

Abstract
The level of serum uric acid in human has been increasing over the last decades, and correlates with an increase prevalence of renal disease and metabolic syndrome. Understanding the role of uric acid in these conditions may provide clues for preventing the current epidemic of renal disease. Controversy still remains if hyperuricemia is simply a consequence or a cause of renal disease although epidemiological studies have attempted to resolve this issue. In this review, we discuss the clinical and experimental evidence for a causal role of hyperuricemia in renal diseases and potential relationships of hyperuricemia with metabolic syndrome.
Keywords: Uric acid, Insulin resistance, Renal disease, Metabolic syndrome
Introduction
There has been a general increase in mean uric acid levels in the USA over the last several decades. Levels have risen in men from <3.5 mg/dl in the 1920s [1], to approximately 5.0 mg/dl in the 1950s [2], and to 6.0–6.5 mg/dl in the 1970s [3]. The increase in serum uric acid levels during the twentieth century correlates not only with the increasing frequency of diabetes and obesity, but also with the progressive increase in hypertension, all of which are common causes of renal disease. Therefore, hyperuricemia could be viewed as a reflection of the current epidemic.
Many investigators have viewed the rise in uric acid to simply be a marker for renal disease since the increase in serum uric acid is in part a consequence of decreased renal function. However, recent epidemiological and experimental evidence have provided compelling evidence that uric acid may also have a role in causing renal disease.
In this review, we discuss the controversial role of hyperuricemia in renal disease, and discuss potential mechanism(s) by which uric acid may cause renal disease.
Regulation of urate in the kidney
The regulation of urinary urate excretion is complex. According to the classic paradigm, urate is freely filtrated at glomerulus, followed by reabsorption of nearly 99% in the proximal tubule (reabsorption), with 50% secretion in the S2 segment (secretion), and then 40% reabsorption in the S3 segment (postsecretory reabsorption). As a consequence, only 10% of urate is excreted. There is no evidence of urate handling in distal tubules.
More recent studies have focused on the specific transporters in the proximal tubules that are involved in reabsorption. Uric acid transporter 1 (URAT1) has been recently identified as one of the major urate transporters present in the luminal border of human proximal tubular cells [4]. URAT1 is an organic anion transporter that exchanges organic anions for urate [4]. In humans mutation of URAT1 is known to result in hypouricemia [4], suggesting a key role of URAT1 in regulation of serum uric acid levels. At the basolateral membrane, other organic anion transporters (OAT), including OAT1 and OAT3, and possibly OAT4, appear to have key roles in urate transport. A voltage-sensitive urate transporter (UAT) is located in apical membrane in proximal tubular cell and may also be involved in the reabsorption of urate [5]. However, in spite of these in vitro evidences, there is still no direct evidence that UAT transports urate in vivo. In addition, the multidrug resistance protein-4 (MRP4) has been also identified in the apical membrane of renal proximal tubular cells as an ATP-dependent unidirectional efflux pump for urate [6].
Uromodulin, also known as Tamm-Horsfall protein, may also be involved in urate regulation in the kidney. Uromodulin is an 80-kDa glycoprotein synthesized in the medullary thick ascending limb cells of Henle’s loop (mTAL) and macula densa. Mutation in the uromodulin gene (UMOD) causes hyperuricemia due to low urinary excretion of urate and is observed in familial juvenile hyperuricemic nephropathy (FJHN)/autosomal dominant medullary cystic kidney disease type2 (MCKD2) [7]. These patients exhibit abnormal uromodulin accumulation in mTAL, but not in proximal tubules, suggesting that uromodulin may have an indirect role on urate excretion. Abnormal accumulation of uromodulin in mTAL might reduce NaCl reabsorption in mTAL, leading to volume contraction, and thereby promoting the proximal reabsorption of urate.
Clinical evidence for hyperuricemia in renal disease
Prior to the availability of treatments for hyperuricemia, 25% of gouty subjects developed proteinuria, 50% suffered from chronic renal insufficiency, and 10–25% developed end stage renal disease [8]. These patients commonly showed renal lesions such as arteriolosclerosis, glomerulosclerosis, and tubulointerstitial fibrosis, which was historically described as “Gouty nephropathy”. The cause of the renal lesion was originally thought to be due to intrarenal urate crystal deposition. Later, however, it was found that focal deposition of urate crystals did not appear to adequately explain the diffuse lesions, and that intrarenal crystal deposition could occasionally be found in autopsy specimens in which no renal disease was present [9]. Subsequently, most investigators viewed mild to moderate increases in uric acid in patients with renal disease as usually of no clinical significance.
Consistent with this observation, the Modification of Diet in Renal Disease (MDRD) study reported in 1997 that uric acid was a marker, but not a predictor of decline in renal function [10]. In contrast, a potential causal role of uric acid in renal disease was supported by evidence that uric acid was a risk factor for renal failure among over 49,000 male in the general Japanese population with a relative risk (RR) of 8.52 [11]. Similarly, hyperuricemia was reported to be a risk factor for renal insufficiency in 3,499 patients in Thailand and 6,300 subjects in Japan [12]. Furthermore, recent studies have shown that hyperuricemia was associated with a decline of renal function, hypertension, diffuse proliferative glomerulonephritis, and tubulointerstitial damage in IgA nephropathy [13], and serum uric acid positively correlates with urinary albumin excretion in type 2 diabetes in Taiwan [14]. In addition, in one study uric acid was found to be a better risk factor than proteinuria for development of renal insufficiency [12]. Thus, the role of uric acid in renal disease still remains to be addressed.
Clinical intervention studies to evaluate the effect of lowering uric acid in subjects with renal disease have been mixed. Studies performed in the 1970s were often confounded by short durations of treatment, ineffective lowering of uric acid level, or complicating attacks of gout. However, more recently, Siu et al. randomized 54 asymptomatic hyperuricemic patients with chronic kidney disease to allopurinol (a xanthine oxidase inhibitor) or placebo and reported that allopurinol significantly preserved renal function over the year period of observation [15]. Using a different approach, Talaat and El-Sheikh evaluated the effect of withdrawing allopurinol from 50 subjects with chronic kidney disease and noted a marked exacerbation of blood pressure with renal progression in the subset that was not receiving agents to block the renin angiotensin system [16]. In contrast, subjects receiving ACE inhibitors or angiotensin receptor blockers appeared to show no change, suggesting that allopurinol treatment was working in part by blocking activation of the renin angiotensin system.
Diuretics are commonly used for treatment of hypertension, but their use may be complicated by an increase in serum uric acid. Diuretics reduce blood pressure due to volume depletion, which stimulates proximal sodium reabsorption as well as the rennin–angiotensin system. Uric acid reabsorption appears to be coupled with sodium reabsorption, resulting in an elevation of serum uric acid. Several clinical studies have demonstrated that diuretics have a protective role in heart failure or stroke [17, 18]. However, it has been reported that diuretic usage did not slow but rather accelerates renal progression in hypertensive patients despite reducing blood pressure. For example, the EWPHE [19], SHEP [20], INSIGHT [21], and ALLHAT [17] studies all reported that the usage of diuretics was statistically associated with a greater decline in renal function compared with the other treatment groups. Thus, while diuretics reduce blood pressure and blood pressure-associated events such as stroke and heart failure, they may be less effective at slowing renal disease or reducing cardiovascular events. The reason why diuretics do not seem protect renal function may be due to diuretic-induced hyperuricemia, which could counteract some of the beneficial effects of diuretics in these latter conditions. While this has not been studied directly, it is interesting that in the Systolic Hypertension in the Elderly Program (SHEP) trial, the use of diuretics resulted in a significant reduction of cardiovascular events, but this protection was largely prevented in those patents whose uric acid levels increased [22]. However, we need randomized clinical trials to address the causal role of hyperuricemia in renal and cardiovascular disease.
Clinical evidence for a role of hyperuricemia in subjects with hypertension
Recent studies suggest that hyperuricemia is common in subjects with essential hypertension. For example Feig et al. reported that approximately 90% of adolescents with essential hypertension had serum uric acid levels greater than 5.5 mg/dl versus 0% in normotensive subjects [23]. Interestingly, the association of uric acid with hypertension was not observed in secondary hypertension or white coat hypertension despite the latter group having similar degrees of obesity. The causal role of uric acid for development of hypertension was suggested by the evidence that lowering of uric acid by allopurinol resulted in normal blood pressure in four of five subjects, followed by a return to a hypertensive state on stopping allopurinol [23]. Care must be taken into the interpretation of this latter data, as there was no placebo group.
A mechanism by which uric acid could potentially raise blood pressure could be due to the ability of uric acid to induce endothelial dysfunction. Hyperuricemic patients exhibit endothelial dysfunction defined as impairment of flow-mediated dilatation of the brachial artery [24]. If hyperuricemic subjects are treated with allopurinol, an improvement in endothelial function has been observed in subjects with chronic heart failure [25]. On the other hand, George et al. reported that improvement of endothelial function in heart failure could be achieved with high-dose allopurinol and not probenecid (the latter drug being a uricosuric), suggesting that allopurinol may improve endothelial function via other mechanisms besides lowering uric acid levels, such as by blocking xanthine oxidase-induced oxidants [26]. Therefore, the precise role of uric acid in endothelial dysfunction and hypertension remains to be determined.
Metabolic syndrome, renal disease and uric acid
Metabolic syndrome, which is characterized by the presence of obesity, insulin resistance, hypertriglyceridemia and elevated blood pressure, is now recognized as being epidemic in our society, affecting 27% of the population in the USA [27]. A strong association of hyperuricemia with metabolic syndrome has been shown in epidemiological studies [28]. On the other hand, a close relationship between metabolic syndrome and chronic renal disease has been also recognized and metabolic syndrome is a strong and independent risk factor for chronic kidney disease and microalbuminuria [29]. There is also a graded relationship between the number of metabolic syndrome components and risk for chronic renal disease [29]. More interestingly, it has been found that a serum Cr > 2.4 mg/dl in Japanese patients with non-diabetic renal disease was associated with insulin resistance [30]. Becker et al. have performed a larger scale study with 227 subjects who were diagnosed as mild renal insufficiency due to glomerulonephritis, adult polycystic kidney disease, and interstitial nephritis and found that the patients with these renal diseases developed insulin resistance even when their renal function was preserved (GFR was >90 ml/min/1.73 m2) [31]. These data suggest an intricate link between renal disease and insulin resistance.
Insulin as an endogenous regulator of uric acid in the kidney (Fig. 1)
Fig. 1.
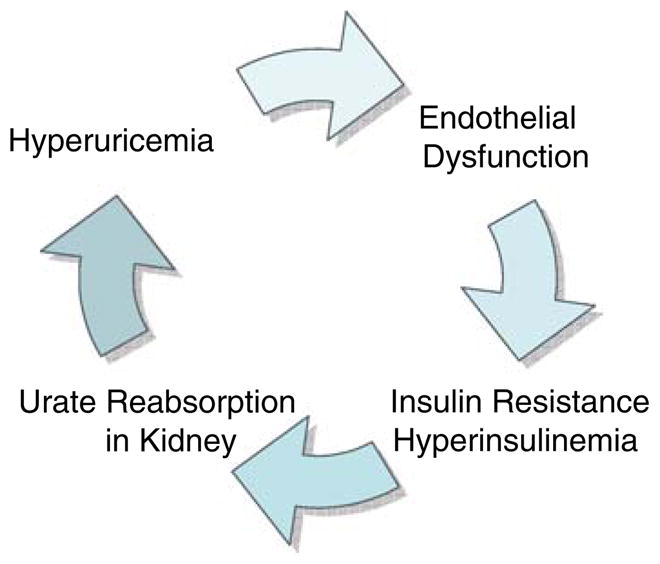
A possible association between hyperuricemia and insulin resistance in the kidney
Metabolic syndrome is characterized by the presence of hyperinsulinemia and an insulin resistant state. Hyperinsulinemia is common in subjects with asymptomatic hyperuricemia, and in individuals with diabetes or hypertension. One potential explanation is that hyperinsulinemia may cause hyperuricemia. For example, insulin acts on the proximal tubule to stimulate urate reabsorption coupled with sodium [32]. Fasting insulin levels inversely correlate with urinary uric acid clearance and are positively associated with serum uric acid in healthy subjects [28]. Furthermore, exogenously administered insulin decreases urinary excretion of uric acid and this is mediated by stimulating urate reabsorption [33] in healthy subjects as well as in hypertensive patients [33]. These studies suggest that the renal proximal tubule remains sensitive to the effects of insulin even in subjects with hypertension (who are often insulin resistant).
On the other hand, there is also evidence that uric acid may cause insulin resistance. Recently, we studied metabolic syndrome induced with fructose in rats, and found that lowering uric acid partially improved the insulin resistance [34]. Taken together, hyperuricemia could potentially be both a cause and a consequence of hyperinsulinemia in metabolic syndrome (Fig. 1).
Experimental evidence demonstrating a causal role for uric acid in renal disease (Fig. 2)
Fig. 2.

Diverse effects of uric acid on endothelial cell. CRP C-reactive protein, NO nitric oxide, ROS reactive oxygen species
In the late 1990s, our group discovered that administration of an uricase inhibitor (2% oxonic acid) in diet can mildly increase uric acid (1.5–3.0 mg/dl) in rats at a level that does not cause urate crystal deposition in the kidney [35]. Surprisingly, hyperuricemic rats developed systemic hypertension, glomerular hypertrophy, glomerular hypertension, afferent arteriolar disease, tubulointerstitial damage, and macrophage infiltration [36–39]. In a model of progressive renal disease (remnant kidney model), hyperuricemia accelerated renal injury, resulting in more glomerulosclerosis, tubulointerstitial fibrosis, and severe vascular injury [39, 40]. These lesions were significantly ameliorated by lowering uric acid with either allopurinol or benziodarone (a uricosuric agent), suggesting a role for uric acid in this process. Similarly, a causal role of uric acid on renal disease was demonstrated in several models of renal disease (Table 1).
Table 1.
The animal models in which uric acid causes renal injury
Mechanism by which uric acid induces renal disease
Uric acid on endothelial cell
As mentioned earlier, there is clinical evidence that hyperuricemia may cause endothelial dysfunction, as lowering uric acid with allopurinol can improve endothelial function as measured by brachial artery vasodilatation [24]. Interestingly, while both uric acid and nitric oxide (NO) exhibit circadian variation, serum uric acid peaks around 6 a.m. when the level of NO is lowest [41]. This relationship can be accounted for by the finding that uric acid also inhibits endothelial cell dependent vasodilatation of rat aortic rings [34] and NO production in endothelial cells [42]. Furthermore, uric acid blunts endothelial cell proliferation in response to serum [42]. The mechanism(s) by which uric acid inhibits NO levels is complex. It may involve scavenging by oxidants, which can be induced by NADPH oxidase under hyperuricemia [43]. A reduced NO bioavailability could also be due in part to inhibition secondary to CRP production [42].
On the other hand, uric acid is also an antioxidant and therefore might be expected to protect against these diseases. In fact, acute administration of uric acid improves endothelial function in diabetic patients [44], and reduces oxidative stress in response to acute exercise in healthy subjects [45]. The anti-oxidant effects of uric acid may be due to the ability of uric acid to scavenge peroxynitrite [46]. Skinner et al have found that the product of uric acid/peroxynitrite reaction results in endothelium-dependent vasorelaxation by releasing NO [47]. The diverse effects of uric acid on the endothelial cell are summarized in Fig. 2.
Uric acid, glomerular hemodynamic, and arteriolar disease
Hyperuricemia also alters glomerular hemodynamics [37]. Hyperuricemia causes cortical renal vasoconstriction in rats as evidenced by a significant increase of afferent and efferent arteriolar resistances. A decrease in glomerular plasma flow and the ultrafiltration coefficient resulted in a 35% decrease in single nephron GFR whereas glomerular pressure was increased. These changes were restored by allopurinol treatment.
Aberrant renal autoregulation appears to be responsible for the glomerular hypertension observed with experimental hyperuricemia. Under normal conditions, an increase in mean systemic arterial pressure causes a reflex vasoconstriction of the afferent arteriole, thus preventing the transmission of the increased pressure to the glomerular circulation. However, in the event that the afferent arteriolar vasoconstriction is insufficient, the transmission of increased pressure to the glomeruli results in glomerular hypertension [37]. While renal vasoconstriction occurs in experimental hyperuricemia, it may be insufficient for the degree of systemic hypertension, therefore glomerular pressures are increased. This may be due to the disease of the afferent arteriole that occurs in the hyperuricemic rats, as evidenced by an increase in the media to lumen ratio. Again, the observation that allopurinol was able to prevent arteriolar hypertrophy leading to a normal renal autoregulatory response indicates a potential role of uric acid on this process [37].
In addition, an increase in renin expression is also observed in hyperuricemic rats [36]. A relationship between serum uric acid and plasma renin activity has been described in humans [48]. Blocking the renin angiotensin system also ameliorates hypertension and renal injury in hyperuricemic rats [35]. Furthermore, studies in humans suggest that uric acid acts on blood pressure and renal injury in part via the renin angiotensin system [16]. This suggests that uric acid may also mediate its effects via activation of the renin angiotensin system.
Summary
In this review, we present epidemiological and experimental evidence suggesting a causal role for uric acid in renal disease. While the data are suggestive, it will be critical to perform well-controlled clinical trials to determine if a benefit can be realized. Since uric acid may work in part through the renin angiotensin system, designing clinical studies may be difficult since most patients are already on ACE inhibitors as part of their renoprotective management. Nevertheless, identifying alternative strategies and new treatments for chronic kidney disease is critical if we are to have an impact on this burgeoning epidemic.
Acknowledgments
Supported by NIH grants HL-68607, DK-52121 and generous funds from Gatorade.
Contributor Information
Takahiko Nakagawa, Email: nakagt@medicine.ufl.edu, Division of Nephrology, Hypertension and Transplantation, University of Florida, PO Box 100224, Gainesville, FL 32610, USA.
Pietro Cirillo, Division of Nephrology, Hypertension and Transplantation, University of Florida, PO Box 100224, Gainesville, FL 32610, USA.
Waichi Sato, Division of Nephrology, Hypertension and Transplantation, University of Florida, PO Box 100224, Gainesville, FL 32610, USA.
Michael Gersch, Division of Nephrology, Hypertension and Transplantation, University of Florida, PO Box 100224, Gainesville, FL 32610, USA.
Yuri Sautin, Division of Nephrology, Hypertension and Transplantation, University of Florida, PO Box 100224, Gainesville, FL 32610, USA.
Carlos Roncal, Division of Nephrology, Hypertension and Transplantation, University of Florida, PO Box 100224, Gainesville, FL 32610, USA.
Wei Mu, Division of Nephrology, Hypertension and Transplantation, University of Florida, PO Box 100224, Gainesville, FL 32610, USA.
L. Gabriela Sánchez-Lozada, Department of Nephrology, Instituto Nacional de Cardiologiá “Ignacio Chávez”, Mexico, Mexico.
Richard J. Johnson, Division of Nephrology, Hypertension and Transplantation, University of Florida, PO Box 100224, Gainesville, FL 32610, USA
References
- 1.Fishberg AM. The interpretation of increased blood uric acid in hyertension. Arch Int Med. 1924;34:503–507. [Google Scholar]
- 2.Hall AP, Barry PE, Dawber TR, et al. Epidemiology of gout and hyperuricemia. A long-term population study. Am J Med. 1967;42:27–37. doi: 10.1016/0002-9343(67)90004-6. [DOI] [PubMed] [Google Scholar]
- 3.Glynn RJ, Campion EW, Silbert JE. Trends in serum uric acid levels 1961–1980. Arthritis Rheum. 1983;26:87–93. doi: 10.1002/art.1780260115. [DOI] [PubMed] [Google Scholar]
- 4.Enomoto A, Kimura H, Chairoungdua A, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002;417:447–452. doi: 10.1038/nature742. [DOI] [PubMed] [Google Scholar]
- 5.Polkowski CA, Grassl SM. Uric acid transport in rat renal basolateral membrane vesicles. Biochim Biophys Acta. 1993;1146:145–152. doi: 10.1016/0005-2736(93)90349-5. [DOI] [PubMed] [Google Scholar]
- 6.Van Aubel RA, Smeets PH, van den Heuvel JJ, et al. Human organic anion transporter MRP4 (ABCC4) is an efflux pump for the purine end metabolite urate with multiple allosteric substrate binding sites. Am J Physiol Renal Physiol. 2005;288:F327–F333. doi: 10.1152/ajprenal.00133.2004. [DOI] [PubMed] [Google Scholar]
- 7.Duncan H, Dixon AS. Gout, familial hypericaemia, and renal disease. Q J Med. 1960;29:127–135. [PubMed] [Google Scholar]
- 8.Talbott JH, Terplan KL. The kidney in gout. Medicine (Baltimore) 1960;39:405–467. [PubMed] [Google Scholar]
- 9.Linnane JW, Burry AF, Emmerson BT. Urate deposits in the renal medulla. Prevalence and associations. Nephron. 1981;29:216–222. doi: 10.1159/000182373. [DOI] [PubMed] [Google Scholar]
- 10.Hunsicker LG, Adler S, Caggiula A, et al. Predictors of the progression of renal disease in the Modification of Diet in Renal Disease Study. Kidney Int. 1997;51:1908–1919. doi: 10.1038/ki.1997.260. [DOI] [PubMed] [Google Scholar]
- 11.Tomita M, Mizuno S, Yamanaka H, et al. Does hyperuricemia affect mortality? A prospective cohort study of Japanese male workers. J Epidemiol. 2000;10:403–409. doi: 10.2188/jea.10.403. [DOI] [PubMed] [Google Scholar]
- 12.Iseki K, Oshiro S, Tozawa M, et al. Significance of hyperuricemia on the early detection of renal failure in a cohort of screened subjects. Hypertens Res. 2001;24:691–697. doi: 10.1291/hypres.24.691. [DOI] [PubMed] [Google Scholar]
- 13.Ohno I, Hosoya T, Gomi H, et al. Serum uric acid and renal prognosis in patients with IgA nephropathy. Nephron. 2001;87:333–339. doi: 10.1159/000045939. [DOI] [PubMed] [Google Scholar]
- 14.Tseng CH. Correlation of uric acid and urinary albumin excretion rate in patients with type 2 diabetes mellitus in Taiwan. Kidney Int. 2005;68:796–801. doi: 10.1111/j.1523-1755.2005.00459.x. [DOI] [PubMed] [Google Scholar]
- 15.Siu Y, Leung K, Tong M, et al. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis. 2006;47:51–59. doi: 10.1053/j.ajkd.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 16.Talaat KM, El-Sheikh AR. The effect of mild hyperuricemia on urinary transforming growth factor beta and the progression of chronic kidney disease. Am J Nephrol. 2007;27:435–440. doi: 10.1159/000105142. [DOI] [PubMed] [Google Scholar]
- 17.Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the antihypertensive, lipid-lowering treatment to prevent heart attack trial (ALLHAT) Jama. 2002;288:2981–2997. doi: 10.1001/jama.288.23.2981. [DOI] [PubMed] [Google Scholar]
- 18.Turnbull F. Effects of different blood-pressure-lowering regimens on major cardiovascular events: results of prospectively-designed overviews of randomised trials. Lancet. 2003;362:1527–1535. doi: 10.1016/s0140-6736(03)14739-3. [DOI] [PubMed] [Google Scholar]
- 19.Fletcher A, Amery A, Birkenhager W, et al. Risks and benefits in the trial of the European Working Party on High Blood Pressure in the Elderly. J Hypertens. 1991;9:225–230. doi: 10.1097/00004872-199103000-00005. [DOI] [PubMed] [Google Scholar]
- 20.Savage PJ, Pressel SL, Curb JD, et al. Influence of long-term, low-dose, diuretic-based, antihypertensive therapy on glucose, lipid, uric acid, and potassium levels in older men and women with isolated systolic hypertension: the systolic hypertension in the Elderly Program. SHEP Cooperative Research Group. Arch Intern Med. 1998;158:741–751. doi: 10.1001/archinte.158.7.741. [DOI] [PubMed] [Google Scholar]
- 21.Brown MJ, Palmer CR, Castaigne A, et al. Morbidity and mortality in patients randomised to double-blind treatment with a long-acting calcium-channel blocker or diuretic in the International Nifedipine GITS study: Intervention as a Goal in Hypertension Treatment (INSIGHT) Lancet. 2000;356:366–372. doi: 10.1016/S0140-6736(00)02527-7. [DOI] [PubMed] [Google Scholar]
- 22.Franse LV, Pahor M, Di Bari M, et al. Serum uric acid, diuretic treatment and risk of cardiovascular events in the Systolic Hypertension in the Elderly Program (SHEP) J Hypertens. 2000;18:1149–1154. doi: 10.1097/00004872-200018080-00021. [DOI] [PubMed] [Google Scholar]
- 23.Feig DI, Johnson RJ. Hyperuricemia in childhood primary hypertension. Hypertension. 2003;42:247–252. doi: 10.1161/01.HYP.0000085858.66548.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zoccali C, Maio R, Mallamaci F, et al. Uric acid and endothelial dysfunction in essential hypertension. J Am Soc Nephrol. 2006;17:1466–1471. doi: 10.1681/ASN.2005090949. [DOI] [PubMed] [Google Scholar]
- 25.Doehner W, Schoene N, Rauchhaus M, et al. Effects of xanthine oxidase inhibition with allopurinol on endothelial function and peripheral blood flow in hyperuricemic patients with chronic heart failure: results from 2 placebo-controlled studies. Circulation. 2002;105:2619–2624. doi: 10.1161/01.cir.0000017502.58595.ed. [DOI] [PubMed] [Google Scholar]
- 26.George J, Carr E, Davies J, et al. High-dose allopurinol improves endothelial function by profoundly reducing vascular oxidative stress and not by lowering uric acid. Circulation. 2006;114:2508–2516. doi: 10.1161/CIRCULATIONAHA.106.651117. [DOI] [PubMed] [Google Scholar]
- 27.Ford ES, Giles WH, Mokdad AH. Increasing prevalence of the metabolic syndrome among U.S. adults. Diabetes Care. 2004;27:2444–2449. doi: 10.2337/diacare.27.10.2444. [DOI] [PubMed] [Google Scholar]
- 28.Facchini F, Chen YD, Hollenbeck CB, et al. Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. Jama. 1991;266:3008–3011. [PubMed] [Google Scholar]
- 29.Chen J, Muntner P, Hamm LL, et al. The metabolic syndrome and chronic kidney disease in U.S. adults. Ann Intern Med. 2004;140:167–174. doi: 10.7326/0003-4819-140-3-200402030-00007. [DOI] [PubMed] [Google Scholar]
- 30.Kobayashi S, Maesato K, Moriya H, et al. Insulin resistance in patients with chronic kidney disease. Am J Kidney Dis. 2005;45:275–280. doi: 10.1053/j.ajkd.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 31.Becker B, Kronenberg F, Kielstein JT, et al. Renal insulin resistance syndrome, adiponectin and cardiovascular events in patients with kidney disease: the mild and moderate kidney disease study. J Am Soc Nephrol. 2005;16:1091–1098. doi: 10.1681/ASN.2004090742. [DOI] [PubMed] [Google Scholar]
- 32.Quinones Galvan A, Natali A, Baldi S, et al. Effect of insulin on uric acid excretion in humans. Am J Physiol. 1995;268:E1–E5. doi: 10.1152/ajpendo.1995.268.1.E1. [DOI] [PubMed] [Google Scholar]
- 33.Muscelli E, Natali A, Bianchi S, et al. Effect of insulin on renal sodium and uric acid handling in essential hypertension. Am J Hypertens. 1996;9:746–752. doi: 10.1016/0895-7061(96)00098-2. [DOI] [PubMed] [Google Scholar]
- 34.Nakagawa T, Tuttle KR, Short RA, et al. Hypothesis:fructose-induced hyperuricemia as a causal mechanism for the epidemic of the metabolic syndrome. Nat Clin Pract Nephorl. 2005;1:80–86. doi: 10.1038/ncpneph0019. [DOI] [PubMed] [Google Scholar]
- 35.Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension. 2001;38:1101–1106. doi: 10.1161/hy1101.092839. [DOI] [PubMed] [Google Scholar]
- 36.Nakagawa T, Mazzali M, Kang DH, et al. Hyperuricemia causes glomerular hypertrophy in the rat. Am J Nephrol. 2003;23:2–7. doi: 10.1159/000066303. [DOI] [PubMed] [Google Scholar]
- 37.Sanchez-Lozada LG, Tapia E, Santamaria J, et al. Mild hyperuricemia induces vasoconstriction and maintains glomerular hypertension in normal and remnant kidney rats. Kidney Int. 2005;67:237–247. doi: 10.1111/j.1523-1755.2005.00074.x. [DOI] [PubMed] [Google Scholar]
- 38.Mazzali M, Kanellis J, Han L, et al. Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism. Am J Physiol Renal Physiol. 2002;282:F991–F997. doi: 10.1152/ajprenal.00283.2001. [DOI] [PubMed] [Google Scholar]
- 39.Kang DH, Nakagawa T, Feng L, et al. A role for uric acid in the progression of renal disease. J Am Soc Nephrol. 2002;13:2888–2897. doi: 10.1097/01.asn.0000034910.58454.fd. [DOI] [PubMed] [Google Scholar]
- 40.Mazzali M, Kim YG, Suga S, et al. Hyperuricemia exacerbates chronic cyclosporine nephropathy. Transplantation. 2001;71:900–905. doi: 10.1097/00007890-200104150-00014. [DOI] [PubMed] [Google Scholar]
- 41.Kanabrocki EL, Third JL, Ryan MD, et al. Circadian relationship of serum uric acid and nitric oxide. Jama. 2000;283:2240–2241. doi: 10.1001/jama.283.17.2240. [DOI] [PubMed] [Google Scholar]
- 42.Feig DI, Nakagawa T, Karumanchi SA, et al. Hypothesis: Uric acid, nephron number, and the pathogenesis of essential hypertension. Kidney Int. 2004;66:281–287. doi: 10.1111/j.1523-1755.2004.00729.x. [DOI] [PubMed] [Google Scholar]
- 43.Sautin YY, Nakagawa T, Zharikov S, et al. Adverse effects of the classical antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am J Physiol Cell Physiol. 2007;293:C584–C596. doi: 10.1152/ajpcell.00600.2006. [DOI] [PubMed] [Google Scholar]
- 44.Waring WS, McKnight JA, Webb DJ, et al. Uric acid restores endothelial function in patients with type 1 diabetes and regular smokers. Diabetes. 2006;55:3127–3132. doi: 10.2337/db06-0283. [DOI] [PubMed] [Google Scholar]
- 45.Waring WS, Webb DJ, Maxwell SR. Systemic uric acid administration increases serum antioxidant capacity in healthy volunteers. J Cardiovasc Pharmacol. 2001;38:365–371. doi: 10.1097/00005344-200109000-00005. [DOI] [PubMed] [Google Scholar]
- 46.Whiteman M, Halliwell B. Protection against peroxynitrite-dependent tyrosine nitration and alpha 1-antiproteinase inactivation by ascorbic acid. A comparison with other biological antioxidants. Free Radic Res. 1996;25:275–283. doi: 10.3109/10715769609149052. [DOI] [PubMed] [Google Scholar]
- 47.Skinner KA, White CR, Patel R, et al. Nitrosation of uric acid by peroxynitrite. Formation of a vasoactive nitric oxide donor. J Biol Chem. 1998;273:24491–24497. doi: 10.1074/jbc.273.38.24491. [DOI] [PubMed] [Google Scholar]
- 48.Saito I, Saruta T, Kondo K, et al. Serum uric acid and the renin-angiotensin system in hypertension. J Am Geriatr Soc. 1978;26:241–247. doi: 10.1111/j.1532-5415.1978.tb02396.x. [DOI] [PubMed] [Google Scholar]
- 49.Roncal CA, Mu W, Croker B, et al. Effect of elevated serum uric acid on cisplatin-induced acute renal failure. Am J Physiol Renal Physiol. 2007;292:F116–122. doi: 10.1152/ajprenal.00160.2006. [DOI] [PubMed] [Google Scholar]
- 50.Sanchez-Lozada LG, Tapia E, Jimenez A, et al. Fructose-induced metabolic syndrome is associated with glomerular hypertension and renal microvascular damage in rats. Am J Physiol Renal Physiol. 2006;292(1):F423–F429. doi: 10.1152/ajprenal.00124.2006. [DOI] [PubMed] [Google Scholar]