

Abstract
The structural assembly of synapses can be accomplished in a rapid time frame, although most nascent synapses formed during early development are not fully functional and respond poorly to presynaptic action potentials. The mechanisms that are responsible for this delay in synapse maturation are unknown. Histone deacetylases (HDACs) regulate the activity state of chromatin and repress gene expression through the removal of acetyl groups from histones. Class I HDACs, which include HDAC1 and HDAC2, are expressed in the CNS, although their specific role in neuronal function has not been studied. To delineate the contribution of HDAC1 and HDAC2 in the brain, we have used pharmacological inhibitors of HDACs and mice with conditional alleles to HDAC1 and HDAC2. We found that a decrease in the activities of both HDAC1 and HDAC2 during early synaptic development causes a robust facilitation of excitatory synapse maturation and a modest increase in synapse numbers. In contrast, in mature neurons a decrease in HDAC2 levels alone was sufficient to attenuate basal excitatory neurotransmission without a significant change in the numbers of detectable nerve terminals. Therefore, we propose that HDAC1 and HDAC2 form a developmental switch that controls synapse maturation and function acting in a manner dependent on the maturational states of neuronal networks.
Introduction
In the mammalian CNS, synaptogenesis is an extremely rapid process (Aghajanian and Bloom, 1967). The structural assembly of synapses is accomplished within hours (Ahmari et al., 2000; Friedman et al., 2000). However, most nascent synapses formed during early development are not fully functional and respond poorly to presynaptic stimulation and typically undergo a maturation process that increases their efficacy (Vaughn, 1989; Fletcher et al., 1994; Matteoli et al., 1995; Hanse and Gustafsson, 2001; Mozhayeva et al., 2002; Mohrmann et al., 2003; Polo-Parada et al., 2004; Shen et al., 2006). These newly formed synapses lack a fully primed, readily releasable pool of vesicles (Mozhayeva et al., 2002; Shen et al., 2006) which underlies their characteristically unreliable responses to presynaptic action potentials (Basarsky et al., 1994). Recent studies have identified mechanisms that switch these presynaptically “silent” synapses into functional ones as this switch may form a robust substrate for synaptic plasticity (Atasoy and Kavalali, 2006). This work has shown that neuronal activity as well as neurotrophin signaling can render immature synapses functional and potentiate synaptic transmission in developing synaptic circuits (Luikart et al., 2005; Shen et al., 2006; Yao et al., 2006).
Despite our increasing understanding of the characteristics of these nascent terminals and the stimuli that can augment their activity, the mechanisms that delay their maturation are unknown. Processes that control synapse maturation play a critical role in establishing excitation/inhibition balance in synaptic networks and impairments in this balance may lead to neurodevelopmental disorders, such as Rett syndrome (Zoghbi, 2003; Dani et al., 2005; Nelson et al., 2006) and other autism-spectrum disorders (Rubenstein and Merzenich, 2003; Morrow et al., 2008).
Histone deacetylases (HDACs) are nuclear enzymes that trigger long-term changes in gene expression by removing acetyl groups from key histone residues, thus promoting an inactive chromatin state (Saha and Pahan, 2006; Hildmann et al., 2007). HDACs also regulate several developmental processes as well as plasticities in mature tissue (Zhang et al., 2002; Fischer et al., 2007; Vecsey et al., 2007; Abel and Zukin, 2008; Nott et al., 2008). HDACs are widely expressed in the brain (Broide et al., 2007), with HDAC1 and HDAC2 expressed in the hippocampus (MacDonald and Roskams, 2008), although the role of individual HDACs in the CNS is largely unknown.
To address whether HDACs impact synapse development and neuronal function, we used pharmacological and genetic approaches. Treatment of immature hippocampal neurons with HDAC inhibitors resulted in increased synapse numbers and a robust augmentation of synaptic function. Deletion of both HDAC1 and HDAC2 in floxed mouse neurons during early synaptic development resulted in a similar facilitation of excitatory synapse maturation and a modest increase in synapse numbers. In contrast, in mature neurons a decrease in HDAC2 alone was sufficient to decrease basal excitatory neurotransmission without a significant change in synapse numbers. Based on these findings, we propose that HDAC1 and HDAC2 form a developmental switch that controls synapse maturation and function, which is dependent on the maturational state of the neuron.
Materials and Methods
Cell culture.
Primary dissociated hippocampal cultures were prepared according to previously published protocols (Kavalali et al., 1999). Briefly, whole hippocampi were dissected from C57BL/6 mice on postnatal days 0–3, or floxed HDAC1, floxed HDAC2, or floxed HDAC1&2 mice on postnatal day 0. Tissue was trypsinized for 10 min at room temperature, mechanically dissociated using siliconized glass pipettes, and then plated onto Matrigel-coated coverslips. At day 1 in vitro (1 DIV), 4 μm cytosine arabinoside (Sigma) was added.
Lentiviral infection.
HEK 293 cells were transfected with the Fugene 6 transfection system (Roche Molecular Biochemicals) with the expression plasmid, pFUGW or pFUGW-Cre, and two helper plasmids, Δ8.9 and vesicular stomatitis virus G-protein, at 3 μg of each DNA per 75 cm2 flask (Dittgen et al., 2004). After 48 h transfection, lentivirus-containing culture medium was harvested, filtered at a 0.45 μm pore size, and immediately used for infection. Hippocampal cultures were infected either at 2 DIV or 7 DIV by adding 300 μl of viral suspension to each well containing 700 μl of media, and recordings were done either at 7–8 DIV or 16–18 DIV. Titer was determined by counting the number of infected neurons per coverslip (high: >80%; low: <20%).
Electrophysiology.
Synaptic activity was recorded from hippocampal pyramidal neurons (in at least three independent cultures) using a whole-cell voltage clamp technique. Data were acquired using an Axopatch 200B amplifier and Clampex 9.2 software (Molecular Devices). Recordings were filtered at 2 kHz and sampled at 200 μs. A modified Tyrode's solution containing the following (in mm): 150 NaCl, 4 KCl, 2 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES, pH 7.4, was used as external bath solution, with 50 μm picrotoxin and 1 μm TTX to isolate miniature EPSCs (mEPSCs) or 10 μm CNQX, 50 μm APV, and 1 μm TTX to isolate miniature IPSCs (mIPSCs). The pipette internal solution for some of the voltage-clamp experiments contained the following (in mm): 115 Cs-MeSO3, 10 CsCl, 5 NaCl, 10 HEPES, 0.6 EGTA, 20 TEA-Cl, 4 Mg-ATP, 0.3 Na3GTP, pH 7.35, and 10 QX-314 (300 mOsm). For some of the mIPSC experiments (supplemental Figs. S2, S5, available at www.jneurosci.org as supplemental material), a different internal solution was used. This internal solution contained the following (in mm): 117.5 CsCl, 8 NaCl, 10 HEPES, 0.2 EGTA, 0.2 MgCl2, 2 Mg-ATP, 0.3 Na3GTP, pH 7.2, and 5 QX-314 (285 mOsm). All electrophysiology experiments were performed in cultures from at least three independent experiments.
Fluorescence imaging.
Synaptic boutons were loaded with FM2-10 (400 μm) in the presence of a 47 mm K+-containing modified Tyrode's solution for 90 s to ensure maximal uptake of the dye into individual synapses. After perfusion with a dye-free Tyrode's solution for 10 min, the degree of loading was determined with three rounds of 60-s-long application of the 90 mm K+/2 mm Ca2+ solution (each separated by 60 s intervals) to reach baseline levels of fluorescence. All experiments were conducted in the presence of 10 μm CNQX and 50 μm AP5 to prevent recurrent activity. Images were obtained by a cooled-intensified digital CCD camera (Roper Scientific) during illumination at 480 ± 20 nm (505 DCLP, 535 ± 25 BP) via an optical switch (Sutter Instruments). Images were acquired and analyzed using Metafluor Software (Universal Imaging).
Immunocytochemistry.
Dissociated hippocampal neurons were fixed for 30 min with methanol, rinsed twice with 1× PBS/glycine, and then blocked in 2% goat serum for 1 h. The cells were then incubated with primary antibodies, anti-postsynaptic density protein 95 (PSD-95) mouse monoclonal (1:200) (Affinity Bioreagents), and anti-synapsin rabbit polyclonal (1:1000) (Synaptic Systems) overnight at 4°C. The next day the cells were washed with 1× PBS, then incubated with fluorescent secondary antibodies, goat-anti-rabbit (1:200) (Invitrogen), and goat-anti-mouse (1:200) (Invitrogen). Coverslips were mounted with Vectashield (Vector Laboratories) and neurons were visualized on a Zeiss confocal microscope. Minimums of four coverslips were used in each condition, and three to five images were taken from each coverslip using the program LSM510 Meta (Carl Zeiss). These pictures were analyzed for the synapsin and PSD-95 puncta as well as colocalization of PSD-95 and synapsin puncta using the program ImageJ (NIH) by an observer blind to the treatment conditions. All immunocytochemistry was performed in triplicate in cultures from three independent experiments.
Western blotting.
Hippocampal neurons were washed with 1× PBS and cell lysates were prepared using Laemmli sample buffer. Cell lysates were run on a 12% SDS-PAGE and proteins were transferred onto a nitrocellulose membrane (Biorad). Blocking of the membrane was performed with either 5% or 3% nonfat dry milk solution in 1× PBS containing 0.05% Tween 20 and then immunoblotted with anti-acetyl H3 (Millipore), anti-acetyl H4 (Millipore), HDAC1 (Sigma), or HDAC2 (Abcam). Secondary antibodies used were either goat anti-mouse or goat anti-rabbit (Vector Laboratories). All the incubations for Western blots were done at room temperature. Membranes were incubated with primary [actin (1:1000; Santa Cruz Biotechnology), acetyl histone H3 (1:2000; Millipore), acetyl histone H4 (1:2000; Millipore), HDAC1 (1:1000; Sigma), and HDAC2 (1:1000; Abcam)] and secondary antibodies for 2 and 1 h, respectively, on a shaker with mild shaking. Enhanced chemiluminescent detection kit (GE Healthcare) was used to visualize the bands. All Western blots experiments were done on three independent cultures.
mRNA isolation and RT-QPCR.
RNA was isolated from hippocampal neurons plated onto a six-well plate using STAT-60 reagents according to the manufacturer's instructions. Isolated RNA was treated with DNase I before cDNA construction. DNase I was inactivated by incubating the sample at 85°C for 15 min. Five hundred nanograms of RNA was used to construct cDNA using Superscript III first-strand synthesis system of RT-PCR (Invitrogen) according to the manufacturer's protocol. Real-time PCR was performed using 1 μl (20 ng/μl) of cDNA added to 10.5 μl of Power SYBR Green PCR Master mix (Applied Biosystems), 7.5 μl of H2O, and 1 μl of each primer. Quantitative real-time PCR of HDAC 1 was performed using Taqman probes (Applied Biosystems) according to the manufacturer's instructions. The following primers were used at a 10 μm concentration: HDAC1 forward, 5′-TCTACCGCCCTCACAAGGC-3′; reverse, 5′-ACAGAACTCAAA-CAAGCCATC-3′; HDAC2 forward, 5′-GCGTACAGTCAAGGAGGCGG-3′; and reverse, 5′-GCTTCATGGGATGACCCTGGC-3′. GAPDH primers were used as controls: forward, 5′-AGGTCGGTGTGAACGGATTTG-3′; and reverse, 5′-TGTAGACCATGTAGTTGAGGTCA-3′. Each sample was run in triplicate. Reactions were run on an 7500 real-time PCR system (Applied Biosystems) with the following cycling program: 50°C for 10 min 95°C for 2 min, 40 cycles of 95°C for 15 s, 60°C for 1 min, and dissociation protocol included 95°C for 15 s, 60°C for 1 min 95°C for 15 s. Detection of fluorescent products was taken at the end of the second step. Dissociation protocol was not used with Taqman probes.
Data and statistical analyses.
mEPSCs (AMPA-type) and mIPSCs were typically recorded for at least 3–5 min and analyzed using Mini Analysis Program version 6.0.3 (Synaptosoft). All data were analyzed using a two-tailed Student's t test, and all error bars represent the SEM with significance set at p < 0.05. The FM2-10 dye unloading experiment was analyzed using the Kolmogorov–Smirnov test.
Results
HDAC inhibitors facilitate activity of nascent synapses
To examine the role of transcriptional repression in the regulation of nascent synaptic transmission onto developing neurons, we treated dissociated hippocampal cultures from C57BL/6 mice with low concentrations (50 nm; 250 nm) of the HDAC inhibitor Trichostatin A (TSA) at 4 DIV, a time point when synaptic contacts are first detectable (Basarsky et al., 1994; Mozhayeva et al., 2002; Shen et al., 2006) (Fig. 1A). Eighteen to twenty-four hours after TSA treatment, we found a significant increase in the frequency but not amplitudes of mEPSCs in 5 DIV cultures (Fig. 1B–D), suggesting an increase in excitatory neurotransmission. TSA treatment also increased acetylation of histone H3 and H4 residues in immature cultures suggesting alterations in gene expression (supplemental Fig. S1A–D, available at www.jneurosci.org as supplemental material). In contrast, mIPSCs were not altered in their frequency or amplitudes by TSA treatment (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). In parallel experiments, treatment with valproic acid (20 μm; 400 μm), another HDAC inhibitor, also significantly increased the frequency of mEPSCs with no change in amplitude or in mIPSC properties (supplemental Fig. S3, available at www.jneurosci.org as supplemental material, and data not shown).
Figure 1.

Increased spontaneous excitatory neurotransmission and synapse formation in immature hippocampal neurons (5 DIV) following 18–24 h of treatment with the histone deacetylase inhibitor TSA. A, Schematic timeline of the experiment. B–D, Spontaneous excitatory synaptic currents upon 250 nm TSA treatments for 18–24 h. B, Representative recording of miniature excitatory events in DMSO and TSA-treated neurons recorded in 1 μm tetrodotoxin and 50 μm picrotoxin. C, Bar graph depicts the significant increase in mEPSC frequency following TSA treatment. The numbers in the bars denote the number of neurons recorded for each condition. D, Bar graph of mEPSC amplitude reveals no change following TSA treatment. E–G, Representative images of immunostaining of young hippocampal culture (5 DIV). Immunostaining was performed using synapsin (red) and PSD-95 (green) antibodies. White arrows indicate the colocalized puncta. H–J, Bar graphs depict the number of synapsin puncta (H), PSD-95 puncta (I), and colocalized synapsin and PSD-95 puncta (J) calculated from confocal images using ImageJ software. The immunocytochemistry was performed in triplicate in cultures from three independent experiments. K, Representative recordings of sucrose response. L, Bar graph depicting the charge/30 s calculated using mEPSC recordings upon application of 500 mm sucrose revealed a significant increase in sucrose response following TSA treatment (*p < 0.05 in this and all subsequent figures). M, Representative recordings depicting EPSCs evoked in response to action potential stimulation in cultures treated with either DMSO or 250 nm TSA. After TSA treatment an increased fraction of neurons exhibited response to stimulation compared with DMSO treatment. N, Cumulative histogram shows that a higher percentage of cells respond to evoked stimulation upon 250 nm TSA treatment compared with control DMSO treatment.
This specific augmentation in miniature excitatory synaptic transmission could originate from an increase in the number of excitatory synapses formed onto a neuron or an increase in their efficacy, presumably by facilitating the maturation of these nascent synaptic contacts. To better understand how HDAC inhibitors may contribute to the increase in mEPSC frequency, we examined the number of presynaptic terminals formed on pyramidal neuron dendrites in culture. Neurons were immunostained for synapsin, a presynaptic vesicle protein, and PSD-95, a marker for mature excitatory postsynaptic sites, in 5 DIV cultures. The incorporation of PSD-95 into nascent synapses has been shown to be a critical factor for their functional maturation (El-Husseini et al., 2000). We found an abundance of presynaptic and postsynaptic markers in vehicle treated cultures indicating the availability of structural markers for initial synaptic contacts (Fig. 1E,H,I). Treatment with TSA produced a modest increase (∼40%) in the number of presynaptic terminals as identified with synapsin immunoreactivity whereas the numbers of postsynaptic sites identified as PSD-95 puncta showed a robust increase (∼300%) (Fig. 1F–I). Analysis of the colocalization of the synapsin and PSD-95 puncta demonstrated the largest increase (∼450%) following TSA treatment, consistent with the notion that HDAC inhibition in immature cultures facilitates maturation of synaptic contacts rather than simply an increase in the number of excitatory synapses per se (Fig. 1J).
To determine whether HDAC inhibition in young cultures facilitated maturation of synaptic responses, we examined the response of TSA-treated nascent synapses to hypertonic stimulation, an indicator for functional maturity in nerve terminals as well as the assembly of the readily releasable pool of vesicles (Mozhayeva et al., 2002). We found that TSA treatment produced a significant increase in synaptic response to hypertonic stimulation suggesting that these new synapses were indeed functional (Fig. 1K,L). Furthermore, we have performed additional experiments to determine whether HDAC inhibition altered evoked transmission. Only a small fraction of cells responded to stimulation at this early time point (Mozhayeva et al., 2002) and the proportion of responding cells was higher in 250 nm TSA treatment (44%) compared with DMSO treatment (28%). Moreover, we found the average response amplitude upon stimulation was higher in TSA-treated cells, although these changes did not reach significance (Fig. 1M,N). This finding suggests that despite the increase in the number of recycling vesicles and spontaneous release, the HDAC inhibition does not facilitate stimulation-secretion coupling significantly presumably due to an additional deficiency in full vesicle priming. Collectively, these findings suggest that inhibition of HDAC activity during early synaptic development produces a pronounced increase in the maturation of synaptic function as well as a modest increase in the number of synapses.
Knockdown of HDAC1&2 in immature hippocampal neurons augments synaptic activity
The pharmacological HDAC inhibitor, TSA, targets both class I and class II HDACs, whereas valproic acid selectively inhibits class I HDACs (Krämer et al., 2003; Göttlicher, 2004; Drummond et al., 2005). HDAC1 and HDAC2 are two prominent class I HDACs that are present in hippocampal neurons and are part of the corepressor complex that interacts with MeCP2, an important regulator of hippocampal synaptic function (Asaka et al., 2006; Nelson et al., 2006, 2008). To examine whether HDAC1 and/or HDAC2 are involved in mediating the enhanced maturation of excitatory synaptic contacts, we used a genetic experimental approach using mice with conditional null alleles for HDAC1, HDAC2, or for both HDAC1 and HDAC2 (HDAC1&2) (Montgomery et al., 2007). We made primary dissociated hippocampal cultures from newborn floxed HDAC1, HDAC2, or HDAC1&2 mice and infected them at 2 DIV with high-titer lentivirus expressing the Cre recombinase gene tagged with green fluorescent protein (GFP) (referred to as CRE) or GFP alone (GFP) and then recorded at 7–8 DIV, which allows sufficient time for Cre-mediated recombination (Fig. 2A). In this paradigm, we found that CRE expression in floxed HDAC1&2 cultures produced a significant knockdown of HDAC1 mRNA (80%), and HDAC2 mRNA (90%) as well as significantly decreased HDAC1 and HDAC2 protein levels (supplemental Fig. S4B–G, available at www.jneurosci.org as supplemental material).
Figure 2.

Deletion of HDAC1&2 increases spontaneous excitatory neurotransmission in immature hippocampal neurons. A, A schematic of the experimental timeline showing that young neurons at 2 DIV were infected with lentivirus (GFP or CRE) and recordings were performed on 7–8 DIV. B, E, H, Representative recordings of miniature excitatory events recorded in 1 μm tetrodotoxin and 50 μm picrotoxin in GFP- and CRE-infected HDAC1, HDAC2, or HDAC1&2 neurons, respectively. C, F, I, Bar graph depicting the mEPSC frequency following the loss of HDAC1, HDAC2, or HDAC1&2. The loss of HDAC1 or HDAC2 had no significant effect on mEPSC frequency, while the loss of HDAC1&2 produced a significant increase in mEPSC frequency. D, G, J, Bar graphs of mEPSC amplitudes showing no change following the loss of HDAC1, HDAC2, or HDAC1&2 in young neurons.
In floxed HDAC1 neurons infected with either GFP or CRE, we did not detect any difference in mEPSC frequency or amplitudes (Fig. 2B–D). In floxed HDAC2 neurons infected with either GFP or CRE, we also did not detect a difference in mEPSC frequency or amplitude (Fig. 2E–G). These data suggest that either HDAC1 or HDAC2 by themselves are not sufficient to mediate the suppression in synapse maturation. In contrast, when we recorded from hippocampal neurons with HDAC1&2 double knockdown, we observed a dramatic increase in mEPSC frequency (Fig. 2H,I) without a significant change in the amplitudes of mEPSCs (Fig. 2H,J) compared with floxed cultures infected with GFP. The knockdown of both HDAC1&2 did not alter mIPSC properties (supplemental Fig. S5, available at www.jneurosci.org as supplemental material), similar to our findings with the pharmacological HDAC inhibitors (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). These results strongly suggest that HDAC1&2 act in concert to delay maturation of excitatory synapses.
We next examined whether the TSA effect on synapse maturation was mediated through HDAC1&2. We made primary dissociated hippocampal cultures from newborn floxed HDAC1&2 mice, infected them with CRE or GFP at 2 DIV, and then applied TSA for 18–24 h at 6–7 DIV (Fig. 3A). We applied the TSA at 6–7 DIV, instead of 5 DIV, to allow sufficient time for Cre-mediated recombination to occur (data not shown). We found a significant increase in mEPSC frequency following the deletion of HDAC1&2 (Fig. 3B,C), with no change in amplitude (Fig. 3B,D), in agreement with our pharmacological data (Fig. 2H–J). This increase, however, was not significantly enhanced with the addition of the HDAC inhibitor TSA (Fig. 3B,C), supporting the premise that inhibition of HDAC1&2 activity permits synapse maturation during early synaptogenesis.
Figure 3.
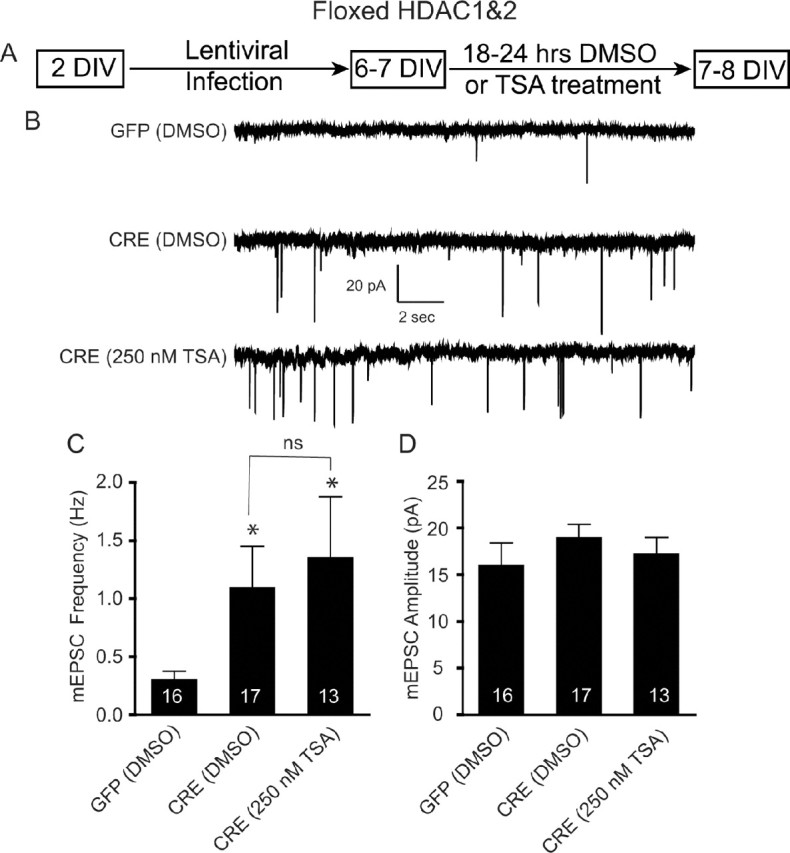
Occlusion of TSA-mediated increase in mEPSC in immature neurons lacking HDAC1&2. A, Schematic timeline of the experiment. Floxed HDAC1&2 neurons were plated, infected with lentivirus expressing either GFP or CRE at 2 DIV, treated with DMSO or TSA (250 nm) at 6–7 DIV, and recorded 18–24 h later. B, Representative recordings of miniature excitatory events in GFP- and CRE-infected neurons treated with either DMSO or TSA. Recordings were made in Tyrode's solution containing 1 μm tetrodotoxin and 50 μm picrotoxin. C, Bar graph showing that the loss of HDAC1&2 results in a significant increase in mEPSC frequency and that TSA treatment did not result in a further increase in mEPSC frequency. D, Bar graph of mEPSC amplitudes revealing no change following TSA treatment on HDAC1&2 knockdown neurons.
Knockdown of HDAC1&2 increases the rate of synaptic vesicle mobilization in nascent nerve terminals
Following initial synapse formation, individual presynaptic nerve terminals typically possess a small number of actively recycling vesicles, most of which are not readily releasable (Mozhayeva et al., 2002; Mohrmann et al., 2003). These immature synaptic boutons can be distinguished by their slow synaptic vesicle mobilization kinetics under high-potassium-mediated stimulation. Mature synapses, on the other hand, show rapid synaptic vesicle mobilization demonstrating the formation of the readily releasable pool. To investigate which characteristics of synaptic vesicle mobilization are apparent following the loss of HDAC1&2 in young neurons, we monitored the activity-dependent uptake and release of the fast-departitioning styryl dye FM2-10 (400 μm) (Fig. 4A). We first loaded synaptic boutons with FM2-10 in the presence of a 47 mm K+/2 mm Ca2+ solution for 90 s to ensure maximal uptake of the dye into individual synapses. We determined the degree of loading after multiple applications of the 90 mm K+/2 mm Ca2+ solution to establish baseline levels of fluorescence. At 7 DIV, when challenged with 90 mm K+ solution, GFP-infected synapses maximally labeled with FM2-10 showed slow dye release typical of immature synapses (Fig. 4B,D). In contrast, the majority of synapses formed in the floxed HDAC1&2 cultures after CRE infection showed robust FM dye loss similar to synaptic boutons seen at later stages of synaptic maturation (Fig. 4C,D). Interestingly, this swift vesicle mobilization, a hallmark of presynaptic maturity, was accompanied by only a small decrease in the size of the total actively recycling vesicle pool (Fig. 4E), quantified by multiple 90 mm K+/2 mm Ca2+ solution applications (Mozhayeva et al., 2002). Collectively, these results suggest that the maturational delay induced by the activities of HDAC1&2 targets presynaptic vesicle dynamics without markedly changing the number of recycling vesicles.
Figure 4.

Knockdown of HDAC1&2 accelerates synaptic vesicle mobilization in immature hippocampal neurons. A, Schematic timeline of the experiment. B, C, Monitoring the activity-dependent uptake and release of the fast-departitioning styryl dye FM2-10 (400 μm). To probe the release kinetics, synaptic boutons were loaded with FM2-10 in the presence of a 47 mm K+/2 mm Ca2+ solution for 90 s to ensure maximal uptake of the dye into individual synapses. The degree of loading was determined after multiple applications of the 90 mm K+/2 mm Ca2+ solution to establish baseline levels of fluorescence. B, At 7 DIV, when challenged with 90 mm K+ solution, GFP-infected cultures maximally labeled with FM2-10 showed slow dye release typical of the immature synapses. C, In contrast, synapses formed in the floxed HDAC1&2 cultures after CRE infection showed robust FM dye loss similar to synaptic boutons seen at later stages of synaptic maturation. The superimposed gray lines correspond to the first-order exponential decay fitted onto raw traces to extract destaining kinetics. D, Normalized cumulative histograms of time constant values for the CRE-infected neurons and the GFP-infected neurons. The difference between the two distributions is statistically significant [Kolmogorov–Smirnov test, p < 0.001, n = 1695 (GFP) and 1015 (CRE) boutons]. E, Total pool sizes measured as the maximum value of fluorescence after loading with FM 2–10, for CRE-infected neurons (n = 9) and GFP-infected neurons (n = 6).
Knockdown of HDAC1&2 in isolated postsynaptic neurons alone is not sufficient to recapitulate the effect of global knockdown
Our data strongly implicate a primary effect of HDAC1&2 at the presynaptic neuron; however, to examine a possible role for postsynaptic neuron in synapse maturation, we used a viral-mediated knockdown approach (Fig. 5A). If the activity of HDAC1&2 in a particular neuron was sufficient to delay maturation of synapses formed onto the same neuron in a cell-autonomous manner, then knockdown of HDAC1&2 in a single neuron should recapitulate the effects of a global knockdown and pharmacological HDAC inhibitor treatment. To address this question, we infected floxed HDAC1&2 hippocampal cultures with low titers of lentivirus expressing CRE allowing the deletion of HDAC1 and HDAC2 in only a small number (∼20%) of cells (data not shown). Under these conditions, when we recorded from CRE-infected neurons (neurons with HDAC1 and HDAC2 deleted), we did not detect a significant increase in the frequency or amplitudes of mEPSCs compared with neurons infected with GFP-expressing virus (Fig. 5B–D). These results demonstrate that knockdown of HDAC1&2 in isolated postsynaptic neurons alone is not sufficient to recapitulate the effect of the global knockdown. This finding is consistent with the premise that accelerated maturation of synapses seen after the knockdown of HDAC1&2 is attributable to the presynaptic inputs onto a neuron.
Figure 5.

HDAC1&2 deletion using low-titer lentiviral infection does not affect spontaneous excitatory neurotransmission. A, Schematic timeline of the experiment showing that 2 DIV after floxed HDAC1&2 neurons were plated and infected with low titer lentivirus expressing either GFP or CRE. B, Representative recordings of miniature excitatory events in GFP- and CRE-infected neurons (low-titer) in 1 μm tetrodotoxin and 50 μm picrotoxin. C, Bar graph showing that mEPSC frequency was not significantly different when recorded from an infected neuron in which the majority of presynaptic cells were uninfected. D, Bar graph of mEPSC amplitudes, also showing no change following the low-titer lentiviral infection of floxed HDAC1&2 neurons.
Knockdown of HDAC2 but not HDAC1 decreases synaptic activity in mature hippocampal neurons
Given that epigenetic mechanisms are known to regulate developmental processes, we wondered if the observed function of HDAC1 and HDAC2 to inhibit excitatory synaptic function is specific to early stages of neuronal development. To address this question, we plated dissociated hippocampal neurons from floxed HDAC1 or HDAC2 mice, infected at 7 DIV, treated the mature cultures at 15 DIV with DMSO or TSA, and recorded at 16 DIV. At 16 DIV, hippocampal synapses share all the structural and functional characteristics of mature synapses formed in vivo, including a sizable recycling vesicle pool and robust responses to action potential stimulation (Mozhayeva et al., 2002). We found that CRE expression in floxed HDAC1 cultures produced a robust knockdown of HDAC1 mRNA and HDAC1 protein without altering HDAC2 mRNA (supplemental Fig. S6B,D, available at www.jneurosci.org as supplemental material). Floxed HDAC2 cultures infected with CRE had a significant decrease in HDAC2 mRNA and HDAC2 protein without any change in HDAC1 mRNA levels (supplemental Fig. S6C,E, available at www.jneurosci.org as supplemental material). In agreement with a previous study (Nelson et al., 2006), we found that treatment with TSA resulted in a significant decrease in mEPSC frequency in control mature hippocampal neurons (Fig. 6A–C). The knockdown of HDAC1 in mature neurons did not induce an alteration in mEPSC frequency or amplitudes (Fig. 6B–D). Moreover, TSA treatment of HDAC1 deleted neurons produced a significant decrease in mEPSC frequency, suggesting that in mature neurons, inhibition of HDAC1 does not mediate the deficit in synaptic transmission (Fig. 6B,C). In contrast, knockdown of HDAC2 in mature neurons significantly reduced mEPSC frequency similar to that seen with TSA treatment of GFP-infected neurons with no effect on mEPSC amplitudes (Fig. 6B–D). In addition, TSA treatment of mature neurons lacking HDAC2 resulted in a trend, but no significant further decrease in the frequency of mEPSCs, suggesting that HDAC2 function is critical for maintenance of unitary synaptic transmission in mature neurons (Fig. 6B,C). Interestingly, knockdown of HDAC1 or HDAC2 in mature hippocampal neurons did not alter mIPSC properties, suggesting that the HDAC-mediated inhibition is specific to excitatory neurotransmission (supplemental Fig. S7, available at www.jneurosci.org as supplemental material).
Figure 6.

In mature neurons, HDAC2 but not HDAC1 deletion affects spontaneous excitatory neurotransmission. A, Schematic of the experimental timeline showing that floxed HDAC1 or HDAC2 neurons were infected with lentivirus expressing either GFP or CRE on 7 DIV then at 15 DIV cultures were treated with either DMSO vehicle or TSA (1 μm) and then recorded at 16 DIV. B, Representative recordings of miniature excitatory events in GFP- and CRE-infected neurons treated with either DMSO or TSA. Recordings were made in Tyrode's solution containing 1 μm tetrodotoxin and 50 μm picrotoxin. C, Bar graph depicting the significant decrease in mEPSC frequency observed by TSA treatment. In HDAC1 knockdown mature neurons, there was no change in mEPSC treatment, however treatment with TSA did significantly decrease mEPSC frequency. In mature neurons, knockdown of HDAC2 resulted in a significant decrease in mEPSC frequency revealing its importance in mediating synaptic transmission in mature neurons. TSA treatment of neurons with HDAC2 knockdown did not produce a further significant decrease in mEPSC frequency. D, A bar graph of mEPSC amplitude did not reveal a significant change in amplitude in any of the conditions examined in mature neurons. E, F, H, I, Representative images of immunostaining of mature hippocampal culture (15 DIV). Immunostaining was performed using PSD-95 and synapsin antibodies. G, J, Bar graphs depicting the number of PSD-95 and synapsin colocalized puncta/20 μm length of the dendrite.
To test whether this specific decrease in excitatory neurotransmission in mature neurons originates from a decrease in the number of excitatory synapses, we examined immunocytochemical colocalization of synapsin and PSD-95 (Fig. 6E–J). Treatment of mature cultures with TSA did not result in a significant decrease in the number of synapsin/PSD-95 coclusters (Fig. 6E–G). Similarly, knockdown of HDAC2 in mature neurons did not alter the distribution of synapsin/PSD-95 coclusters detected per dendritic length (Fig. 6H–J). Together, these findings indicate that maintenance of miniature excitatory transmission in mature neurons requires the activity of HDAC2, which targets the presynaptic release machinery but not the number or maturation state of synapses.
To further investigate the role of HDAC2-mediated effects on synaptic transmission in mature neurons, we infected C57BL/6 hippocampal neurons at 7 DIV with lentivirus expressing HDAC2 (Fig. 7A). We found that overexpression of HDAC2 in mature neurons produced a significant increase in mEPSC frequency with no change in their amplitudes (Fig. 7B–D). The significant increase in mEPSC frequency with HDAC2 overexpression, coupled with the significant decrease in mEPSC in HDAC2 null neurons, suggests that HDAC2 plays a critical role in mediating synaptic transmission in mature neurons and changes in its expression can profoundly impact synaptic function.
Figure 7.

Overexpression of HDAC2 increases spontaneous excitatory neurotransmission in mature hippocampal neurons A, Schematic of the experimental timeline showing that hippocampal neurons were infected with lentivirus expressing either GFP or GFP-HDAC2 on 7 DIV and then recorded at 16–18 DIV. B, Representative recordings of miniature excitatory events in GFP and GFP-HDAC2-infected neurons. Recordings were made in Tyrode's solution containing 1 μm tetrodotoxin and 50 μm picrotoxin. C, Bar graph depicting the significant increase in mEPSC frequency observed by HDAC2 overexpression. D, A bar graph of mEPSC amplitude did not reveal a significant change in amplitudes between GFP- and GFP-HDAC2-infected mature neurons.
HDAC inhibition by TSA treatment decreases MeCP2 expression in young but not mature hippocampal cultures
To examine the molecular mechanisms that may underlie the synaptic effects caused by HDAC inhibition, we treated 4 DIV and 19 DIV hippocampal culture with 250 nm TSA for 18–24 h (supplemental Fig. S8A,F, available at www.jneurosci.org as supplemental material). Brain-derived neurotrophic factor (BDNF) has been implicated in multiple roles in synaptogenesis and synaptic transmission (Huang and Reichardt, 2001). We therefore examined BDNF expression in the TSA-treated cultures. Using quantitative real-time PCR, we did not detect a change in BDNF mRNA following 250 nm TSA treatment in either young or mature hippocampal cultures (supplemental Fig. S8B,G, available at www.jneurosci.org as supplemental material).
Previous work has demonstrated that MeCP2, an important regulator of synaptic transmission, interacts with a corepressor complex that contains HDAC1&2 (Nan et al., 1998). Therefore, we examined the expression of MeCP2 upon HDAC inhibition in young and mature hippocampal culture. Rather surprisingly, we found a significant decrease in MeCP2 mRNA, as well as protein levels, from young but not mature hippocampal culture treated with 250 nm TSA (supplemental Fig. S8C–E,H–J). These data suggest that in young neurons gene repression may be further relieved by the loss of MeCP2, which in turn allows cells to increase transcription of necessary genes required for the maturation of nascent synapses.
Discussion
In this study, using electrophysiological, immunocytochemical and optical methods, we have examined the role of HDAC inhibition, specifically HDAC1 and HDAC2, in the maturation and function of hippocampal synapses. Our results demonstrate that during early synapse development, HDAC1- and HDAC2-mediated chromatin silencing restricts the progression of excitatory synapse maturation. In contrast, in mature neurons, HDAC2 alone is required to maintain excitatory drive without altering synapse numbers. Our findings suggest that when synapse formation is sparse and neurons are immature, the chromatin state maintained by HDACs suppresses excitatory neurotransmission, and once neuronal networks mature and synapses are in abundance, it augments excitation. Under the same conditions, however, HDAC activity does not impact inhibitory transmission, which showed an ∼50-fold increase over development (0.02–0.03 Hz at 5 DIV to 1.0–1.5 Hz at 16 DIV), suggesting network specificity in the epigenetic control of synaptic circuits. Together, these results indicate that HDAC1 and HDAC2 are critical regulators of excitation–inhibition balance in developing synaptic networks through their control of excitatory drive.
From the results presented, we propose that histone deacetylation acts as a developmentally regulated feedback mechanism for the maturation of excitatory synapses. This hypothesis rests on several key observations. First, treatment with pharmacological inhibitors of HDACs, such as TSA, significantly augmented miniature excitatory neurotransmission ∼10-fold. However, TSA treatment produced only a modest increase (∼40%) in the number of presynaptic terminals as identified by synapsin immunoreactivity, whereas under the same conditions the numbers of postsynaptic PSD-95 puncta and their colocalization with synapsin puncta showed a robust increase (∼300%), indicating a significant increase in excitatory synapses. Second, knockdown of HDAC1&2 in immature hippocampal neurons mimicked the effect of TSA and specifically augmented excitatory synaptic activity. Third, knockdown of HDAC1&2 in immature hippocampal neurons increased the rate of synaptic vesicle mobilization in nerve terminals, consistent with facilitation of synapse maturation. Fourth, knockdown of HDAC1&2 in isolated postsynaptic neurons alone was not sufficient to recapitulate the effect of global knockdown, indicating that HDAC activity had a non-cell-autonomous impact on synaptic maturation presumably originating from presynaptic neurons.
In contrast to our findings in immature neurons, knockdown of HDAC2 but not HDAC1 in mature hippocampal cultures specifically decreased excitatory synaptic activity in a manner that occluded the similar effect of TSA on excitatory neurotransmission. This result is in agreement with the recent finding that HDAC1 is predominantly expressed in neural progenitor cells and glia, whereas HDAC2 expression is initiated in neural progenitors and is upregulated in postmitotic neurons but not in fully differentiated glia (MacDonald and Roskams, 2008).
During early development, the stability of synaptic networks faces significant challenges due to ongoing synaptogenesis (Davis and Bezprozvanny, 2001; Turrigiano and Nelson, 2004). Immature neurons typically possess high membrane resistance due to the scarcity of channels and the modest extent of their dendritic arborization (Overstreet-Wadiche and Westbrook, 2006). These characteristics of immature neurons render them prone to instability and highlight the importance of potential negative feedback mechanisms that can control synaptic inputs onto these neurons. Our data suggest that histone deacetylases, important regulators of transcriptional repression, are critical for repressing the maturation of excitatory synapses during early development, thereby promoting stable synaptic networks. Moreover, our findings indicate that transcriptional repression mediated via HDAC2 controls excitatory drive in mature synapses, suggesting a developmental specificity of the role of HDACs on synaptic function. HDAC inhibitor drugs, currently under development for the treatment of various diseases, including cancer, have not yet been examined for their potential roles in the brain. Our data suggest that inhibition of HDAC1&2 during development, and HDAC2 in mature brain, may have potential unexpected neurological side effects.
The HDAC-mediated effect on synapse development was specific for controlling the maturation of excitatory synapses, which may contribute to a dysregulation of excitation–inhibition balance. Interestingly, imbalances in excitation–inhibition have been purported to underlie autism-spectrum disorders (Rubenstein and Merzenich, 2003), including Rett syndrome (Dani et al., 2005; Nelson et al., 2006). Our data implicating the involvement of HDAC1 and HDAC2 in this balancing act are particularly intriguing in regards to Rett syndrome, since MeCP2, the gene whose loss of function causes the disorder, is part of a protein complex that includes HDAC1 and HDAC2 that mediates effects on gene expression. The findings we present here are consistent with a previous study from our group that reported a significant selective decrease in mEPSC frequency in recordings from cultured hippocampal neurons from either constitutive MeCP2 null mice or floxed MeCP2 neurons that were transfected with Cre recombinase at 7 DIV and recorded at 14 DIV (Nelson et al., 2006). In the present study, we found that in mature neurons the loss of HDAC2 produced a similar specific change in mEPSC frequency. We also found that the loss of HDAC1 did not alter synaptic properties in mature neurons, suggesting that HDAC2 is the important contributor to excitatory synaptic function. The loss of MeCP2 may impact the ability of HDAC1 and HDAC2 to exert effects on excitatory synapse maturation, resulting in alterations in the development of synaptic networks that contribute to the imbalance in synaptic transmission that ultimately may underlie the neurological deficits associated with this disorder. Indeed, a recent study has reported a direct correlation between the number of glutamatergic synapses in hippocampal autaptic cultures and the levels of MeCP2, suggesting that MeCP2 may play a critical role in regulating glutamatergic synapse formation during early development (Chao et al., 2007). We therefore hypothesize that the link between synaptogenesis and imbalances in synaptic transmission observed following alterations in MeCP2 levels might be the result of impairments in transcriptional repression mediated by HDAC1 and HDAC2. Furthermore, BDNF, a bona fide target of MeCP2-dependent transcription, may play a critical role in regulation of this pathway, as it has recently been demonstrated that in cortical neurons BDNF signaling can lead to S-nitrosylation of HDAC2, which causes its dissociation from chromatin-bound complex, thereby allowing the transcription of various genes responsible for neural development (Nott et al., 2008).
Recent studies have demonstrated that transcriptional regulators, such as the MEF2 (myocyte enhancer factor 2) family and NPAS4 (Flavell et al., 2006; Barbosa et al., 2008; Lin et al., 2008), tightly regulate synaptic drive and influence the balance of excitation–inhibition in developing neuronal networks. Our findings provide direct evidence that class I HDACs regulate these synaptic processes, suggesting that not only transcription factors but also epigenetic mechanisms play crucial roles in the development of synaptic networks. A major effort of future research is to identify the genes that HDAC1 and HDAC2 control to restrict the progression of excitatory synapse maturation (Kalinovsky and Scheiffele, 2004; Akins and Biederer, 2006).
In summary, we provide direct evidence that HDACs, specifically HDAC1 and HDAC2, are critical in the development of synaptic networks by their control of excitatory drive. Our data also demonstrate an important role for HDAC2 in synaptic transmission in mature neurons, suggesting that HDAC1 and HDAC2 are not functionally redundant in mature neurons but instead have unique roles in the brain. Collectively, our findings identify a crucial role for epigenetic mechanisms as key regulators of synapse maturation and transmission.
Footnotes
This work is supported by National Institutes of Health Grant MH081060 to L.M.M. E.T.K. is an Established Investigator of the American Heart Association. We thank M. Adachi, A. Autry, C. Chung, M. Ertunc, M. Mahgoub, S. Ashimi, M. Morris, and E. Na for their helpful discussions and comments.
References
- Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol. 2008;8:57–64. doi: 10.1016/j.coph.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghajanian GK, Bloom FE. The formation of synaptic junctions in developing rat brain: a quantitative electron microscopic study. Brain Res. 1967;6:716–727. doi: 10.1016/0006-8993(67)90128-x. [DOI] [PubMed] [Google Scholar]
- Ahmari SE, Buchanan J, Smith SJ. Assembly of presynaptic active zones from cytoplasmic transport packets. Nat Neurosci. 2000;3:445–451. doi: 10.1038/74814. [DOI] [PubMed] [Google Scholar]
- Akins MR, Biederer T. Cell-cell interactions in synaptogenesis. Curr Opin Neurobiol. 2006;16:83–89. doi: 10.1016/j.conb.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Asaka Y, Jugloff DG, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis. 2006;21:217–227. doi: 10.1016/j.nbd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Atasoy D, Kavalali ET. Presynaptic unsilencing: searching for a mechanism. Neuron. 2006;50:345–346. doi: 10.1016/j.neuron.2006.04.018. [DOI] [PubMed] [Google Scholar]
- Barbosa AC, Kim MS, Ertunc M, Adachi M, Nelson ED, McAnally J, Richardson JA, Kavalali ET, Monteggia LM, Bassel-Duby R, Olson EN. MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc Natl Acad Sci U S A. 2008;105:9391–9396. doi: 10.1073/pnas.0802679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basarsky TA, Parpura V, Haydon PG. Hippocampal synaptogenesis in cell culture: developmental time course of synapse formation, calcium influx, and synaptic protein distribution. J Neurosci. 1994;14:6402–6411. doi: 10.1523/JNEUROSCI.14-11-06402.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broide RS, Redwine JM, Aftahi N, Young W, Bloom FE, Winrow CJ. Distribution of histone deacetylases 1–11 in the rat brain. J Mol Neurosci. 2007;31:47–58. doi: 10.1007/BF02686117. [DOI] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, Bezprozvanny I. Maintaining the stability of neural function: a homeostatic hypothesis. Annu Rev Physiol. 2001;63:847–869. doi: 10.1146/annurev.physiol.63.1.847. [DOI] [PubMed] [Google Scholar]
- Dittgen T, Nimmerjahn A, Komai S, Licznerski P, Waters J, Margrie TW, Helmchen F, Denk W, Brecht M, Osten P. Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proc Natl Acad Sci U S A. 2004;101:18206–18211. doi: 10.1073/pnas.0407976101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol. 2005;45:495–528. doi: 10.1146/annurev.pharmtox.45.120403.095825. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, Greenberg ME. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science. 2006;311:1008–1012. doi: 10.1126/science.1122511. [DOI] [PubMed] [Google Scholar]
- Fletcher TL, De Camilli P, Banker G. Synaptogenesis in hippocampal cultures: evidence indicating that axons and dendrites become competent to form synapses at different stages of neuronal development. J Neurosci. 1994;14:6695–6706. doi: 10.1523/JNEUROSCI.14-11-06695.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron. 2000;27:57–69. doi: 10.1016/s0896-6273(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Göttlicher M. Valproic acid: an old drug newly discovered as inhibitor of histone deacetylases. Ann Hematol. 2004;83(Suppl 1):S91–S92. doi: 10.1007/s00277-004-0850-2. [DOI] [PubMed] [Google Scholar]
- Hanse E, Gustafsson B. Vesicle release probability and pre-primed pool at glutamatergic synapses in area CA1 of the rat neonatal hippocampus. J Physiol. 2001;531:481–493. doi: 10.1111/j.1469-7793.2001.0481i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildmann C, Riester D, Schwienhorst A. Histone deacetylases—an important class of cellular regulators with a variety of functions. Appl Microbiol Biotechnol. 2007;75:487–497. doi: 10.1007/s00253-007-0911-2. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinovsky A, Scheiffele P. Transcriptional control of synaptic differentiation by retrograde signals. Curr Opin Neurobiol. 2004;14:272–279. doi: 10.1016/j.conb.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Klingauf J, Tsien RW. Activity-dependent regulation of synaptic clustering in a hippocampal culture system. Proc Natl Acad Sci U S A. 1999;96:12893–12900. doi: 10.1073/pnas.96.22.12893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J, Peters MA, Brill B, Groner B, Bach I, Heinzel T, Göttlicher M. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 2003;22:3411–3420. doi: 10.1093/emboj/cdg315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Bloodgood BL, Hauser JL, Lapan AD, Koon AC, Kim TK, Hu LS, Malik AN, Greenberg ME. Activity-dependent regulation of inhibitory synapse development by Npas4. Nature. 2008;455:1198–1204. doi: 10.1038/nature07319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart BW, Nef S, Virmani T, Lush ME, Liu Y, Kavalali ET, Parada LF. TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. J Neurosci. 2005;25:3774–3786. doi: 10.1523/JNEUROSCI.0041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JL, Roskams AJ. Histone deacetylases 1 and 2 are expressed at distinct stages of neuro-glial development. Dev Dyn. 2008;237:2256–2267. doi: 10.1002/dvdy.21626. [DOI] [PubMed] [Google Scholar]
- Matteoli M, Verderio C, Krawzeski K, Mundigl O, Coco S, Fumagalli G, De Camilli P. Mechanisms of synaptogenesis in hippocampal neurons in primary culture. J Physiol Paris. 1995;89:51–55. doi: 10.1016/0928-4257(96)80551-1. [DOI] [PubMed] [Google Scholar]
- Mohrmann R, Lessmann V, Gottmann K. Developmental maturation of synaptic vesicle cycling as a distinctive feature of central glutamatergic synapses. Neuroscience. 2003;117:7–18. doi: 10.1016/s0306-4522(02)00835-7. [DOI] [PubMed] [Google Scholar]
- Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21:1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, Al-Saad S, Ware J, Joseph RM, Greenblatt R, Gleason D, Ertelt JA, Apse KA, Bodell A, Partlow JN, Barry B, et al. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321:218–223. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozhayeva MG, Sara Y, Liu X, Kavalali ET. Development of vesicle pools during maturation of hippocampal synapses. J Neurosci. 2002;22:654–665. doi: 10.1523/JNEUROSCI.22-03-00654.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol. 2006;16:710–716. doi: 10.1016/j.cub.2006.02.062. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J Neurosci. 2008;28:395–406. doi: 10.1523/JNEUROSCI.3796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Watson PM, Robinson JD, Crepaldi L, Riccio A. S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature. 2008;455:411–415. doi: 10.1038/nature07238. [DOI] [PubMed] [Google Scholar]
- Overstreet-Wadiche LS, Westbrook GL. Functional maturation of adult-generated granule cells. Hippocampus. 2006;16:208–215. doi: 10.1002/hipo.20152. [DOI] [PubMed] [Google Scholar]
- Polo-Parada L, Bose CM, Plattner F, Landmesser LT. Distinct roles of different neural cell adhesion molecule (NCAM) isoforms in synaptic maturation revealed by analysis of NCAM 180 kDa isoform-deficient mice. J Neurosci. 2004;24:1852–1864. doi: 10.1523/JNEUROSCI.4406-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha RN, Pahan K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006;13:539–550. doi: 10.1038/sj.cdd.4401769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Wu B, Zhang Z, Dou Y, Rao ZR, Chen YR, Duan S. Activity-induced rapid synaptic maturation mediated by presynaptic cdc42 signaling. Neuron. 2006;50:401–414. doi: 10.1016/j.neuron.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Vaughn JE. Fine structure of synaptogenesis in the vertebrate central nervous system. Synapse. 1989;3:255–285. doi: 10.1002/syn.890030312. [DOI] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Qi J, Chen G. Actin-dependent activation of presynaptic silent synapses contributes to long-term synaptic plasticity in developing hippocampal neurons. J Neurosci. 2006;26:8137–8147. doi: 10.1523/JNEUROSCI.1183-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]