average percent of Vβ6+CD4high cells (1.3%) in the lymphocyte gate in the old transgenic mice was virtually identical to that seen in the young transgenic mice (1.2%), and in both cases, was significantly decreased from the levels in the nontransgenic animals (∼9% average in both young and old responses) (Fig. 5B). Overall, it was clear that age did not diminish the effects of tolerance in limiting the clonal expansion of antigen-responsive Vβ6+CD4high T cells in the transgenic mice.
Figure 5.
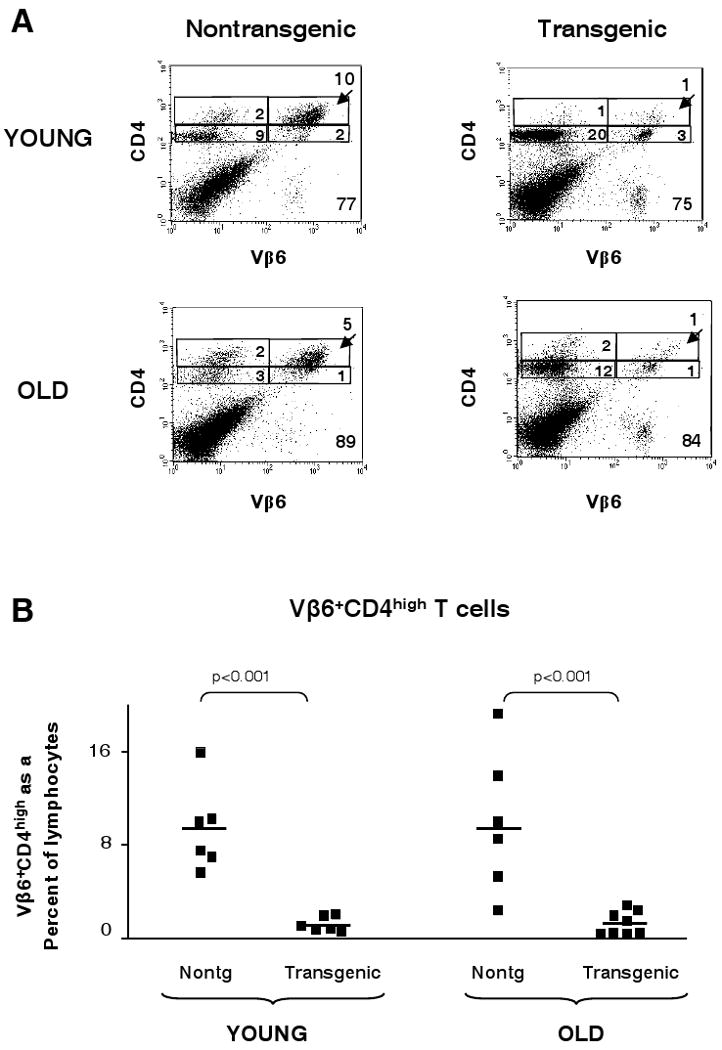
Effects of age on CD4 upregulation in p146-162-responsive Vβ6+CD4+ T cells. Young (2-4 months of age) and old (20-23 months of age) transgenic and nontransgenic mice were immunized with p146-162; draining lymph nodes were extracted and expanded in vitro with the immunizing peptide. The LNCs were stained to detect CD4, CD25, and Vβ6 using antibodies conjugated to FITC, APC, and PE, respectively. The cells were analyzed by FACS to determine whether CD4 had been upregulated in the Vβ6+ population. The experiment was then repeated with an equivalent number of mice of the same age groups. Representative dot plots are shown (Panel A). The frequency of Vβ6+CD4high T cells within the lymphocyte population is shown for the individual mice from the combined experiments (Panel B). The solid line represents the mean value within each group.
B-cell tolerance to the TAChR α chain is not lost with age
It has been reported that B-cell tolerance is compromised with aging, resulting in higher levels of circulating autoantibodies in the elderly (1). However, this possibility has not been interrogated directly for AChR or for any other muscle autoantigen; it is possible to do so in the TAChR α transgenic mice by immunizing with the TAChR α chain, which is seen as self (21). Towards this end, mice were immunized twice with recombinant Talpha (aa1-210) and the resulting antibody titers were assessed by ELISA. As shown in Figure 6, immunized young nontransgenic mice have significant titers to the immunogen, as expected. On the other hand, young transgenic animals fail to respond to TAChR α immunization, consistent with B-cell tolerance. In older mice it was anticipated that antibody titers would be lower, even after two immunizations (22, 34, 35). Nonetheless, detectable antibody responses were seen in ∼50% of the old nontransgenic mice, while older transgenic animals produced negligible levels of anti-TAChR α chain antibody (Fig. 6). Thus, B-cell tolerance to neo-self AChR does appear to be maintained with aging.
Figure 6.

Effects of age on antibody responses to the “neo-self” Talpha antigen. Young (5 months of age) and old (23-27 months of age) mice were immunized twice at one month intervals with 10 μg of Talpha (aa1-210) as described in Methods. Blood samples were taken one week after the secondary immunization, serum was prepared, and the antibody titer to Talpha (aa1-210) was measured in serum dilutions by ELISA. GraphPad Prism 5 was used to fit the titer points from each mouse to a sigmoidal curve and to determine the reciprocal dilution of serum required to obtain an absorbance of 0.1 O.D. above background.
T regulatory cell frequencies are elevated in the old transgenic mice
The somewhat unexpected finding that T-cell and B-cell tolerance to the muscle autoantigen AChR were not impacted by aging impelled us to examine other immune regulatory mechanisms in old mice. Specifically, it had been reported that the frequency of Treg cells (CD4+CD25+foxp3+) is increased with age in both rodents and humans although their function is not always maintained (15-18). Thus, we asked whether Tregs in the elderly transgenic mice could be responsible for continued tolerance to TAChR α chain determinants. As expected, in unimmunized old transgenic mice, >24% of CD4+ lymph node T cells were CD25+foxp3+ Tregs as compared with about 13% for young transgenic mice (Fig. 7A, B). This increase in Tregs with age was very similar to that seen in the nontransgenic littermates (Fig. 7B). To determine the effects of aging on Treg levels following antigen-specific expansion, young and old transgenic mice were immunized with the TAChR p146-162 and the draining lymph node T cells were stimulated in vitro with p146-162. The foxp3+ T cells in this population expressed high levels of CD25 and the Tregs were gated accordingly. The percentages of CD25high Tregs in the cultures derived from old transgenic mice were still higher on average (9%) than those of the young transgenics (4.4%) (Fig. 7C, D). By comparison, the Treg differential between old and young immunized nontransgenic animals was smaller, probably due to the expansion of antigen-specific cells in the nontransgenics (Fig. 7D). Thus, Treg frequency was elevated in the old transgenic mice and this differential was maintained after exposure to the p146-162 TAChR antigen.
Figure 7.
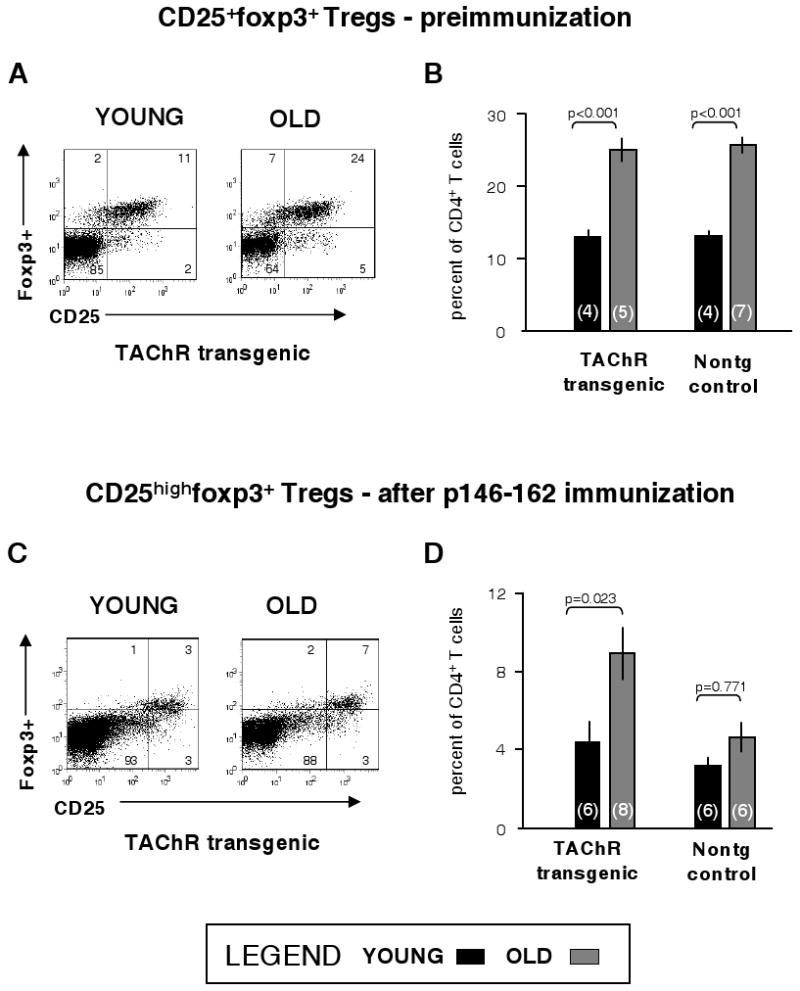
Effects of age on Treg frequency. Inguinal, axillary, brachial, and periaortic lymph node cells were extracted from unimmunized young (3 months of age) and old (21-22 months of age) mice. Cells from each animal were stained for CD4, CD25, and foxp3 or an anti-foxp3 antibody isotype control (IgG2a) using fluorochrome-conjugated antibodies as described in Methods and were analyzed by FACS. The experiment was then repeated with mice of the same ages and the results combined for analysis. Tregs were defined as foxp3+CD25+CD4+; they were quantified from the FACS plots for transgenic and nontransgenic animals. Representative dot plots for one transgenic mouse of each age are shown (Panel A) and the data for all analyzed animals are summarized (Panel B). The number of mice per group is given parenthetically. Another group of young (2-4 months old) and old (20-23 months of age) mice was immunized with p146-162; draining lymph node cells were extracted, expanded in vitro with p146-162 and stained for FACS analysis. The experiment was then repeated with additional mice of the same ages. Sample plots are shown (Panel C) and the percent of CD25highTregs in the CD4+ T-cell population is summarized graphically for all animals (Panel D). Error bars represent ±S.E.M.
Discussion
For the first time, the effects of age on immune tolerance have been examined in a well-defined model, a transgenic mouse in which the TAChR α chain is expressed primarily in muscle. This model is ideal since it can be used to assess both B-cell and T-cell tolerance. In addition, the T-cell fine specificity of the TAChR response has been mapped to a single determinant and tolerance to this T-cell epitope in the young transgenic model was previously demonstrated (21). However, it was possible that this fine specificity would be altered with age. We reasoned that a reduced frequency of dominantly responding clones due to a decline in receptor diversity (10) might reveal reactivities against other less dominant TAChR epitopes. Alternatively, mechanisms that regulate the fine specificity of the T-cell responses in young animals might be altered in the old. For example, epitope spreading, reported in induced T-cell tolerance to specific antigens (36) and in the progression of T-cell responses (37), could conceivably be increased with age. However, this was not observed in the AChR response, the immunodominance of p146-162 was well maintained with age in TAChR-immunized nontransgenic mice (Fig. 1). This result is in agreement with a study of T-cell responses to hen egg lyzozyme in 2 and 20 month old mice; no change in the fine specificity of the peptide response was seen when measured as the frequency of IFN-γ producing T cells (38). Our results suggest that factors controlling T-cell fine specificity of the TAChR response are still intact in 20 month old mice and verify the importance of addressing tolerance to p146-162 in the old transgenic mice.
In view of the reported age-associated alterations in T-cell development and function (10, 12-13), the maintenance of T-cell tolerance to the AChR in old mice might not have been expected. Nonetheless, tolerance was evidenced by diminished T-cell proliferative responses to p146-162 as well as other epitopes of the α chain extracellular domain and in low antibody titers to the TAChR α chain. Moreover, another measure of tolerance in young TAChR α transgenic mice, the reduction in frequency of high avidity CD4high antigen-specific cells (28), was observed in the old transgenic mice as well. Thus, in seeming contradiction to the paradigm that reactions to self-antigens increase with age (1, 39-40), we found that both T-cell and B-cell tolerance to the AChR neo-self antigen were maintained.
Given this persistence of tolerance in the old transgenic mice, it appears that mechanisms thought to promote the loss of T-cell tolerance with age are not applicable in this model. For example, it has been proposed that thymic involution would compromise negative selection, possibly resulting in an increase in the escape of autoreactive T cells to the periphery (40). However, this was clearly not seen in our model or alternatively, peripheral mechanisms of tolerance remained active and effectively eliminated any escapees. It also has been suggested that age-related loss of T-cell tolerance could be due to T-cell signaling alterations that impinge upon apoptotic mechanisms or that promote anergy of Th2-type regulatory cells (reviewed in 39). Alterations in T-cell regulation are of special interest since differences in foxp3+ Treg frequencies (41) or function (42, 43) have been reported in thymus and blood from patients with MG. Patients in these studies were mostly young (41, 43). However, in one study reporting diminished suppressive capacity of Tregs from MG patients (42), twelve of twenty-one individuals exhibited disease onset after the age of 50 years. Thus, it is conceivable that alterations in Treg number and/or function could be a contributing factor in late-onset MG, as well as early-onset MG.
Significantly, the effects of age on Treg profiles in healthy humans and mice have been addressed and could have implications for late-onset MG. While age-related alterations in certain Treg functions were seen in some studies (15, 44), suppressive capabilities were generally maintained (15-18). In addition, the frequencies of Tregs are widely reported to be increased in old individuals (15-18). In support of this finding, we found that the Treg frequency within the CD4+ T-cell population from old unimmunized TAChR α transgenic mice was significantly elevated over that of the young transgenic mice and this enhanced frequency was also evident in CD4+ T cells derived from p146-162 immunized mice. While it is far from clear whether the increased proportion of Tregs in the old transgenic mice helps to maintain tolerance to the TAChR studied here, this is an important area for future investigation.
Importantly, our results indicating that tolerance to the neo-self AChR is not broken in the old mice, raise a more basic question. In the face of this tolerance maintenance, how is tolerance disturbed in autoimmune diseases where symptoms are first seen in old age? One possible explanation for loss of tolerance in late-onset MG is that in some individuals, deficiencies in T-cell regulation are present at a young age but are not sufficient to result in clinical manifestations of autoimmunity. Thus, autoimmune challenges may be held in check although autoimmune memory T cells are generated. Upon re-exposure to the self antigen in old age, due to age-related tissue damage or exposure to mimicking foreign antigens, “recall” autoimmune memory cells established at a young age would respond vigorously and rapidly. T-cell tolerance, already weak in these individuals, would be “broken” by the strong memory response. In support of this hypothesis, in B-cell studies, Nobrega et al. have demonstrated that although auto-antibody levels were higher in old C57BL/6 mice, the autoantigen recognition profile was the same in young and old (45). Moreover, our own work has shown that autoreactive B cells first established by immunization of young mice could be recalled in old age to give a vigorous antibody response and produce autoimmune MG (22, 46). Thus, similar to autoreactive B cells, it may be that certain self-reactive T cells in the elderly reflect specificities first established in youth.
Acknowledgments
We thank Crystal Davenport and Bruce Gelb for technical help. We are also grateful to Irma Gonzales for isolating the AChR from electroplaque tissues, to Peggy Rifleman for peptide analysis and to Dr. William Morgan for his valuable advice on the statistical analyses of these data. The Genetics Institute generously provided the IL-12 for these experiments. FACS analysis was provided by the Flow Cytometry Core of UTHSCSA with technical assistance from Charles Thomas and Karla Gorena.
This work was supported in part by an Executive Research Committee New Investigator award from UTHSCSA (SS); an NIA Training grant (T32AG021890), NIH grants [R03 AG 14557 (EK), R36 AG 029667 (EW), R03 AG 22675 (SS)], the Autoimmune Prevention Center (NIAID), the Semp Russ Foundation of the San Antonio Area Foundation, and the Nathan Shock Aging Center. The FACS facility is supported in part by the San Antonio Cancer Institute Cancer Center Support Grant, P30 CA54174.
Abbreviations used in this paper
- MG
myasthenia gravis
- TAChR
Torpedo acetylcholine receptor
- Talpha
recombinant TAChR α chain (aa 1-210)
- Treg
T regulatory cells
Footnotes
Publisher's Disclaimer: “This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at http://www.jimmunol.org.”
References
- 1.Manoussakis MN, Tzioufas AG, Silis MP, Pange PJ, Goudevenos J, Moutsopoulos HM. High prevalence of anti-cardiolipin and other autoantibodies in a healthy elderly population. Clin Exp Immunol. 1987;69:557–565. [PMC free article] [PubMed] [Google Scholar]
- 2.Christensen PB, Jensen TS, Tsiropoulos I, Sorensen T, Kjaer M, Hojer-Pedersen E, Rasmussen MJ, Lehfeldt E, de Fine Olivarius B. Incidence and prevalence of myasthenia gravis in western Denmark: 1975 to 1989. Neurology. 1993;43:1779–1783. doi: 10.1212/wnl.43.9.1779. [DOI] [PubMed] [Google Scholar]
- 3.Aarli JA, Romi F, Skeie GO, Gilhus NE. Myasthenia gravis in individuals over 40. Ann NY Acad Sci. 2003;998:424–431. doi: 10.1196/annals.1254.055. [DOI] [PubMed] [Google Scholar]
- 4.Symmons DP. Epidemiology of rheumatoid arthritis: determinants of onset, persistence and outcome. Best Prac Res Clin Rheumatol. 2002;16:707–722. doi: 10.1053/berh.2002.0257. [DOI] [PubMed] [Google Scholar]
- 5.Thomas E, Hay EM, Hajeer A, Silman AJ. Sjögren syndrome: a community-based study of prevalence and impact. Br J Rheumatol. 1998;37:1069–1076. doi: 10.1093/rheumatology/37.10.1069. [DOI] [PubMed] [Google Scholar]
- 6.Crisi GM, Tsiagbe VK, Russo C, Basch RS, Thorbecke GJ. Evaluation of presence and functional activity of potentially self-reactive T cells in aged mice. Int Immunol. 1996;8:387–395. doi: 10.1093/intimm/8.3.387. [DOI] [PubMed] [Google Scholar]
- 7.Ishimaru N, Yoneda T, Saigusa K, Yanagi J, Haneji N, Moriyama K, Saito I, Hayashi Y. Severe destructive autoimmune lesions with aging in murine Sjogren's Syndrome through fas-mediated apoptosis. Am J Path. 2000;156:1557–1564. doi: 10.1016/S0002-9440(10)65027-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monsonego A, Aota V, Karni A, Krieger JI, Bar-Or A, Bitan G, Budson AE, Sperling R, Selkoe D, Weiner HL. Increased T cell reactivity to amyloid β protein in older humans and patients with Alzheimer disease. J Clin Invest. 2003;112:415–422. doi: 10.1172/JCI18104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aspinall R, Andrew D. Thymic involution in aging. J Clin Immunol. 2000;20:250–256. doi: 10.1023/a:1006611518223. [DOI] [PubMed] [Google Scholar]
- 10.Naylor K, Li G, Vallejo AN, Lee WW, Koetz K, Bryl E, Witkowski J, Fulbright J, Weyand CM, Goronzy JJ. The influence of age on T-cell generation and TCR diversity. J Immunol. 2005;174:7446–7452. doi: 10.4049/jimmunol.174.11.7446. [DOI] [PubMed] [Google Scholar]
- 11.Van der Put E, Sherwood EM, Blomberg BB, Riley RL. Aged mice exhibit distinct B cell precursor phenotypes differing in activation, proliferation and apoptosis. Exp Gerontol. 2003;38:1137–1147. doi: 10.1016/j.exger.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Pawelec G, Hirokawa K, Fulop T. Altered T-cell signaling in ageing. Mech Ageing Dev. 2001;122:1613–1637. doi: 10.1016/s0047-6374(01)00290-1. [DOI] [PubMed] [Google Scholar]
- 13.Linton PJ, Haynes L, Klinman NR, Swain SL. Antigen-independent changes in naive CD4 T cells with aging. JExp Med. 1996;184:1891–1900. doi: 10.1084/jem.184.5.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Costantino CM, Baecher-Allan C, Hafler DA. Multiple sclerosis and regulatory T cells. J Clin Immunol. 2008;28:697–706. doi: 10.1007/s10875-008-9236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao L, Sun L, Wang H, Ma H, Liu G, Zhao Y. Changes of CD4+CD25+Foxp3+ regulatory T cells in aged Balb/c mice. J Leukoc Biol. 2007;81:1386–1394. doi: 10.1189/jlb.0506364. [DOI] [PubMed] [Google Scholar]
- 16.Chiu BC, Stolberg VR, Zhang H, Chensue SW. Increased Foxp3(+) Treg cell activity reduces dendritic cell co-stimulatory molecule expression in aged mice. Mech Ageing Dev. 2007;128:618–627. doi: 10.1016/j.mad.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 17.Gregg R, Smith CM, Clark FJ, Dunnion D, Khan H, Chakraverty R, Nayak L, Moss PA. The number of human peripheral blood CD4+CD25high regulatory T cells increases with age. Clin Exp Immunol. 2005;140:540–546. doi: 10.1111/j.1365-2249.2005.02798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lages CS, Suffia I, Velilla PA, Huang B, Warshaw G, Hildeman DA, Belkaid Y, Chougnet C. Functional regulatory T cells accumulate in aged hosts and promote chronic infectious disease reactivation. J Immunol. 2008;181:1835–1848. doi: 10.4049/jimmunol.181.3.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindstrom J, Shelton D, Fuji Y. Myasthenia gravis. Adv Immunol. 1988;42:233–284. doi: 10.1016/s0065-2776(08)60847-0. [DOI] [PubMed] [Google Scholar]
- 20.Tournier-Lasserve E, Bach JF. The immunogenetics of myasthenia gravis, multiple sclerosis and their animal models. J Neuroimmunol. 1993;47:103–114. doi: 10.1016/0165-5728(93)90020-y. [DOI] [PubMed] [Google Scholar]
- 21.Stacy S, Gelb BE, Koop BA, Windle JJ, Krolick KA, Infante AJ, Kraig E. Split tolerance in a novel transgenic model of autoimmune myasthenia gravis. J Immunol. 2002;169:6570–6579. doi: 10.4049/jimmunol.169.11.6570. [DOI] [PubMed] [Google Scholar]
- 22.Stacy S, Wall KA, Krolick KA, Infante AJ, Kraig E. Recall immune memory: a new tool for generating late onset autoimmune myasthenia gravis. Mech Ageing Dev. 2003;124:931–940. doi: 10.1016/s0047-6374(03)00165-9. [DOI] [PubMed] [Google Scholar]
- 23.Wall KA, Hu JY, Currier P, Southwood S, Sette A, Infante AJ. A disease-related epitope of Torpedo acetylcholine receptor. Residues involved in I-Ab binding, self-nonself discrimination, and TCR antagonism. J Immunol. 1994;152:4526–4536. [PubMed] [Google Scholar]
- 24.Infante AJ, Thompson PA, Krolick KA, Wall KA. Determinant selection in murine experimental autoimmune myasthenia gravis. Effect of the bm12 mutation on T-cell recognition of acetylcholine receptor epitopes. J Immunol. 1991;146:2977–2982. [PubMed] [Google Scholar]
- 25.Olsberg CA, Maxwell LC, Mikiten TM, Krolick KA. Analysis of contractile properties of muscles from rats immunized with purified acetylcholine receptor. J Neuroimmunol. 1987;14:253–266. doi: 10.1016/0165-5728(87)90013-0. [DOI] [PubMed] [Google Scholar]
- 26.Yokoi T, Mulac-Jericevic B, Kurisaki J, Atassi MZ. T lymphocyte recognition of acetylcholine receptor: localization of the full T cell recognition profile on the extracellular part of the alpha chain of Torpedo californica acetylcholine receptor. Eur J Immunol. 1987;17:1697–1702. doi: 10.1002/eji.1830171204. [DOI] [PubMed] [Google Scholar]
- 27.Karachunski PI, Ostlie N, Bellone M, Infante AJ, Conti-Fine BM. Mechanisms by which the I-ABM12 mutation influences susceptibility to experimental myasthenia gravis: a study in homozygous and heterozygous mice. Scand J Immunol. 1995;42:215–225. doi: 10.1111/j.1365-3083.1995.tb03648.x. [DOI] [PubMed] [Google Scholar]
- 28.Standifer N, Stacy S, Kraig E, Infante AJ. Discrete T-cell populations with specificity for a neo-self antigen bear distinct imprints of tolerance. J Immunol. 2007;178:3544–3550. doi: 10.4049/jimmunol.178.6.3544. [DOI] [PubMed] [Google Scholar]
- 29.Yager EJ, Ahmed M, Lanzer K, Randall TD, Woodland DL, Blackman MA. Age-associated decline in T cell repertoire diversity leads to holes in the repertoire and impaired immunity to influenza virus. J Exp Med. 2008;205:711–723. doi: 10.1084/jem.20071140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicoletti C, Yang X, Cerny J. Repertoire diversity of antibody response to bacterial antigens in aged mice. III. Phosphorylcholine antibody from young and aged mice differ in structure and protective activity against infection with Streptococcus pneumoniae. J Immunol. 1993;150:543–549. [PubMed] [Google Scholar]
- 31.Yang X, Stedra J, Cerny J. Relative contribution of T and B cells to hypermutation and selection of the antibody repertoire in germinal centers of aged mice. J Exp Med. 1996;183:959–970. doi: 10.1084/jem.183.3.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krieger NR, Fathman CG, Shaw MK, Ridgway WM. Identification and characterization of the antigen-specific subpopulation of alloreactive CD4+ T cells in vitro and in vivo. Transplantation. 2000;69:605–609. doi: 10.1097/00007890-200002270-00023. [DOI] [PubMed] [Google Scholar]
- 33.Ridgway W, Fasso M, Fathman CG. Following antigen challenge, T cells up-regulate cell surface expression of CD4 in vitro and in vivo. J Immunol. 1998;161:714–720. [PubMed] [Google Scholar]
- 34.Eaton SM, Burns EM, Kusser K, Randall TD, Haynes L. Age-related defects in CD4 T cell cognate helper function lead to reductions in humoral responses. J Exp Med. 2004;200:1613–1622. doi: 10.1084/jem.20041395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frasca D, Riley RL, Blomberg BB. Humoral immune response and B-cell functions including immunoglobulin class switch are downregulated in aged mice and humans. Semin Immunol. 2005;17:378–384. doi: 10.1016/j.smim.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 36.Wu B, Deng C, Goluszko E, Christadoss P. Tolerance to a dominant T cell epitope in the acetylcholine receptor molecule induces epitope spread and suppresses murine myasthenia gravis. J Immunol. 1997;159:3016–3023. [PubMed] [Google Scholar]
- 37.Ellmerich S, Takacs K, Mycko M, Waldner H, Wahid F, Boyton RJ, Smith PA, Amor S, Baker D, Hafler DA, Kuchroo VK, Altmann DM. Disease-related epitope spread in a humanized T cell receptor transgenic model of multiple sclerosis. Eur J Immunol. 2004;34:1839–1848. doi: 10.1002/eji.200324044. [DOI] [PubMed] [Google Scholar]
- 38.Ebhardt MB, Shive CL, Guardia R, Gapin L, Boehm BO, Forsthuber TG. Immunological adjuvants efficiently induce antigen-specific T cell responses in old mice: implication for vaccine adjuvant development in aged individuals. Cell Immunol. 2002;215:87–97. doi: 10.1016/s0008-8749(02)00005-9. [DOI] [PubMed] [Google Scholar]
- 39.Fulop T, Jr, Larbi A, Dupuis G, Pawelec G. Ageing, autoimmunity and arthritis: Perturbations of TCR signal transduction pathways with aging – a biochemical paradigm for the ageing immune system. Arthritis Res Ther. 2003;5:290–302. doi: 10.1186/ar1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Urban L, Bessenyei B, Marka M, Semsei I. On the role of aging in the etiology of autoimmunity. Gerontology. 2002;48:179–184. doi: 10.1159/000052839. [DOI] [PubMed] [Google Scholar]
- 41.Li X, Bao-Guo X, Jian-Ying X, Lu C, Lu J. Decrease of CD4+CD25highFoxp3+ regulatory T cells and elevation of CD19+ BAFF-R+ B cells and soluble ICAM-1 in myasthenia gravis. Clin Immunol. 2008;126:180–188. doi: 10.1016/j.clim.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Wang H, Chi L, Wang W. The role of FoxP3+CD4+CD25hi Tregs in the pathogenesis of myasthenia gravis. Immunol Lett. 2009;122:52–57. doi: 10.1016/j.imlet.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 43.Le Panse R, Cizeron-Clairac G, Cuvelier M, Truffault F, Bismuth J, Nancy P, De Rosbo NK, Berrih-Aknin S. Regulatory and pathogenic mechanisms in human autoimmune myasthenia gravis. Ann NY Acad Sci. 2008;1132:135–142. doi: 10.1196/annals.1405.019. [DOI] [PubMed] [Google Scholar]
- 44.Tsaknaridis L, Spencer L, Culbertson N, Hicks K, LaTocha D, Chou YK, Whitham RH, Bakke A, Jones RE, Offner H, Bourdette DN, Vanderbark AA. Functional assay for human CD4+CD25+ Treg cells reveals an age-dependent loss of suppressive activity. J Neuroscience Res. 2003;74:296–308. doi: 10.1002/jnr.10766. [DOI] [PubMed] [Google Scholar]
- 45.Nobrega A, Haury M, Gueret R, Coutinho A, Weksler ME. The age-associated increase in autoreactive immunoglobulins reflects a quantitative increase in specificities detectable at lower concentrations in young mice. Scand J Immunol. 1996;44:437–443. doi: 10.1046/j.1365-3083.1996.d01-335.x. [DOI] [PubMed] [Google Scholar]
- 46.Stacy S, Krolick KA, Infante AJ, Kraig E. Immunological memory and late onset autoimmunity. Mech Ageing Dev. 2002;123:975–985. doi: 10.1016/s0047-6374(02)00035-0. [DOI] [PubMed] [Google Scholar]