

Abstract
Long‐term neurological sequela is common among herpes simplex encephalitis (HSE) survivors. Animal models for HSE are used to investigate mechanisms of acute disease, but little has been done to model chronic manifestations of HSE. The current study presents a detailed, systematic analysis of chronic neuropathology, including characterization of topography and sequential progression of degenerative lesions and inflammation. Subsequent to intranasal HSV‐1 infection, inflammatory responses that were temporally and spatially distinct persisted in infected cortical and brain stem regions. Neutrophils were present exclusively within the olfactory bulb and brain stem regions during the acute phase of infection, while the chronic inflammation was marked by plasma cells, lymphocytes and activated microglia. The chronic lymphocytic infiltrate, cytokine production, and activated microglia were associated with the loss of cortical neuropile in the entorhinal cortex and hippocampus. Animals surviving the acute infection showed a spectrum of chronic lesions from decreased brain volume, neuronal loss, activated astrocytes, and glial scar formation to severe atrophy and cavitations of the cortex. These lesions were also associated with severe spatial memory deficits in surviving animals. Taken together, this model can be utilized to further investigate the mechanisms of neurological defects that follow in the wake of HSE.
Keywords: Herpes encephalitis, chronic inflammation, neuropathology, memory deficits
INTRODUCTION
Herpes simplex virus 1 (HSV‐1) is the most common cause of fatal sporadic viral encephalitis of immunocompetent individuals in the United States, accounting for about 10%–20% of all cases of encephalitis (59). The virus is prevalent in 80%–90% of the population worldwide, with majority of primary infection occurring during the first decade of life up to 40 years of age, depending on socioeconomic and geographic factors (51). Viral brain infection occurs either as a primary infection, commonly in neonates (30), or is caused by reactivation of latent virus in immunocompetent adults (49). Acyclovir therapy increases the survival rate (30%) for HSV‐1 encephalitis (HSE) without treatment to 70%–80% when administered early. Despite antiviral therapy, neurological morbidities associated with HSE are observed in >50% of patients 30, 59. Neurological outcomes from HSV‐1 infection in neonates include mild ocular dysfunction or blindness, speech delay, motor abnormalities like hemiparesis or spastic quadriplegia, persistent seizures, and microencephaly (30). In contrast, HSE‐associated sequelae in adults is predominantly manifested as anterograde memory loss, anosmia (loss of smell), and dysphasia (loss of language) 4, 27, 35, 57, 59. Little is known about the pathogenesis of the long‐term neurological outcomes ensuing HSE.
Classically, HSE in adult patients manifests as bilateral cerebrocortical lesions in the temporal lobes, with or without parietal and frontal lobe involvement (19). The long‐term damage produced by infection is functionally and anatomically confined to the limbic system, in particular, the amygdaloid nucleus, hippocampus, insulae, parahippocampus, and the orbital, fusiform, and cingulate gyrii of the brain are affected (27). Based on the location of the lesions, it has been postulated that the virus enters the central nervous system (CNS) through the olfactory bulb and/or trigeminal nerves, possibly after a reactivation event in the trigeminal ganglion 9, 19. Murine models have shown more demonstrable evidence that both the olfactory and trigeminal nerve routes are likely conduits to the CNS 15, 52. Viral antigens in the brain are demonstrable during the acute phase of the infection 20, 58. However, it is not known if experimental infection results in chronic neuropathology and neurological deficits commonly seen during human infection.
Recent studies in our laboratory have demonstrated persistent T lymphocyte infiltration and activated microglial cells in the brain up to 30 days postinfection (33). However, the consequence of persistent inflammation, particularly in the development of chronic brain lesions, the neuroanatomical location of inflammatory cells, and the long‐term effects on neural systems affected by HSV‐1, brain infection remain unknown. In the present study, we hypothesized that similar to humans, prolonged neuroinflammation during herpes encephalitis in Balb/c mice (33) would result in neural tissue damage and detectable neurological deficits. A detailed, systematic analysis of long‐term neuropathological alterations, including characterization of topography and sequential progression of degenerative brain lesions, nature of inflammatory infiltrates associated with sites of acute viral replication, and memory deficits in an experimental murine model of HSE are presented.
MATERIALS AND METHODS
Viral infection
HSV‐1 strain 17 syn+ was propagated and titrated using plaque assay on rabbit skin fibroblasts (CCL68; American Type Culture Collection®, Manassas, VA, USA). Eight to ten‐week‐old female BALB/c mice (Charles River Laboratories™, Boston, MA, USA) were infected via intranasal (i.n) administration with 1.25 × 105 HSV‐1 plaque‐forming units (PFU)/nostril (2.5 × 105 PFU/mouse). Animals were euthanized under deep anesthesia by cardiac puncture at various times postinfection and perfused with 10% neutral buffered formalin (NBF). Seven animals died between 7 and 9 days post infection (p.i) and thus could not be perfused with NBF. This study was approved by The University of Minnesota Institutional Animal Care and Use Committee.
Tissue preparation
Following perfusion with 10% formalin, brains were removed immediately and immersed in NBF for a minimum of 48 h. Post fixation, 1 mm coronal sections were cut using a precision brain matrice (Braintree Scientific, Braintree, MA, USA), and processed using standard procedures for paraffin embedding. To examine the nasal cavity, the skull was decalcified in hydrochloric acid for a maximum of 6 h before completely cutting cross and sagittal sections at 2 mm intervals. Briefly, paraffin‐embedded tissues were sectioned at 5 µm and stained with hematoxylin/eosin (HE) or luxol fast blue/cresyl violet (LFB/CV). For mice that died, in addition to the nervous system (NS), major organs, including liver, lungs, heart, spleen, and intestine, were sectioned and stained with HE and examined for evidence of HSV‐1 related lesions.
Immunohistochemistry
Briefly, 5 µm brain sections were deparaffinized in xylene and hydrated through graded alcohols and finally in deionized water. Tissue sections were subjected to antigen retrieval with either 10 mM citrate buffer (pH 6.0) under pressure or heated tris‐EDTA buffer (pH 9.0). Endogenous peroxidase was quenched with 3% H2O2 diluted in methanol. Primary antibodies were rat antihuman CD3 (Serotec, Raleigh, NC, USA) at 1:1500 dilution, rabbit antihuman cleaved caspase‐3 (Cell Signalling Technology Inc., Danvers, MA, USA) at 1:200 dilution, rat antimouse Mac‐2 (Dako, Carpinteria, CA, USA) at 1:10 000 dilution, rabbit antihuman myeloperoxidase (MPO; Thermo Scientific, Rockford, IL, USA) at 1:100 dilution, and rabbit antimouse glial fibrillary acid protein (GFAP; Dako) goat anti‐HSV‐1 (Chemicon International, Inc. acquired by Millipore, Billerica, MA). Secondary antibodies were either antirabbit horseradish peroxidase (HRP) conjugated (Envision™ rabbit, Dako), rat‐on‐mouse polymer detection kit with HRP (Biocare, Concord, CA, USA), or biotinylated rabbit antigoat (Vector Laboratories, Burlingame, CA, USA). The rabbit antigoat biotinylated antibody was reacted with an avidin‐HRP conjugate. The HRP chromagen was developed using diaminobenzidine (DAB) substrate (Dako). All sections except that of the HSV‐1 were counterstained with Mayer's hematoxylin. The HSV‐1 sections were counterstained with Papanicolaou stain.
Characterization of lesions in the nervous system
For the detection of inflammatory and degenerative lesions in the brain, two observers were used and each slide was blinded during assessment. The analysis of lesion distribution and characterization were performed by examination of 10 different coronal brain sections per mouse taken from brain regions relative to bregma at approximately +2.96, +1.46, +1.18, −1.46, −3.3, −3.08, −3.80, −5.02, −5.40, and −6.48 mm (22). At each neuroanatomical location, a semi‐quantitative scale was used to grade the lesion based on both inflammatory and degenerative changes: (−), no lesions; (+), minimal lesions; +, mild lesions; ++, moderate lesions; +++, severe lesions. In addition, qualitative assessment for lesion distribution (focal, multifocal, extensive, or diffuse), inflammation, degeneration, and viral inclusion bodies for each location was recorded. Inflammatory cells were identified by HE staining as lymphocytes, neutrophils, macrophages, and plasma cells. Identities of immune cells were confirmed by immunohistochemistry using specific monoclonal or polyclonal antibodies to CD3 (T lymphocytes), MPO (neutrophils), and Mac‐2 (macrophages). In addition, apoptotic cells were identified using the monoclonal antibody to caspase‐3.
The distribution of HSV‐1 antigen was assessed throughout the brain on at least 10 different brain coronal sections and identifying unequivocal immunopositive cells. Comparable uninfected brain sections were used as staining controls.
Fluorescence‐activated cell sorter (FACS) sorting and cytokine analysis of brain leukocytes
Leukocytes were isolated from HSV‐1‐infected brains using a previously described procedure 21, 33. Briefly, single cell preparations from infected or uninfected age‐matched controls brains were resuspended in 30% Percoll and banded on a 70% Percoll cushion at 900 Xg at 15°C. Brain leukocytes obtained from the 30% to 70% Percoll interface were treated with Fc Block (anti‐CD32/CD16, 2% normal rat and 2% normal mouse serum) to inhibit nonspecific antibody binding, and were stained with antimouse immune cell surface markers for 45 minutes at 4°C [anti‐CD45‐Allophycocyanin (APC), anti‐CD11b‐FITC (BD Biosciences, San Jose, CA, USA)], and analyzed by flow cytometry. Control isotype antibodies were used for all isotype and fluorochrome combinations to assess nonspecific antibody binding. Live leukocytes were gated using forward scatter and side scatter parameters. Non‐overlapping populations of brain leukocytes that were immunostained with antimouse CD45 and antimouse CD11b‐FITC (BD Biosciences) were separated using a FACS (BD FACSAria™, BD Biosciences). Postsort analysis indicated that the separated populations were >93% positive for their associated markers. Total RNA isolated from sorted cell populations was analyzed by quantitative real‐time reverse transcription polymerase chain reaction (RT‐PCR) for interferon (IFN)‐γ and tumor necrosis factor (TNF)‐α expression.
Morris water maze
The water maze was constructed in a circular trough (4 ft. in diameter) with a translucent removable escape platform placed in the center of the maze. The four coordinates of the maze were marked with distinct black and white patterns, providing the spatial cues distal to the platform. The trough was filled with water kept at 24 ± 1°C rendered opaque with white nontoxic paint. At each trial, the animal was placed in the maze at the same coordinate and given 90 s to find the hidden platform. At the end of 90 s, animals were placed on the platform for 15 s to allow them to process the distal cues. Animals were allowed to remain on the platform for 15 s if they found the platform before the 90 s time allotted. The time taken to find the platform was recorded for four trials per day for 12 consecutive days. An intertrial period of 15 minutes was given to each animal when they were placed in their holding cage under a heat lamp. After all the trials were completed, animals were administered a visual probe test, where the escape platform was raised and a visual cue placed on the platform. Animals that did not find the visible platform in the maze in at least two consecutive trials (out of four) were excluded from the analysis.
RESULTS
Infected animals began to show signs of infection at 5–6 days p.i. The predominant clinical signs were dehydration, dermatitis, kerato‐conjunctivitis, tremors, excitability, hunched posture, disoriented or high stepping gait, followed by either stupor and death (between 7 and 12 days p.i.) in animals showing advanced neurological signs (like seizures or paralysis) or recovery in ∼60% of the animals. Animals presenting with neurological signs died within 6–24 h after onset or were euthanized when neurological manifestations were severe. Postmortem examination showed no systemic lesions related to HSV‐1 infection. Hence, cause of death was most likely due to HSV‐1‐induced encephalitis. Animals that survived viral infection at 14 days p.i. were apparently normal, demonstrating normal feeding and grooming behaviors.
Virus distribution
Distribution of virus in the acute phase
HSV‐1 immunopositive cells were detected in infected mouse brains as early as 2 days p.i. Numerous HSV‐1 immunopositive cells were observed in both the glomerular and granular layer of the olfactory bulb at 2 and 5 days p.i. At 5 days p.i., the infection extended into the median eminence, medial parvicellular area of the paraventricular hypothalamic nuclei, supraoptic nucleus, medial amygdaloid nucleus, and within the medulla oblongata in the trigeminal nerve root (Table 1).
Table 1.
Progression of HSV‐1 infection and degenerative lesions in the brain. Abbreviations: HSV‐1 = herpes simplex virus 1; Ob = olfactory bulb; Pir = piriform cortex; Ent = entorhinal cortex; Th = thalamus; C = Cerebellum; Hi = hippocampus; fi = fimbria of the hippocampus; Sol = solitary nucleus; ME = median eminence; Sp5 = trigeminal nucleus; 7N = facial nucleus; 10N = vagus nucleus; 12N = hypoglossal nucleus; sp5 = spinal trigeminal tract; 7n = facial nerve; 1n = Olfactory nerve.
| Animals no. | Viral antigen | Cortical necrosis | Focal noncortical necrosis | Cortical cell loss and atrophy | Focal non‐cortical cell loss | Axonal degeneration | Demyelination | Astrogilosis |
|---|---|---|---|---|---|---|---|---|
| 2 days (n = 2) | + f † Ob | ++ f Ob | − | − | − | − | Myelin swelling | − |
| 5 days (n = 2) | + f Ob, Th, ME, Pir, Sp5, | ++ f Ob | − | − | − | − | Myelin swelling | + d |
| 7 days (n = 4) | + /+++f Ob, Pir, Ent, Hi, Th, Sp5, 7N, Sol, 10N | +++ Ob, Pir, Hi, Ent | +/+++ mf Th, 5N,7N, NSol, Xn, XIIN | − | − | lysis | +/++ mf fi, 1n, 5sp, 7n | ++f / +++d edema |
| 7–9 days‡ (n = 10) | +++ mf Ob, Pir, Ent, Hi, Th, Sp5, 7N, Sol, 10N | +/ +++ Ob, Pir, Ent, C | +++ mf Th, 5N,7N, NSol, Xn, 12N | − | − | lysis | +/++ mf fi, 1n, 5sp, 7n | +++ d edema |
| 14 days (n = 3) | (+)f Ob, ME | − | − | − | +/++ mf Th, 5N,7N, NSol, 10n, 12N | + mf fi, 1n, 5sp, 7n | ++/+++ mf fi, 1n, 5sp, 7n | +/++ mf Edema |
| 30 days (n = 3) | − | − | − | +++ Pir, Ent | +/++ mf Th, H, 5N,7N, NSot, 10n, 12N | + mf fi, 1n, 5sp, 7n | +/++ mf fi, 1n, 5sp, 7n | + /++ mf–ext, hypertrophy |
| 60 days (n = 4) | − | − | − | +++ Pir, Ent, Hi | +/++ mf Th, H, 5N,7N, NSot, 10n, 12N | − | (+) | + /+++ mf–ext, hypertrophy |
− = no lesions; (+) = minimal lesions; + = mild lesions; ++ = moderate lesions; +++ = severe lesions; f = focal lesions; mf = multifocal lesions; ext = extensive lesions; d = diffuse lesions.
Lethal infection.
A marked increase in viral antigen positive neuronal cells was observed by 7 days p.i. in all the animals tested compared with earlier time points, although the extent of virus distribution varied among the individual animals. HSV‐1 antigen positive neurons were observed in the optic tract, thalamus, trigeminal and facial nerve tracts, and their corresponding nuclei in 75% (3 of 4) of animals. HSV‐1 antigen was also expressed in the olfactory bulb, temporal cortex, hippocampus, trigeminal, solitary, and vagus nuclei (Table 1). In most animals, small numbers of inclusion bodies were visible in all areas showing HSV‐1 immunostaining with multifocal distribution within the olfactory bulb, piriform cortex, temporal cortex, amylagdoid nuclei, thalamus, hypothalamic nuclei, lateral hypothalamus, ventromedial thalamic nuclei, and trigeminal, vagal, and solitary nuclei.
Among surviving animals at 14 days p.i., there was a significant reduction in the number of HSV‐1 immunopositive cells. One animal in this group had no detectable viral antigens, while the others exhibited scattered HSV‐1 immunopositive neurons in the granular layer of the olfactory bulb and medial eminence. At 30 days p.i., HSV‐1 antigen staining was absent from all the brain regions affected during the acute phase. However, rare occurrences of HSV‐1 immunopositive cells were observed (1–3 among all brain sections) within the piriform cortex at both 30 and 60 days p.i. in non‐neural cells that could not be identified.
Progression and topography of degenerative lesions and inflammation ensuing HSE
Acute phase of infection (2–7 days p.i.)
At the earliest time point tested, 2 days p.i., the only detectable lesion in the brain was a small focal area of cells with eosinophilic cytoplasm, pyknosis, and karyorrhexis (necrosis) seen in one of the animals in the periglomerular and mitral cells of the olfactory bulb (Table 1), and associated small numbers of neutrophils in the olfactory bulb and trigeminal nerve root (Figure 1). In addition, scattered lymphocytes and neutrophils were observed within the leptomeninges and focal areas of mild myelin swelling in the median eminence. At 5 days p.i., all animals in the group demonstrated multiple focal areas of necrosis affecting the periglomerular and mitral cells of the olfactory bulb (Table 1). In addition, myelin swelling associated with scattered lymphocytes and macrophages was seen within both the trigeminal and facial tracts in the medulla oblongata. Subtle signs of myelin swelling were also present in the optic tract (Figure 1). Small numbers of neutrophils were also observed with necrosis of the olfactory bulb and trigeminal nerve root.
Figure 1.
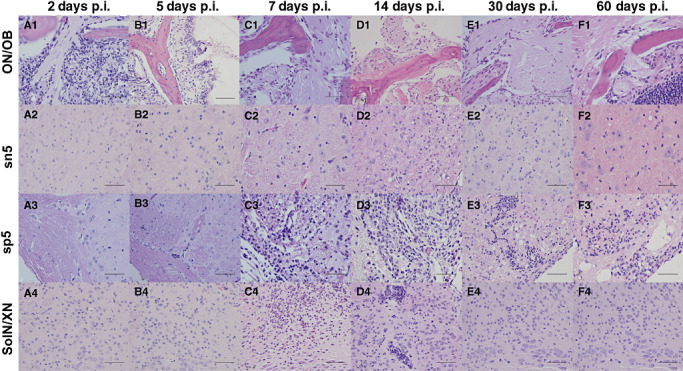
Progression of inflammatory lesions in the brain associated with the olfactory and trigeminal nerve tracts. Representative photomicrographs of hematoxylin/eosin (HE)‐stained coronal brain sections from infected animals at 2, 5, 7, 14, 30, and 60 days p.i. Specific areas of the brain are shown, including the first cranial (olfactory) nerve/olfactory bulb [(ON)/OB; A1–F1], sensory nucleus of the trigeminal nerve (sn5; B2–F2), spinal root of the trigeminal nerve (sp5, A3–F3), and the solitary and vagus nuclei (SolN/XN; A1–A4; bar = 50 µm). The progression of inflammation in the various brains stem regions and their associated nerves demonstrating a predominant neutrophil infiltrate up to 7 days p.i., which changed to a lymphocytic and plasmacytic infiltrate at 14 days p.i.
Majority of animals at 7 days p.i. demonstrated areas of necrosis primarily within the cranial nerve tracts and nuclei (Figure 1). This necrosis was accompanied by inflammatory cells, composed predominantly of neutrophils with few lymphocytes and macrophages. These cells were immunopositive for MPO, CD3, and Mac‐2, respectively. There were a small number of Caspase‐3 immunopositive cells observed primarily within the trigeminal nucleus. Multifocal areas of the neuropile showed edema particularly within the periventricular areas throughout the brain. Neuropile edema was generally mild within the medulla oblongata, but more severe and focal around the trigeminal, facial, and optic tracts, and was present multifocally throughout the cortex. In addition to increased HSV‐1 antigen staining in the brain, one animal (no. 1826) showed extensive focal neuronal necrosis within the piriform and temporal cortices (Figure 2), as well as in the vagus and solitary nuclei (Figure 3). Large numbers of MPO (neutrophils), Mac‐2 (macrophages), and Caspase‐3 immunopositive cells were also detected in the olfactory bulb, vagus, and solitary nuclei of the brain stem of this animal (Figure 3). Interestingly, MPO positive cells, when present, were exclusively localized to the olfactory bulb and brain stem nuclei in all animals.
Figure 2.
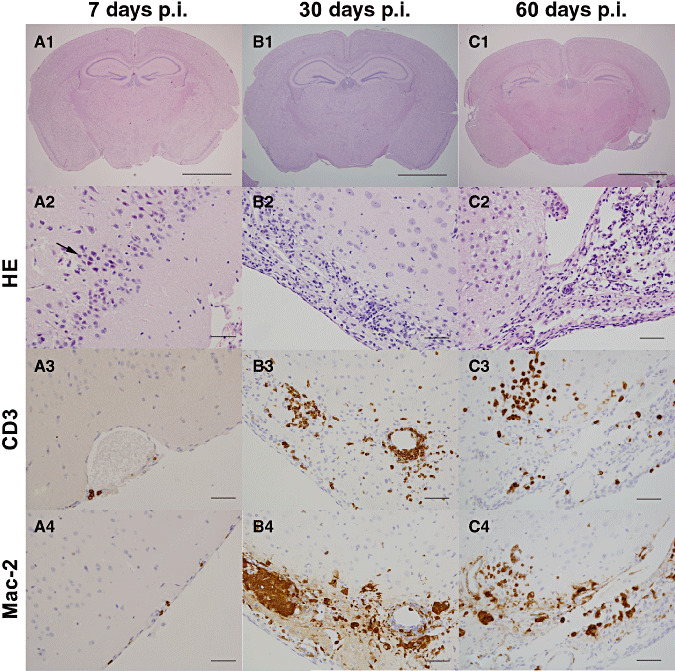
Progression of inflammatory lesions in the Piriform cortex. Photomicrographs of representative hematoxylin/eosin (HE)‐stained coronal brain sections at 1.82 mm caudal to the Bregma. A1, B1, and C1 are shown as low magnification at 7, 30, and 60 days p.i., respectively, from which descriptive lesions are presented in the subsequent figures (bar = 2 mm). (A2–A4) During the initial stages of acute infection (7 days p.i.), signs of severe degeneration and ultimately necrosis of infected neurons were observed (A2; arrow). Scattered CD3 (A3) and Mac‐2 (A4) positive cells were present in the leptomeninges (bar = 50 µm). (B2–B4) During the chronic inflammatory phase, 30 days p.i., the piriform cortex exhibited severe atrophy with a marked loss of neurons and neuropile, which is replaced by inflammatory infiltrate (B2). The infiltrate was composed of moderate numbers of CD‐3 (B3) and Mac‐2 (B4) expressing cells (lymphocytes and macrophages, respectively; bar = 50 µm). (C2–C4) At 60 days p.i., the piriform cortex and amygdaloid nuclei showed extensive loss of neural tissue with cavitations (C2). The inflammatory infiltrates that demarcated the lesion (C2) were composed of large numbers of CD‐3 (C3) and Mac‐2 (C4) expressing cells (lymphocytes and macrophages, respectively; bar = 50 µm).
Figure 3.
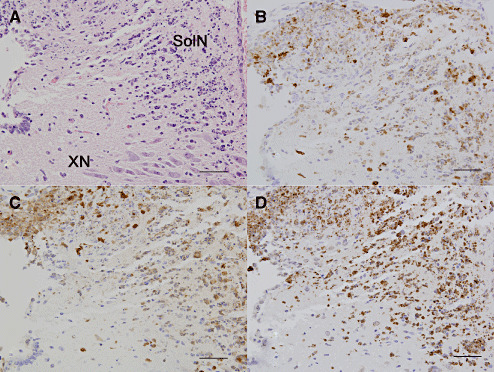
Inflammatory changes in the brain stem during herpes encephalitis. During the acute infection (7 days p.i.), inflammatory infiltration within the solitary nuclei (SolN) and vagus nucleus (XN) were composed of mostly neutrophils, and demonstrated abundant nuclear debris, cell lysis, and neurons with intranuclear viral inclusion bodies. Coronal section at 7.20 mm caudal to the bregma stained with (A) hematoxylin/eosin (HE) or immunostained for (B) myeloperoxidase, (C) Mac‐2, and (D) caspase 3 demonstrated the presence of neutrophils, activated macrophages, and apoptotic leukocytes in this region (bar = 50 µm).
Lesions in the animals that succumbed to HSV‐1 infection (7–10 days p.i.) demarcate the extent to which brain regions are affected by HSV‐1 encephalitis (Table 1). Degenerative lesions in lethally infected animals were characterized by cellular changes that extended from the olfactory bulb up to the cervical spinal cord, affecting all functional neural systems associated with the olfactory, trigeminal, facial, solitary, vagal, and hypoglossal nerves (Table 1). Multifocal necroses of cortical areas varying from mild to moderate degrees of severity were observed in the piriform, entorhinal, and occipital cortices, in the thalamus, and occasionally in the cerebellum (Table 1). Glial cell necrosis and myelin degeneration were observed within the fimbria of the hippocampus and lateral tegmental nucleus. (Table 1) Inflammatory lesions were characterized by severe, focal, and mixed cellular infiltrates, including small numbers of diffusely scattered lymphocytes in the cortex and midbrain, all of which immunostained for CD3. Throughout the affected cortical regions were moderate numbers of macrophages that frequently appeared foamy and vacuolated, indicating active phagocytosis of myelin and immunostained for Mac‐2. In addition, extensive Mac‐2 immunopositive cells were observed between the ependymal cells and periventricular area.
Similar to lesions in the cerebrum and midbrain, the medulla had small numbers of diffusely distributed lymphocytes. The most extensive and intense Mac‐2 immunostaining was seen in the medulla, uniformly distributed across 80% of this brain region, with discrete regions of focally intense Mac‐2 immunostaining seen within the trigeminal nerve tract and nucleus, as well as the facial nucleus. This activation of macrophages was also associated with an extensive array of necrosis and myelin swelling or axonal degeneration, characterized by scattered spheroids (axonal degeneration) within the trigeminal and facial nerves roots.
MPO immunopositive, neutrophils were seen scattered in focal areas within the periventricular region and trigeminal motor nucleus. The number and location of MPO positive cells correlated not only with the observation of neutrophils in HE sections, but also caspase‐3 immunopositive cells, indicating that neutrophils were likely undergoing apoptosis in the infected brain.
Astrogliosis during the early acute phase was relatively mild with small numbers of reactive astrocytes spread diffusely through the midbrain and medulla, indicated by their distinct appearance in both HE sections and GFAP immunostaining. In severely necrotic areas, astrocytes were swollen with loss of GFAP staining, astrocyte/oligodendrocyte nuclei were replaced by karyorrhextic figures, and the brain areas surrounding these necrotic lesions showed intense GFAP staining.
Kinetics and topography of chronic lesions (14–60 days p.i.)
Day 14
Degeneration and necrosis in cortical and noncortical regions of the brain in the group that survived infection at 14 days p.i. were not as evident as those observed in animals that succumbed to infection (7–9 days p.i.). However, there was an obvious decrease in neuron density, potentially caused by neuronal loss, in the trigeminal and facial nerve tracts, solitary, vagus and hypoglossal nuclei, hippocampus, and the periaquaductal gray matter (Table 1, Figure 2). Axonal degeneration was seen in the form of swelling or lysis and extensive secondary demyelination was observed within the nerve root and tracts of the trigeminal and facial nerve. Mild to moderate secondary demyelination was also seen within the fimbria of the hippocampus (Table 1).
A small number of CD3 immunopositive lymphocytes were located within the olfactory bulb, multiple cortical regions (piriform and temporal cortex, amygdaloid nuclei, and hippocampus), and thalamus. Moderate numbers of lymphocytes (CD3+ cells), plasma cells, and macrophages (Mac‐2+ cells) were observed within most of the cranial nerve nuclei and nerve roots in the brainstem, which include facial nerve root, solitary nuclei, vagus nuclei, locus coeruleus, hypoglossal nucleus, superior vestibular nucleus, trigeminal nucleus, and lateral paragigantocellular nucleus (Figure 4).
Figure 4.
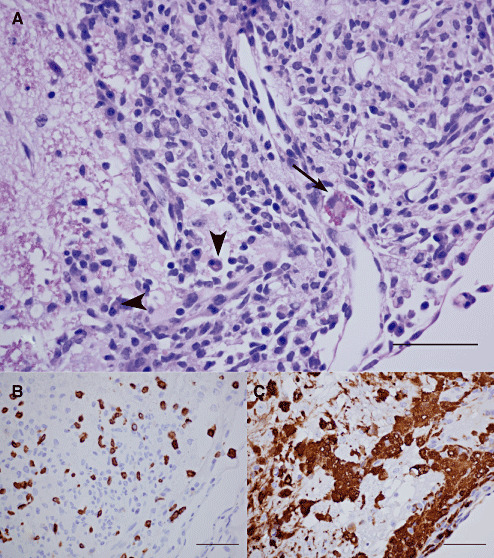
Inflammatory changes in the brain stem (14 days p.i.). Changes in the brain stem at 14 days p.i. are shown in a representative coronal section at 5.52 mm caudal to the Bregma, demonstrating a severe subchronic inflammatory response in the spinal trigeminal tract/facial nerve root. (A) A hematoxylin/eosin (HE)‐stained preparation demonstrating the presence of moderate numbers of plasma cells (arrow head) and Russell bodies (arrow), (B) moderate numbers CD‐3 lymphocytes, and (C) large numbers of Mac‐2 positive phagocytes (bar = 50 µm).
Most of the brain regions affected in the animals examined during subchronic infection developed moderate focal to extensive astrogliosis. Moderate astrogliosis was observed, with GFAP immunostaining, within the olfactory bulb, piriform, temporal and parietal cortex, hippocampus, amygdaloid nuclei, hypothalamus, optic tract, and periaquaductal mecencephalic gray matter (Figure 5). Extensive astrogliosis was seen in the brain stem (Figure 5). Astrogliosis at this time point was characterized by the presence of moderate numbers of astrocytes with elongated and robust, strong GFAP inmunopositive cell processes (hypertrophic or reactive astrocyte). Within the medulla, reactive astroglia were evident in the spinal trigeminal tract, principal and sensory trigeminal nuclei, as well as in the periventricular areas.
Figure 5.
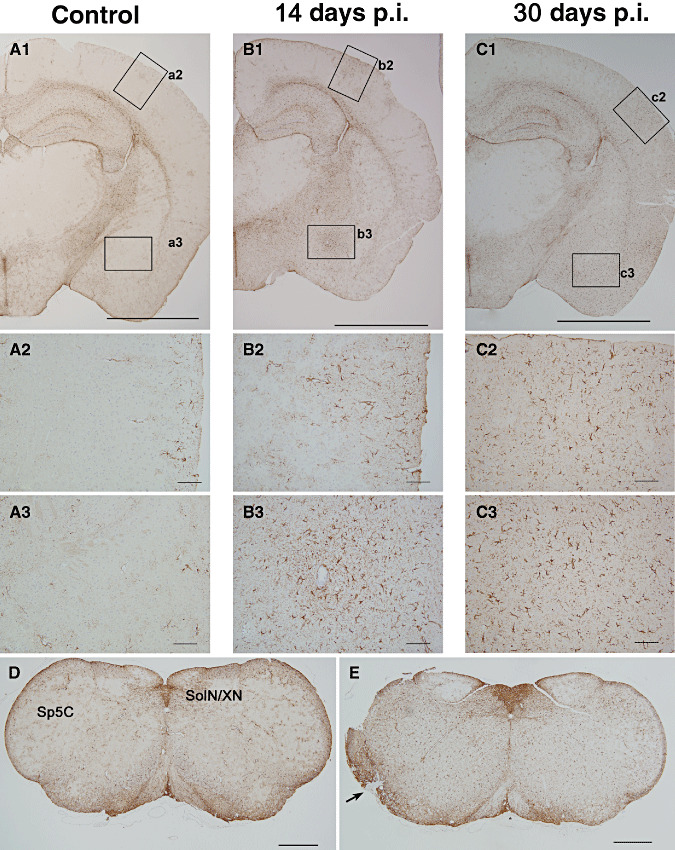
Progressive astrogliosis and glial scars formation in the inflamed cortex. Photomicrographs of cortical brain sections, 1.82 mm caudal to the Bregma, obtained from infected animals at various days p.i. demonstrating the progressive astroglial activation in the cerebral cortex with no parenchymal loss and scar formation in the brainstem consequent to herpes simplex virus 1 (HSV‐1) infection. Low magnification sections immunostained for glial fibrillary acid protein (GFAP) expression are presented from (A1) uninfected animals and (A2–A3) corresponding high magnification micrographs denoting normal structure and numbers of GFAP positive cells in the uninfected brain. Similar low magnification coronal sections taken at (B1) 14 days p.i. and (C1) 30 days p.i. showed an chronic increase in GFAP signal across many brain structures (bar = 2 mm). (B2–B3) At 14 days p.i., magnification of the parietal cortex (B2) and amygdaloid nuclei (B3) showed a locally extensive astrocyte reaction (bar = 50 µm), particularly in the amygdaloid nuclei, astrogliosis was located around areas of lymphocytic perivasculitis. (C2–C3) Astrogliosis at 30 days p.i. (bar = 2 mm) was observed as hypertrophic astrocytes (C2) possessing prominent cell processes and strong GFAP‐expression indicative of glial scar formation (C3, bar = 50 µm). (D–E) Similar astrogliosis was observed in the brain stem regions. Brain cross sections at 7.76 mm caudal to the Bregma from uninfected animals (D) compared with infected brains at 30 days p.i. (E) showed a diffuse astrogliosis in the infected brain stem. Severe astrogliosis was observed in the caudal part of the spinal trigeminal nucleus and glial scar formation (arrow) in the spinal trigeminal track (bar = 0.5 mm).
Day 30
Total brain volume was grossly reduced at 30 days p.i., reflecting a 20% loss in gray matter compared with uninfected age‐matched controls. Focal loss of neurons was also observed in all animals examined, particularly evident in the hippocampus. Profound atrophy of the piriform and entorhinal cortices and amygdaloid nuclei was seen in at least one out of three (33%) animals analyzed at this time point (Table 1; Figure 2).
Characteristics of inflammatory lesions were also altered at this time point. Plasma cells were observed in all animals examined at this time point. Lymphocytes and macrophages were also observed in all affected cortical regions (Table 1). In addition, mild to moderate demyelination, neuronal loss, and moderate lymphoplasmacytic infiltration were observed within the trigeminal, facial, solitary, vagal, and hypoglossal tracts and nuclei (Table 1; Figure 1).
Previous studies from our laboratory have reported that expression of interferon gamma (IFN‐γ) persisted into the chronic phase of HSV‐1 infection (33). In the present study, we extended the analysis to investigate the source of inflammatory cytokines during the chronic phase. Using FACS, distinct immunophenotypic populations (Figure 6A), which represent lymphocytes (CD45hiCD11b‐) and macrophages (CD45hiCD11b+ or CD45intCD11b+), were isolated. Analysis of the individual FACS‐sorted populations for IFN‐γ, and TNF‐α mRNA expression showed that IFN‐γ expression among lymphocytes (CD45hiCD11b‐) peaked at 15 days p.i. and was sustained up to 30 days p.i. at levels that were threefold higher than uninfected animals (Figure 6B). Additionally, TNF‐α expression, which was detected both in the CD11b+ and CD11b‐ populations at 8 days p.i. (33), was also observed at 30 days p.i.
Figure 6.
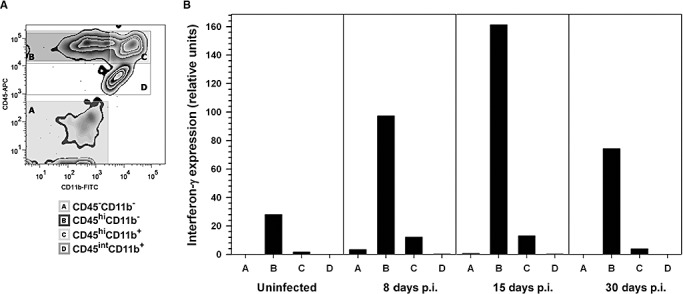
Interferon gamma expression by brain‐infiltrating leukocytes. Single cell suspensions of brain tissue obtained from herpes simplex virus 1 (HSV‐1)‐infected mice (five animals, 30 days p.i.) were layered on a 70% Percoll cushion. (A) Brain leukocytes banded at the 30%–70% Percoll interface were collected, labeled with mouse antibodies specific for CD45 and CD11b, and sorted into four populations using fluorescence‐activated cell sorter (FACS): A‐ CD45‐CD11b‐ non‐myeloid cells, B‐ CD45hiCD11b‐ predominantly lymphocytes, C‐ CD45hiCD11b+ are infiltrating macrophages or neutrophils, and D‐CD45intCD11b+, microglial cells. Post‐sort analysis indicated that the separated populations were >93% positive for their associated surface markers. (B) Total RNA extracted from each of the separated populations (A through D) at 30 days p.i. was analyzed by real‐time reverse transcription polymerase chain reaction (RT‐PCR) for interferon (IFN)‐γ expression. Relative transcript levels in cells isolated from brain tissues of five animals normalized to the housekeeping gene, HPRT, expression are presented.
The magnitude and presentation of astrogliosis increased dramatically at 30 days p.i. (Figure 5). Mild to moderate and focal to extensive astrogliosis, characterized by large astrocytes possessing elongated, robust, interlacing cell processes, was seen within the cortex, basal nuclei, thalamus, mecencephalon, brainstem, and cerebellum. In areas of atrophy within the piriform cortex, reactive astrocytes formed a dense intricate network of GFAP‐rich processes with a concomitant distinct lack of neurons in the neuropile (glia scar) surrounded by inflammatory cells. The optic tract, hypothalamus, amygdaloid nuclei, and hippocampus had markedly higher numbers of reactive astrocytes. Reactive astrogliosis within the medulla oblongata was increasingly severe at 30 days p.i. than all previous time points tested. Moderate astrogliosis was also observed within the trigeminal nerve tract, sensory and motor nuclei, as well as within the cerebellar peduncles and nuclei (Figure 5). Interestingly, cranial nerves fibers showed evidence of remyelination and Schwann cell proliferation at 30 days p.i.
Day 60
The HSV‐1 infected brain at 60 days p.i. was also visibly reduced in size. Degenerative lesions at 60 days p.i. were similar to those seen at 30 days p.i. However, in most affected areas of the brain stem, the nature and degree of inflammatory infiltrates were less obvious compared with those seen at 30 days p.i. (Figure 1). In one of the animals tested, loss of approximately 40% the temporal–occipital cortex was observed and was marked with cavitations and cortical atrophy (Figure 7). This animal also showed mild diffuse lymphocytic infiltration in the temporal cortex, basal nuclei, and thalamus (1, 2). In addition, lymphocyte and macrophage infiltration was moderate to severe in areas showing loss of neuropile, particularly the temporal cortex and hippocampus. Specifically, neuronal loss was evident in the pyramidal layer of the hippocampus (Figure 7).
Figure 7.
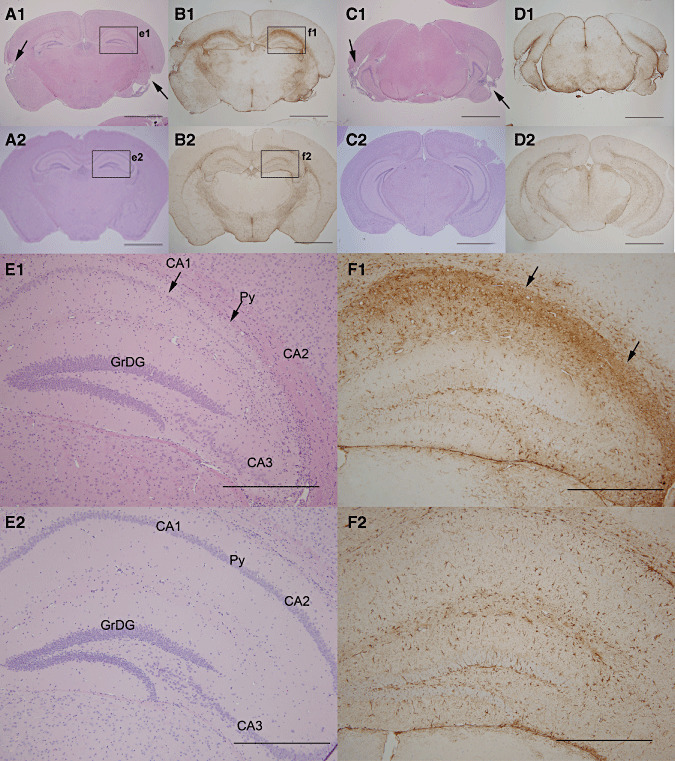
Chronic cortical and hippocampal lesions at 60 days p.i. Low magnification photomicrographs presenting the brain of a mouse showing severe atrophy and cavitations of cortical structures at 60 days p.i. (A1) Hematoxylin/eosin (HE)‐stained coronal section, 1.82 mm caudal to Bregma, from infected and (A2) uninfected age‐matched animal, demonstrating bilateral chronic temporal cortical atrophy with cavitations (arrows). (B1) Adjacent sections (–1.82 mm) stained with GFAP shown a diffuse severe astrogliosis and hypertrophic astrocytes. Notice the intense and extensive GFAP‐expression demonstrating hypertrophic astrocytes in a glial scar formation (B2) compared with the same region in uninfected animals. (C1) Similar HE‐stained coronal sections at 3.16 mm caudal to bregma show cavitations and atrophy of the entorhinal cortex (arrows) compared with (C2) uninfected age‐matched controls. (D1) Adjacent sections stained with GFAP showed a diffuse severe astrogliosis compared (D2) with similar region in uninfected brains (Bar = 2.0 mm). (E1) High power examination of the HE stained coronal brain section, presented in A1, exhibited severe loss of neurons of the CA1, CA2, and CA3 regions of hippocampus compared with (E2) age‐matched, uninfected control controls (bar = 0.5 mm). (F1) This loss of neuronal structure in the hippocampus was associated with severe astrogliosis, characterized by a strong expression of GFAP in concurring CA areas compared with (F2) similar regions in uninfected, age‐matched control brains. (Bar = 0.5 mm). Py = pyramidal neurons, GrDG = granular cell layer of the dentate gyrus.
Cranial nerves fibers showed evidence of moderate lymphoplasmacytic infiltration in conjunction with signs of remyelination and Schwann cell proliferation. The remyelination process, which was characterized by a compact and thin myelin sheath around axons, observed using Luxol fast blue stain, extended to the trigeminal, facial, vagal, solitary, and hypoglossal nuclei (shown in HE stained sections, Figure 1). Scattered neutrophils were also detected in cranial nerve nuclei within the medulla oblongata.
In general, astrogliosis in animals at 30–60 days p.i. appeared to be stronger than those observed during the acute (7–9 days p.i.) and subchronic (14 days p.i.) stages of HSV‐1 encephalitis. Furthermore, astrogliosis at 30 and 60 days p.i. was predominantly localized to the regions affected by HSV‐1‐induced damage and where persistent inflammatory lesions were observed (Table 1, Figure 7).
Long‐term memory deficit consequent to experimental herpes encephalitis
Based on chronic neuropathologic changes seen ensuing HSV‐1 brain infection, we set out to determine if the surviving animals at 30 days p.i. would manifest behavioral defects. Infected animals and age‐matched controls were tested using a Morris water maze task designed to measure spatial memory function. Animals were tested for their ability to learn the location of a hidden platform based visual cues in the maze over a 12 days period. Uninfected animals (n = 6) exhibited a steady decrease in average latency to find a hidden platform in the water maze, decreasing from 78.8 ± 4.6 s on the first day to 13.5 ± 1.8 s at day 12 (Figure 8). However, all HSV‐1 infected animals (n = 10) performed poorly on the water maze task, indicating a severe defect in spatial memory, demonstrated by unchanged latency values (69.5 ± 3.7 s) after 12 days of testing (Figure 8). The differences in latency were statistically significant by Student's t‐test (P < 0.001). Animals were then tested under conditions where the platform was visible. Among the infected mice, 4 out of 14 were not able to find the visible platform in two out of the four consecutive trials and were excluded from the analysis. Under these conditions, both infected and non‐infected animals were able to locate a visible platform with similar latency (47.9 ± 6.1 s and 42.8 ± 6.9 s, respectively; P = 0.5) indicating that deficits exhibited by the infected animals in locating the submerged platform was not caused by visual dysfunction.
Figure 8.
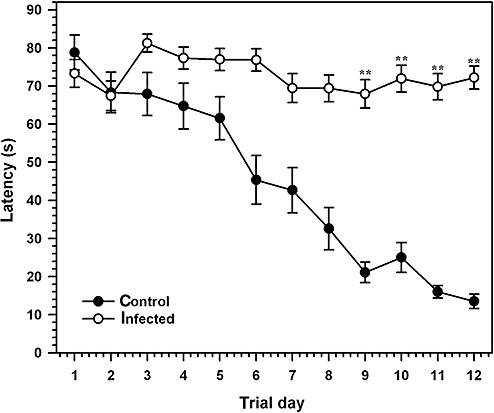
Herpes simplex encephalitis (HSE) results in a spatial memory defect on a Morris maze task. Performance of herpes simplex virus 1 (HSV‐1) ‐infected mice at 30 days p.i. and age‐matched uninfected controls were tested on a Morris water maze designed to measure spatial memory formation. Average latency associated with finding an invisible platform based on visual cues was measured daily for 12 days, with each animal receiving 4 trials/days from the same quadrant of the maze. Uninfected Balb/c (open circles) mice demonstrated lower latency rates than HSV‐1‐infected animals, indicating a severe spatial memory defect. A visual probe test was used to eliminate animals that could not find a visible platform after 4 trials. Data presented are mean latency (±Standard error of mean (SEM)) from six uninfected animals and 10 HSV‐1 infected animals with no visual deficits (**P < 0.001).
DISCUSSION
Following intranasal inoculation of Balb/c mice, HSV‐1 reaches the CNS via the olfactory and trigeminal routes. Viral antigens were demonstrable in the brain between 2–14 days p.i., but were rarely detected at later times during infection. Once the virus reaches the CNS, the progression through different associated routes were detected as focal lesions, localized along neural circuits that can be traced back to their source at the olfactory (the piriform cortex, entorhinal cortex, thalamus, amygdaloid nucleus, and fimbria of the hippocampus) and trigeminal nerve endings (trigeminal, vagal, and solitary nucleus and respective nerve roots). Similar observations of brain infection have been described by several groups in separate studies and using different viral strains in susceptible mice during the acute phase of infection 1, 20, 36, 39, 52, 55, 58. In the present study, a systematic analysis of HSV‐1‐induced neuropathology demonstrated that HSV‐1‐infected animals surviving the acute phase of infection exhibited sustained inflammation, neuronal loss, and neuropathological sequelae (>60 days p.i.). Infected animals demonstrated a severe spatial memory deficit, which was consistent with the functions associated with inflammatory lesions in the piriform and entorhinal cortical regions 17, 50. This model of experimental herpes encephalitis, as defined by the present study, now broadens the scope for investigating mechanisms of long‐term neurological sequelae ensuing HSV‐1 brain infection.
The presence of viral antigens in the olfactory bulb and associated brain regions generally preceded CNS infection via the trigeminal route. Infection in the brain stem was not fully evident until 7 days p.i. The ability of HSV‐1 to utilize the olfactory nerve as a conduit to infect the brain is relevant in that virus‐induced damage from the olfactory nerve tracks progresses along neuronal pathways to the piriform cortex, thalamus, medial amygdaloid nucleus, and medial hypothalamic nuclei, brain regions involved in formation of memory 12, 17. Studies using the KH strain of HSV‐1 demonstrated that this virus preferentially infects the vomeronasal chemosensory system, which progresses to limbic and cortical structures. On the other hand, HSV‐2 preferentially precipitated brainstem encephalitis (8). Interestingly, the ability of HSV‐1 to invade the CNS effectively is also associated with inhibition of apoptosis by the virus (40). In the present study, limited Caspase‐3 staining was observed in infected brain regions, corroborating the possibility that brain damage may be influenced by mechanisms other than virus‐induced apoptosis. It is possible that infection via the olfactory receptors may influence the neurological outcomes of HSE. Among the various models of experimental HSE, this intranasal infection most accurately reflects the histopathological patterning of HSE in humans 27, 36.
Approximately half of the infected animals succumbed between 7 and 12 days p.i., as previously described (33). We propose two possible mechanisms for mortality and the neuropathology associated with HSV‐1 brain infection. First, the primary mechanism may be associated with damage to critical brain areas controlling vital physiologic functions, viz. the vagus and solitary nuclei and the thalamus, resulting from lytic viral replication in the affected neurons. Second, bystander damage associated with neuroinflammation resulting in additive tissue damage, which may also contribute to both mortality and neuropathological sequelae. Although it is difficult to experimentally separate these two processes, recent studies have shown that the early inflammatory response, composed largely of macrophages and neutrophils, induced widespread damage in the brainstem, which resulted in HSE‐induced mortality (32). However, abrogating either the immune response or viral replication did not completely reverse mortality rates in these studies (32). Additional studies from our laboratory have demonstrated that transfer of activated immune cells escalates the mortality rate due to intranasal HSV‐1 infection (33), suggesting that inflammatory responses, as well as direct viral destruction of the brain, may contribute to HSV‐1 induced disease.
Previous studies from our laboratory have shown that neutrophil infiltration into the CNS occurs as early as 5 days p.i. (33). The neuroanatomical location of neutrophils observed in the present study is particularly interesting in that these cells were located predominantly at the entry points of the infection, that is, in the olfactory bulb, optic, trigeminal, and facial nerve tracts, and their associated nuclei in the brain stem regions. They were significantly absent or sparsely populated in deeper cortical regions. Early infiltration of neutrophils has been reported to be essential for controlling virus replication 3, 53, 56 and spread of virus into the CNS (61) in a murine model of HSV retinitis. On the other hand, the accumulation of neutrophils has also been associated with corneal damage (16) and brain stem encephalitis (32). Neutrophils produce numerous factors that cause neuronal damage and exacerbate brain injury, including reactive oxygen species, metalloproteinases, and myeloperoxidase 10, 42. The observation that some of neutrophils also immunostained for Caspase 3 and showed nuclear changes consistent with apoptosis, concurs with a previous report that HSV‐1 induces apoptosis in neutrophils (18), where the resolution of corneal inflammation in mice with necrotizing HSV keratitis was associated with apoptotic neutrophils. The impact of neutrophils on HSE and the significance of their presence at viral entry sites in the CNS are currently unknown.
A switch in the nature of the inflammatory infiltrate was observed between the acute and chronic phases of infection. Previous studies in our laboratory showed that the predominant macrophage/neutrophil infiltration at 8 days p.i., changed into a largely lymphocytic infiltrate at 14 days p.i., which was sustained over a period of at least 30 days p.i. (33). Interestingly, the appearance of plasma cells was a distinct feature observed only during the chronic phase of infection in all animals. T lymphocytes and macrophages largely co‐localized in the infected (during the acute phase) or damaged brain regions (chronic phase). The presence of activated microglia appears to be sustained in damaged areas during the chronic phase of infection. These cells were intensely immunostained with Mac‐2, a marker known to be upregulated in macrophages involved in myelin phagocytosis (46). Studies from our laboratory (33) and others 13, 28, 29 have indicated that activated immune cells and cytokine production persist in infected neural tissue. In the present study, persistent lymphocytes showed sustained IFN‐γ expression at 30 days p.i. compared with uninfected controls. The role of cytokines in mediating the long‐term neurological damage observed during HSV‐1 encephalitis is currently unknown.
Contribution of inflammation to the chronic neuropathological processes during HSV encephalitis may be inferred from the data presented. Although the overall histopathological manifestations were less severe in animals that survived HSV‐1 encephalitis (14–60 days p.i.), compared with those that died or were severely ill, decreased neuronal densities in affected cortical areas were evident among all the survivors. Of particular interest is the loss of hippocampal neurons observed at 30 and 60 days p.i. Hippocampal injury is common during viral infections of the brain and has been reported consequent to picornavirus (11), HIV‐1 14, 34, Bornavirus 25, 45, and Japanese encephalitis virus (23) infections. Damage to hippocampal neurons results from either a direct consequence of viral infection of these cells and subsequent apoptosis 11, 45, or indirectly by the inflammatory milieu generated in response to brain infection 14, 23. Although some HSV‐1‐infected cells were observed in the CA1 and CA2 regions of the hippocampus, much of the infection was concentrated along the entorhinal cortex and fimbria of the hippocampus, sparing the Ammon's horn from viral infection. The presence of activated microglia and T lymphocytes in proximity to all lesioned sites and their sustained expression of pro‐inflammatory cytokines suggest that inflammatory responses may influence both neuronal loss and subsequent astrogliosis observed during the chronic phase of infection. It is well known that suppression of inflammation can abrogate neuronal loss and glial scar formation in many models of neurodegeration 26, 31, 47. However, it is unclear what factors determine the extent of cortical lesions, which range from mild atrophic changes to severe loss of cortical tissue.
Deficits in spatial memory among surviving animals was observed in the context of chronic neuropathological changes in the entorhinal cortex, hippocampus, and an overall loss in cortical volume. Similar observations have been reported ensuing experimental encephalitis using picornavirus, where the level of hippocampal damage correlated with memory deficits in infected animals (11). Previous studies have reported that intranasal HSV‐1 infection in Lewis rats results in similar acute lesions seen in the present study, which were resolved in surviving animals 5, 6. However, these animals demonstrated a severe spatial memory deficit on an 8‐arm radial maze test where the animal uses cues to remember the sequence of locations in a maze. Infected animals were unable to retain pre‐infection performance, and were unable to learn new paradigms (6). The most common neurological impairment in patients with HSE is memory impairment, particularly short‐term memory (35). In support of these neurological deficits observed in HSV‐1 infected patients and mice, single‐unit electrophysiological recording studies in laboratory animals have demonstrated that hippocampal neurons exhibit place field properties that encode location of the animal within a defined space 7, 41, 43. Damage of these hippocampal neurons or loss of inputs from entorhinal afferents would therefore result in deficits in spatial navigation function. The present model thus provides a valuable resource for evaluating the mechanisms of memory loss following HSE.
We demonstrate using a murine model that infection of the brain with HSV‐1 results in long‐term neuropathological outcomes that culminate in neurological deficits. Numerous studies have dissected the immunological and pathological consequences of the acute phase of infection, but very few have shown chronic neurological alterations using experimental models of herpes encephalitis. Meyding‐Lamade et al reported that intranasal HSV‐1 infection in SJL/N mice produced chronic progressive MRI changes despite clinical recovery 36, 37. Both in the experimental model and in human patients surviving HSE, neuropathological changes were demonstrated corresponding to the medial temporal cortex and hippocampus 27, 37, 38, and was associated with long‐term inflammation 2, 33, 48. The current study extends these findings to demonstrate the topography, progression, and character of cortical lesions, delineate the neuroanatomical location of the inflammatory response, and demonstrate behavioral deficits associated with this pathology.
Although the findings presented here replicate lesions observed in humans surviving this devastating disease, one must note that mechanisms of viral pathogenesis may be different in mice. In particular, differences reported in viral evasion mechanisms by ICP47 between murine and human cells are noteworthy. ICP47, an immediate‐early viral gene product, actively inhibits MHC I mediated activation of CD8+ lymphocytes by retaining the transporter associated with antigen presentation (TAP) in the endoplasmic reticulum of infected cells 24, 60. Inhibition of murine TAP by HSV‐1 ICP47 is reported to be 50–100 times less efficient than human TAP activity in infected cells (54). However, ICP47 enhances neurovirulence of HSV‐1 infection in mice, in that an ICP47 null‐mutant shows decreased ability to induce neurological disease. This reduction of virulence in the ICP47 mutant is associated with the presence of functional CD8+ T cells (24), indicating additional mechanisms may confer neurovirulence in mice. It is clear that while mechanisms that inhibit MHC I priming of CD8 lymphocytes increase HSV‐1 neurovirulence in mice, the nature and context of the immune response play a significant role in the pathogenesis of HSV‐1 encephalitis in the murine model (44). The availability of a murine model of HSV‐1 infection that displays chronic neuropathology will propel investigation into the mechanisms that result in neurological sequelae, which frequently follows herpes encephalitis.
ACKNOWLEDGMENTS
We wish to thank Joseph Palmquist for excellent technical assistance. The authors also acknowledge the University of Minnesota's Veterinary Diagnostic Laboratory and the Masonic Cancer Center Comparative Pathology core for excellent histopathology, immunohistochemistry, and electron microscopy support. This work was supported in part by an Academic Health Center Faculty Development Grant (to AGA, WCL, and MC‐JC) and NIH grant R01 MH‐066703 (JL).
REFERENCES
- 1. Anderson JR, Field HJ (1983) The distribution of herpes simplex type 1 antigen in mouse central nervous system after different routes of inoculation. J Neurol Sci 60:181–195. [DOI] [PubMed] [Google Scholar]
- 2. Aurelius E, Andersson B, Forsgren M, Skoldenberg B, Strannegard O (1994) Cytokines and other markers of intrathecal immune response in patients with herpes simplex encephalitis. J Infect Dis 170:678–681. [DOI] [PubMed] [Google Scholar]
- 3. Banerjee K, Biswas PS, Kim B, Lee S, Rouse BT (2004) CXCR2‐/‐ mice show enhanced susceptibility to herpetic stromal keratitis: a role for IL‐6‐induced neovascularization. J Immunol 172:1237–1245. [DOI] [PubMed] [Google Scholar]
- 4. Baringer JR (2008) Herpes simplex infections of the nervous system. Neurol Clin 26:657–674. [DOI] [PubMed] [Google Scholar]
- 5. Beers DR, Henkel JS, Schaefer DC, Rose JW, Stroop WG (1993) Neuropathology of herpes simplex virus encephalitis in a rat seizure model. J Neuropathol Exp Neurol 52:241–252. [DOI] [PubMed] [Google Scholar]
- 6. Beers DR, Henkel JS, Kesner RP, Stroop WG (1995) Spatial recognition memory deficits without notable CNS pathology in rats following herpes simplex encephalitis. J Neurol Sci 131:119–127. [DOI] [PubMed] [Google Scholar]
- 7. Best PJ, White AM (1999) Placing hippocampal single‐unit studies in a historical context. Hippocampus 9:346–351. [DOI] [PubMed] [Google Scholar]
- 8. Boivin G, Coulombe Z, Rivest S (2002) Intranasal herpes simplex virus type 2 inoculation causes a profound thymidine kinase dependent cerebral inflammatory response in the mouse hindbrain. Eur J Neurosci 16:29–43. [DOI] [PubMed] [Google Scholar]
- 9. Booss J, Esiri MM (2003) Herpes Simplex Encephalitis. ASM Press: Washington, DC. [Google Scholar]
- 10. Breckwoldt MO, Chen JW, Stangenberg L, Aikawa E, Rodriguez E, Qiu S et al (2008) Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc Natl Acad Sci USA 105:18584–18589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Buenz EJ, Rodriguez M, Howe CL (2006) Disrupted spatial memory is a consequence of picornavirus infection. Neurobiol Dis 24:266–273. [DOI] [PubMed] [Google Scholar]
- 12. Canto CB, Wouterlood FG, Witter MP (2008) What does the anatomical organization of the entorhinal cortex tell us? Neural Plast 2008:381243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheng H, Tumpey TM, Staats HF, Van Rooijen N, Oakes JE, Lausch RN (2000) Role of macrophages in restricting herpes simplex virus type 1 growth after ocular infection. Invest Ophthalmol Vis Sci 41:1402–1409. [PubMed] [Google Scholar]
- 14. Cheng X, Mukhtar M, Acheampong EA, Srinivasan A, Rafi M, Pomerantz RJ, Parveen Z (2007) HIV‐1 Vpr potently induces programmed cell death in the CNS in vivo. DNA Cell Biol 26:116–131. [DOI] [PubMed] [Google Scholar]
- 15. Davis LE, Johnson RT (1979) An explanation for the localization of herpes simplex encephalitis? Ann Neurol 5:2–5. [DOI] [PubMed] [Google Scholar]
- 16. Divito SJ, Hendricks RL (2008) Activated inflammatory infiltrate in HSV‐1‐infected corneas without herpes stromal keratitis. Invest Ophthalmol Vis Sci 49:1488–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eichenbaum H, Lipton PA (2008) Towards a functional organization of the medial temporal lobe memory system: role of the parahippocampal and medial entorhinal cortical areas. Hippocampus 18:1314–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ennaciri J, Menezes J, Proulx F, Toledano BJ (2006) Induction of apoptosis by herpes simplex virus‐1 in neonatal, but not adult, neutrophils. Pediatr Res 59:7–12. [DOI] [PubMed] [Google Scholar]
- 19. Esiri MM (1982) Herpes simplex encephalitis. An immunohistological study of the distribution of viral antigen within the brain. J Neurol Sci 54:209–226. [DOI] [PubMed] [Google Scholar]
- 20. Esiri MM, Drummond CW, Morris CS (1995) Macrophages and microglia in HSV‐1 infected mouse brain. J Neuroimmunol 62:201–205. [DOI] [PubMed] [Google Scholar]
- 21. Ford AL, Goodsall AL, Hickey WF, Sedgwick JD (1995) Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein‐reactive CD4+ T cells compared. J Immunol 154:4309–4321. [PubMed] [Google Scholar]
- 22. Franklin KBJ, Paxinos G (2008) The Mouse Brain in Stereotaxic Coordinates, 3rd edn. Academic Press, Elsevier: Oxford. [Google Scholar]
- 23. Ghoshal A, Das S, Ghosh S, Mishra MK, Sharma V, Koli P et al (2007) Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis. Glia 55:483–496. [DOI] [PubMed] [Google Scholar]
- 24. Goldsmith K, Chen W, Johnson DC, Hendricks RL (1998) Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J Exp Med 187:341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hausmann J, Pagenstecher A, Baur K, Richter K, Rziha HJ, Staeheli P (2005) CD8 T cells require gamma interferon to clear borna disease virus from the brain and prevent immune system‐mediated neuronal damage. J Virol 79:13509–13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hirsch EC, Breidert T, Rousselet E, Hunot S, Hartmann A, Michel PP (2003) The role of glial reaction and inflammation in Parkinson's disease. Ann N Y Acad Sci 991:214–228. [DOI] [PubMed] [Google Scholar]
- 27. Kapur N, Barker S, Burrows EH, Ellison D, Brice J, Illis LS et al (1994) Herpes simplex encephalitis: long term magnetic resonance imaging and neuropsychological profile. J Neurol Neurosurg Psychiatry 57:1334–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Khanna KM, Bonneau RH, Kinchington PR, Hendricks RL (2003) Herpes simplex virus‐specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity 18:593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Khanna KM, Lepisto AJ, Decman V, Hendricks RL (2004) Immune control of herpes simplex virus during latency. Curr Opin Immunol 16:463–469. [DOI] [PubMed] [Google Scholar]
- 30. Kimberlin DW (2007) Management of HSV encephalitis in adults and neonates: diagnosis, prognosis and treatment. Herpes 14:11–16. [PubMed] [Google Scholar]
- 31. Liu J, Gong N, Huang X, Reynolds AD, Mosley RL, Gendelman HE (2009) Neuromodulatory activities of CD4+CD25+ regulatory T cells in a murine model of HIV‐1‐associated neurodegeneration. J Immunol 182:3855–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lundberg P, Ramakrishna C, Brown J, Tyszka JM, Hamamura M, Hinton DR et al (2008) The immune response to herpes simplex virus type 1 infection in susceptible mice is a major cause of central nervous system pathology resulting in fatal encephalitis. J Virol 82:7078–7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marques CP, Cheeran MC, Palmquist JM, Hu S, Urban SL, Lokensgard JR (2008) Prolonged microglial cell activation and lymphocyte infiltration following experimental herpes encephalitis. J Immunol 181:6417–6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mattson MP, Haughey NJ, Nath A (2005) Cell death in HIV dementia. Cell Death Differ 12(Suppl.1):893–904. [DOI] [PubMed] [Google Scholar]
- 35. McGrath N, Anderson NE, Croxson MC, Powell KF (1997) Herpes simplex encephalitis treated with acyclovir: diagnosis and long term outcome. J Neurol Neurosurg Psychiatry 63:321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meyding‐Lamade U, Lamade W, Kehm R, Knopf KW, Hess T, Gosztonyi G et al (1998) Herpes simplex virus encephalitis: cranial magnetic resonance imaging and neuropathology in a mouse model. Neurosci Lett 248:13–16. [DOI] [PubMed] [Google Scholar]
- 37. Meyding‐Lamade U, Lamade W, Kehm R, Oberlinner C, Fath A, Wildemann B et al (1999) Herpes simplex virus encephalitis: chronic progressive cerebral MRI changes despite good clinical recovery and low viral load—an experimental mouse study. Eur J Neurol 6:531–538. [DOI] [PubMed] [Google Scholar]
- 38. Meyding‐Lamade UK, Lamade WR, Wildemann BT, Sartor K, Hacke W (1999) Herpes simplex virus encephalitis: chronic progressive cerebral magnetic resonance imaging abnormalities in patients despite good clinical recovery. Clin Infect Dis 28:148–149. [DOI] [PubMed] [Google Scholar]
- 39. Mori I, Nishiyama Y, Yokochi T, Kimura Y (2005) Olfactory transmission of neurotropic viruses. J Neurovirol 11:129–137. [DOI] [PubMed] [Google Scholar]
- 40. Mori I, Goshima F, Watanabe D, Ito H, Koide N, Yoshida T et al (2006) Herpes simplex virus US3 protein kinase regulates virus‐induced apoptosis in olfactory and vomeronasal chemosensory neurons in vivo. Microbes Infect 8:1806–1812. [DOI] [PubMed] [Google Scholar]
- 41. Morris RG, Garrud P, Rawlins JN, O'Keefe J (1982) Place navigation impaired in rats with hippocampal lesions. Nature 297:681–683. [DOI] [PubMed] [Google Scholar]
- 42. Nguyen HX, O'Barr TJ, Anderson AJ (2007) Polymorphonuclear leukocytes promote neurotoxicity through release of matrix metalloproteinases, reactive oxygen species, and TNF‐alpha. J Neurochem 102:900–912. [DOI] [PubMed] [Google Scholar]
- 43. O'Keefe J, Nadel L, Keightley S, Kill D (1975) Fornix lesions selectively abolish place learning in the rat. Exp Neurol 48:152–166. [DOI] [PubMed] [Google Scholar]
- 44. Orr MT, Edelmann KH, Vieira J, Corey L, Raulet DH, Wilson CB (2005) Inhibition of MHC class I is a virulence factor in herpes simplex virus infection of mice. PLoS Pathog 1:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ovanesov MV, Moldovan K, Smith K, Vogel MW, Pletnikov MV (2008) Persistent Borna Disease Virus (BDV) infection activates microglia prior to a detectable loss of granule cells in the hippocampus. J Neuroinflammation 5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rotshenker S (2009) The role of Galectin‐3/MAC‐2 in the activation of the innate‐immune function of phagocytosis in microglia in injury and disease. J Mol Neurosci 39:99–103. [DOI] [PubMed] [Google Scholar]
- 47. Ryu JK, McLarnon JG (2006) Minocycline or iNOS inhibition block 3‐nitrotyrosine increases and blood‐brain barrier leakiness in amyloid beta‐peptide‐injected rat hippocampus. Exp Neurol 198:552–557. [DOI] [PubMed] [Google Scholar]
- 48. Sellner J, Dvorak F, Zhou Y, Haas J, Kehm R, Wildemann B, Meyding‐Lamade U (2005) Acute and long‐term alteration of chemokine mRNA expression after anti‐viral and anti‐inflammatory treatment in herpes simplex virus encephalitis. Neurosci Lett 374:197–202. [DOI] [PubMed] [Google Scholar]
- 49. Smith TL, Nathan BR (2002) Central nervous system infections in the immune‐competent adult. Curr Treat Options Neurol 4:323–332. [DOI] [PubMed] [Google Scholar]
- 50. Squire LR, Wixted JT, Clark RE (2007) Recognition memory and the medial temporal lobe: a new perspective. Nat Rev Neurosci 8:872–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Steiner I, Kennedy PG, Pachner AR (2007) The neurotropic herpes viruses: herpes simplex and varicella‐zoster. Lancet Neurol 6:1015–1028. [DOI] [PubMed] [Google Scholar]
- 52. Stroop WG, Rock DL, Fraser NW (1984) Localization of herpes simplex virus in the trigeminal and olfactory systems of the mouse central nervous system during acute and latent infections by in situ hybridization. Lab Invest 51:27–38. [PubMed] [Google Scholar]
- 53. Thomas J, Gangappa S, Kanangat S, Rouse BT (1997) On the essential involvement of neutrophils in the immunopathologic disease: herpetic stromal keratitis. J Immunol 158:1383–1391. [PubMed] [Google Scholar]
- 54. Tomazin R, Van Schoot NE, Goldsmith K, Jugovic P, Sempe P, Fruh K, Johnson DC (1998) Herpes simplex virus type 2 ICP47 inhibits human TAP but not mouse TAP. J Virol 72:2560–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tomlinson AH, Esiri MM (1983) Herpes simplex encephalitis. Immunohistological demonstration of spread of virus via olfactory pathways in mice. J Neurol Sci 60:473–484. [DOI] [PubMed] [Google Scholar]
- 56. Tumpey TM, Chen SH, Oakes JE, Lausch RN (1996) Neutrophil‐mediated suppression of virus replication after herpes simplex virus type 1 infection of the murine cornea. J Virol 70:898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Utley TF, Ogden JA, Gibb A, McGrath N, Anderson NE (1997) The long‐term neuropsychological outcome of herpes simplex encephalitis in a series of unselected survivors. Neuropsychiatry Neuropsychol Behav Neurol 10:180–189. [PubMed] [Google Scholar]
- 58. Webb SJ, Eglin RP, Reading M, Esiri MM (1989) Experimental murine herpes simplex encephalitis: immunohistochemical detection of virus antigens. Neuropathol Appl Neurobiol 15:165–174. [DOI] [PubMed] [Google Scholar]
- 59. Whitley RJ (2006) Herpes simplex encephalitis: adolescents and adults. Antiviral Res 71:141–148. [DOI] [PubMed] [Google Scholar]
- 60. York IA, Roop C, Andrews DW, Riddell SR, Graham FL, Johnson DC (1994) A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell 77:525–535. [DOI] [PubMed] [Google Scholar]
- 61. Zheng M, Fields MA, Liu Y, Cathcart H, Richter E, Atherton SS (2008) Neutrophils protect the retina of the injected eye from infection after anterior chamber inoculation of HSV‐1 in BALB/c mice. Invest Ophthalmol Vis Sci 49:4018–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]