

Abstract
Cystic kidney disease represents a major cause of end-stage renal disease, yet the molecular mechanisms of pathogenesis remain largely unclear. Recent emphasis has been placed on a potential role for canonical Wnt signaling, but investigation of this pathway in adult renal homeostasis is lacking. Here we provide evidence of a previously unidentified canonical Wnt activity in adult mammalian kidney homeostasis, the loss of which leads to cystic kidney disease. Loss of the Jouberin (Jbn) protein in mouse leads to the cystic kidney disease nephronophthisis, owing to an unexpected decrease in endogenous Wnt activity. Jbn interacts with and facilitates β-catenin nuclear accumulation, resulting in positive modulation of downstream transcription. Finally, we show that Jbn is required in vivo for a Wnt response to injury and renal tubule repair, the absence of which triggers cystogenesis.
Cystic kidney disorders include autosomal recessive and dominant polycystic kidney diseases as well as nephronophthisis1,2. Although the exact causes of these related disorders are not clear, various signaling pathways have been implicated. Specifically, several of the proteins encoded by the nephronophthisis-associated genes have been identified as negative modulators of the canonical Wnt pathway while activating the noncanonical Wnt pathway (planar cell polarity (PCP))3,4, suggesting a specific link between cystic kidney disease and Wnt signaling. The emerging model is that cystogenesis is at least partly due to overactivation of canonical Wnt signaling. This is supported by work with mutants of negative Wnt regulators, which show embryonic kidney cysts5,6. However, similar embryonic kidney cysts have also been documented in mutants of positive regulators, as well7,8, pointing to a unique dichotomy that may reflect more complex mechanisms than have previously been recognized. Because none of the cystic disease proteins has been examined for altered Wnt signaling in vivo in the mammalian kidney, and because Wnt activity has not been examined in the healthy adult kidney, the exact role of canonical Wnt signaling in adult-onset cystic kidney disease is still poorly understood.
Results
Ahi1−/− mice show pathology consistent with nephronophthisis
Jbn is the protein product of the AHI1 gene that is mutated in Joubert syndrome9,10, a disorder associated with cerebellar hypoplasia, retinitis pigmentosa and nephronophthisis11. We used Ahi1-null mice generated by homologous recombination, which showed a complete loss of the Jbn protein (Supplementary Fig. 1a) but overall normal embryonic development. However, the mice showed postnatal runting, and the majority (approximately 80%) did not survive to adulthood (data not shown). We investigated defects in postnatal kidney morphology as a potential cause of mortality. Ahi1-mutant mice showed no renal abnormalities at any neonatal time points examined from 3 to 21 d after birth (Supplementary Fig. 1b and data not shown). However, AHI1 mutations have been reported in some humans with late-onset nephronophthisis12. Additionally, we identified specific localization of a GFP-Jbn fusion construct in mouse kidney inner medullary collecting duct cells13 at the ciliary basal body (Fig. 1a and Supplementary Fig. 2a,b) and as sparsely distributed puncta along the ciliary axoneme (Fig. 1a and Supplementary Fig. 2a), similar to the ciliary localization recently described14. In vivo examination of endogenous Jbn in adult mouse kidney similarly revealed basal body and axoneme localization in collecting ducts as also described by the recent study14, as well as in proximal and distal tubules (Fig. 1b and Supplementary Fig. 2c), with particular intensity at the cortico-medullary junction (Supplementary Fig. 2d) that was not present in Ahi1-mutant kidney. Furthermore, western blotting for Jbn revealed a steady increase in its abundance with age from postnatal day 8 to 5 months (Supplementary Fig. 2e), suggesting a potential role for Jbn in the adult kidney.
Figure 1.
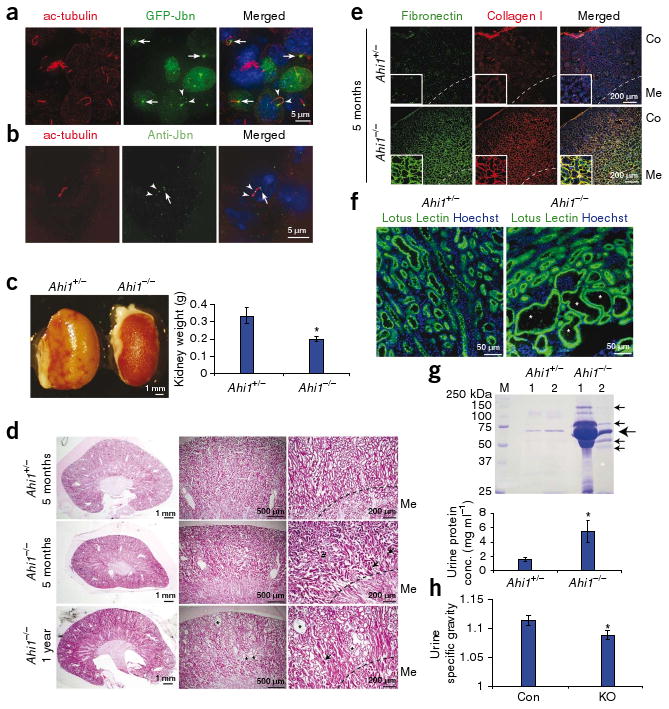
Loss of Jbn leads to nephronophthisis pathology. (a) Mouse kidney inner medullary collecting duct cells transfected with GFP-Jbn (green) and stained for acetylated tubulin (ac-tubulin, red) to visualize cilia. Hoechst (blue) labels nuclei. (b) Endogenous Jbn-specific antibody staining (Anti-Jbn, green) in vivo in tubular epithelial cells of mouse kidney with acetylated tubulin costaining (red). Note basal body localization (arrows), and punctate axonemal staining (arrowheads). (c) Kidneys from Ahi1+/− and Ahi1−/− littermates (left) and average kidney weight measurement at 5 months of age (right) (Student's t test, *P < 0.05, n = 3). (d) H&E staining of 5-month-old Ahi1−/− kidney sections compared to littermate Ahi1+/− kidney sections. Tubular collapse (arrows) and mononuclear cell infiltrate (#) are indicated with tubule dilatation (*) at 1 year. Dashed line demarcates medullary (Me) boundary. (e) Antibody staining of 5-month-old littermate kidneys for fibronectin (green) and collagen I (red). Co, cortex. (f) Lotus lectin staining (green) in 1-year-old littermate kidneys revealing cysts (*) within proximal tubules of Ahi1−/− kidney. (g) Top, Coommassie-stained SDS-PAGE analysis of equal volumes of urine from two pairs of each 18-month-old littermates, one Ahi1+/− and one Ahi1−/−. The predicted size of albumin is shown (large arrow) as well as additional bands present in Ahi1−/− samples (small arrows). Bottom, histogram of average urine protein concentration (n = 5, *P < 0.05, Student's t-test). (h) Average urine specific gravity after 16 h of dehydration in control (Ahi1+/+ or Ahi1+/−, Con) or Ahi1 knockout (KO) littermates at 21 months of age (*P < 0.05, Student's t test, n = 5). Error bars are means ± s.e.m.
We therefore examined surviving adult Ahi1−/− mice for potential late-onset renal phenotypes. By 5 months, null kidneys seemed smaller than those of littermate controls (Fig. 1c) and showed the characteristic histological triad of nephronophthisis15: tubular basement membrane abnormalities, including thickening and disintegration with tubular collapse; interstitial cell infiltrate and fibrosis; and, finally, a later appearance (at 1 year) of multiple microcysts and tubular dilatation (Fig. 1d and Supplementary Fig. 3a,b). Masson's trichrome staining also showed these abnormalities (Supplementary Fig. 3c), and antibodies to collagen I and fibronectin revealed increased staining for these proteins, indicating interstitial fibrosis in kidneys of Ahi1−/− mice (Fig. 1e), consistent with nephronophthisis pathology. Tubule abnormalities were particularly concentrated in the cortico-medullary region. We therefore examined which tubules were most affected and identified colabeling of cystic tubules with lotus lectin, a proximal tubule marker (Fig. 1f)16, indicating that, similar to the human disease, in the Ahi1-knockout mice proximal tubules of the cortico-medullary region were most affected.
We next quantitatively examined cyst progression by measuring cyst index in littermates at various ages17. Ahi1-null mice at least 1 year of age showed significantly increased average cyst area as a ratio of total kidney area (n = 3, P < 0.05, Supplementary Fig. 3d). Next, we examined kidney function impairment by testing for proteinuria and urinary concentrating defects. Bradford assay as well as Coomassie staining of SDS-PAGE gels revealed increased urine protein concentrations in Ahi1-mutant mice at 18–21 months of age (Fig. 1g), similar to those described in the related Glis2 late-onset nephronophthisis mutant mouse18,19. Ahi1-null mice 21 months of age additionally showed low serum albumin concentrations (control littermates' average albumin concentration, 4.0 g dl−1 (n = 3); Ahi1−/− average albumin concentration, 2.9 g dl−1 (n = 3); P < 0.05, Student's t test). Furthermore, we examined urine specific gravities at various time points to assess the progression of renal impairment. Ahi1-null mice at age 2.5 months showed no impairments, whereas all Ahi1-null mice at 21 months of age showed marked defects in urine-concentrating ability compared with littermate controls (Fig. 1h and Supplementary Fig. 4a). As urinary concentrating defects represent a primary diagnostic test in nephronophthisis20, these findings are further consistent with a nephronophthisis phenotype in the mutant mice. Furthermore, all mutant mice examined at 1 year of age or older had cysts (n = 6) and kidney function impairment (n = 8), suggesting a fully penetrant phenotype. Finally, serum creatinine concentration analysis showed that two Ahi1-null mice aged 21 months had moderately elevated creatinine (>0.4 mg dl−1; controls: <0.2 mg dl−1). Notably, the late onset of nephronophthisis in the Ahi1−/− mice mimics that seen in humans with nephronophthisis caused by AHI1 mutations, whose symptoms are not evident until their late teens or early twenties12,21.
The cystic kidney disorders have now been identified as part of a larger class of diseases known as the ciliopathies, which all share a common theme: potential abnormal ciliary structure or signaling22. We therefore hypothesized that loss of Jbn may lead to cilia defects. We examined primary cilia of 5-month-old affected Ahi1−/− and control littermate kidneys and found that, despite the nephronophthisis phenotype, the same percentage of cells showed evident primary cilia in Ahi1−/− kidneys as in control kidneys, and the cilia showed indistinguishable morphology in all kidney regions, at least at the resolution examined (Supplementary Fig. 4b). Furthermore, primary mouse embryonic fibroblasts from Ahi1−/− mice showed indistinguishable primary cilia with comparable length and morphology and in the same percentage of cells as littermate-derived wild-type mouse embryonic fibroblasts (Supplementary Fig. 4c). Finally, electron micrographs from retinal connecting cilia of these mutants revealed grossly normal 9+0 doublet microtubule architecture (data not shown). We therefore concluded that Jbn is not necessary for proper ciliogenesis, suggesting alternative mechanisms for the defects in these mice.
Ahi1−/− mice show abrogated adult renal Wnt activity
Several cystic disease proteins have been identified as direct regulators of the Wnt pathway. Nephronophthisis protein-2 (NPHP-2, or inversin), NPHP-3 and Glis2 have all been shown to inhibit canonical Wnt signaling3,4,23. In contrast, polycystin-1 has been shown to activate the canonical Wnt pathway24–26, although recent data suggests its carboxy terminus may negatively regulate the pathway27. To test whether Jbn has a role in canonical Wnt signaling, we crossed Ahi1-heterozygous mice with TOPGAL transgenic mice, a well documented canonical Wnt transgenic reporter line28. We first examined the mice at embryonic day 13.5, postnatal day 10 and 2 weeks and did not see a defect in β-galactosidase levels in Ahi1−/− TOPGAL+ kidneys, where strong Wnt activity was present in both control and mutant kidneys (data not shown). We next examined whether Wnt signaling defects were present in adult (5-month-old) Ahi1−/− TOPGAL+ mouse kidneys with early signs of nephronophthisis and stained for β-galactosidase using X-gal. The adult mouse kidney had previously been reported to lack canonical Wnt activity29. Likewise, we saw almost no staining upon initial examination of control TOPGAL+ kidneys with brief X-gal staining (data not shown). However, when we performed extended X-gal staining of Ahi1+/− TOPGAL+ control kidneys, while controlling for endogenous activity by using an X-gal solution pH 7.7–8.0 (ref. 30), we identified a previously unrecognized specific staining pattern in the kidney cortex, with particularly high levels in the cortico-medullary region (Fig. 2a). To our surprise, Ahi1−/− TOPGAL+ littermate kidney showed an almost complete absence of activity, even when developed to saturation (Supplementary Fig. 5a).
Figure 2.
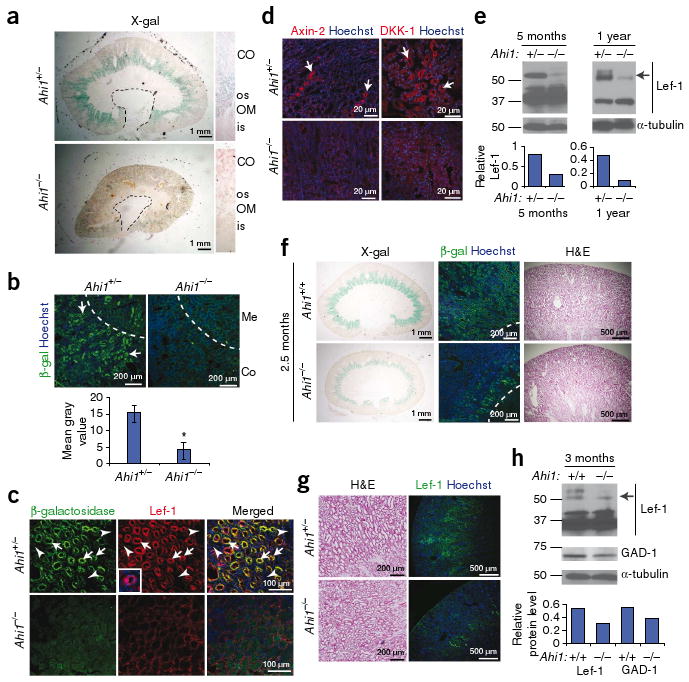
Jbn is required for Wnt activity in adult mouse kidney. (a) X-gal staining in 5-month-old littermate kidneys. Cross-section reveals outer stripe (os) staining. OM, outer medulla; CO, cortex; is, inner stripe. Dashed line, calyx boundary. (b) Top, antibody staining for β-galactosidase (green) in 5-month-old Ahi1+/− TOPGAL+ or Ahi1−/− TOPGAL+ kidneys. Hoechst labels nuclei (blue). Bottom, β-galactosidase average fluorescence. *P < 0.05, Student's t test, n = 3 images. Error bars represent means ± s.e.m. (c) β-galactosidase (green) and Lef-1 (red) protein staining (arrows) in Ahi1+/− TOPGAL+ and Ahi1−/− TOPGAL+ mice. Inset provides higher magnification of Lef-1 staining. Arrowheads indicate negative tubules for reference. (d) Axin-2 and DKK-1 staining (red, arrows) in the corticomedullary region of littermate kidneys. (e) Top, Lef-1 western blot from whole kidney lysates of littermates at 5 months and 1 year of age, revealing decreased expression of the full-length isoform (55–57 kDa, arrow). Bottom, full-length Lef-1 measurement relative to α-tubulin (loading control). (f) X-gal and β-galactosidase antibody staining in littermate kidneys at 2.5 months of age, before the onset of pathology (H&E at right). Dashed line demarcates the medullary boundary. (g) Lef-1 target gene staining (green) of Ahi1+/− and Ahi1−/− mice before nephronophthisis pathology (H&E at left). (h) Top, Lef-1 and GAD-1 western blots from whole-kidney lysates of Ahi1+/+ and Ahi1−/− littermates at 3 months of age. Bottom, full-length Lef-1 and GAD-1 levels relative to α-tubulin.
Given the existence of endogenous β-galactosidase activity in the adult kidney31, we confirmed these results by immunostaining with a bacterial β-galactosidase–specific antibody, which we then quantified (Fig. 2b). This approach revealed absent staining in Ahi1−/− TOPGAL+ kidney, whereas control TOPGAL+ littermate kidney showed broader staining throughout the kidney cortex compared with the X-gal staining, probably owing to the requirement for pentameric β-galactosidase complex formation for its enzymatic activity. Overall, the antibody staining seemed to be more specific, suggesting that this approach is preferable when staining adult kidney for exogenous β-galactosidase reporters. To further control for background endogenous β-galactosidase activity, we performed antibody staining in wild-type TOPGAL− and TOPGAL+ kidneys as well as X-gal staining in TOPGAL+ samples alongside TOPGAL− littermates, which did not have the specific staining seen in TOPGAL+ samples (Supplementary Fig. 5b). Finally, we additionally performed β-galactosidase staining in an alternate Wnt reporter line, BATGAL32, which showed more intense staining in a pattern similar to the TOPGAL pattern, supporting the validity of this Wnt activity (Supplementary Fig. 5c). Although the BATGAL reporter showed a more robust activity, we were unable to generate either Ahi1−/− BATGAL+ or Ahi1+/+ BATGAL− mice, suggesting that the two loci are linked.
To further test this Wnt effect, we examined an independent and endogenous Wnt reporter, lymphocyte enhancer–binding factor (Lef-1)33. In littermate control kidney, tubules positive for β-galactosidase also stained for Lef-1; however, Ahi1−/− kidney showed markedly lower Lef-1 expression concurrent with a lower β-galactosidase expression (Fig. 2c). Furthermore, examination of the expression of two additional Wnt targets, Axin-2 (ref. 34) and dickkopf homolog-1 (DKK-1)35, revealed markedly lower cortico-medullary expression of both proteins in Ahi1−/− kidneys (Fig. 2d). Finally, we examined Lef-1 expression by western blotting of whole kidney lysates as an alternate method of detection. This approach revealed lower expression of the Wnt responsive full-length isoform36 of Lef-1 in Ahi1-null kidneys compared to littermate controls at 5 months of age and 1 year of age, whereas the smaller isoform was unaffected (Fig. 2e). Thus, these data indicate that loss of Jbn leads to abrogation of basal Wnt activity in the adult mouse kidney.
To determine which specific tubules showed Wnt activity, we stained with a variety of lectins and identified colabeling of β-galactosidase tubules with lotus lectin, indicating that Wnt active tubules represent a subset of proximal tubules (Supplementary Fig. 5d), the same tubule type that later shows dilatation in Ahi1 mutants (Fig. 1f). Lotus lectin staining in Ahi1−/− 5 month kidneys revealed normal proximal tubule numbers (Supplementary Fig. 5d), suggesting that the proximal tubule epithelium was not affected by the genetic deficiency of Ahi1.
To further test the specificity of the Wnt defect in these mice and to examine whether the Wnt defect was secondary to early nephronophthisis changes, we performed X-gal staining, as well as staining for β-galactosidase and Lef-1 protein expression, before the appearance of nephronophthisis pathology. At 2.5 months of age, Ahi1−/− mice showed normal kidney morphology, yet X-gal staining and β-galactosidase–specific antibody staining revealed an already noticeably lower Wnt activity when compared to littermate controls (Fig. 2f). In addition, Ahi1−/− kidneys showed less Lef-1 protein staining, further indicating decreased Wnt activity before nephronophthisis onset (Fig. 2g). Finally, we also examined Lef-1 and an alternate Wnt target, glutamate decarboxylase-1 (GAD-1)37, by western blotting of whole kidney lysates from 3-month-old mice with no preexisting renal dysfunction (Supplementary Fig. 4a). Expression of both target proteins was lower in these mice (Fig. 2h). Overall, all Ahi1−/− mice tested (n = 4 before nephronophthisis onset, n = 5 after disease onset) showed a reproducible decrease in Wnt activity compared with control littermates, as measured using a variety of assays. Together, these data indicate the loss of Wnt activity precedes the pathological appearance of nephronophthisis.
The timing of Wnt activity abrogation in Ahi1−/− mice and the onset of nephronophthisis pathology suggests loss of basal Wnt activity may contribute to nephronophthisis pathogenesis in these mice. To test for a genetic interaction with the Wnt pathway, we generated mice doubly heterozygous for Ahi1 and Lrp6 (encoding low-density lipoprotein receptor-related protein-6), as Lrp6 has previously been identified as a necessary component in the canonical Wnt pathway8. Although neither Ahi1+/− mice nor Lrp6+/− control littermates showed kidney pathology, the combination of the two genotypes partially phenocopied the Ahi1−/− mouse pathology, pointing to nonallelic noncomplementation (Fig. 3). Ahi1+/−;Lrp6+/− mice had significantly smaller kidneys compared to single heterozygotes (Fig. 3a) and had tubular abnormalities consistent with nephronophthisis (Fig. 3b,c). Additionally, later-stage cysts and tubule dilatation were evident, similar to that seen in Ahi1−/− kidneys, which we quantified by measuring cyst index (Fig. 3d). Finally, we observed a similar decrease in urine specific gravity as that seen in Ahi1−/− mice (Fig. 3e). We also measured serum creatinine, and although the majority of Ahi1+/−;Lrp6+/− mice showed normal creatinine concentrations (<0.2 mg dl−1), one double heterozygote did show elevated levels (0.6 mg dl−1). These data suggest that Jbn and Lrp6 function in the same pathway and that the nephronophthisis phenotype in Ahi1−/− mice is Wnt dependent.
Figure 3.
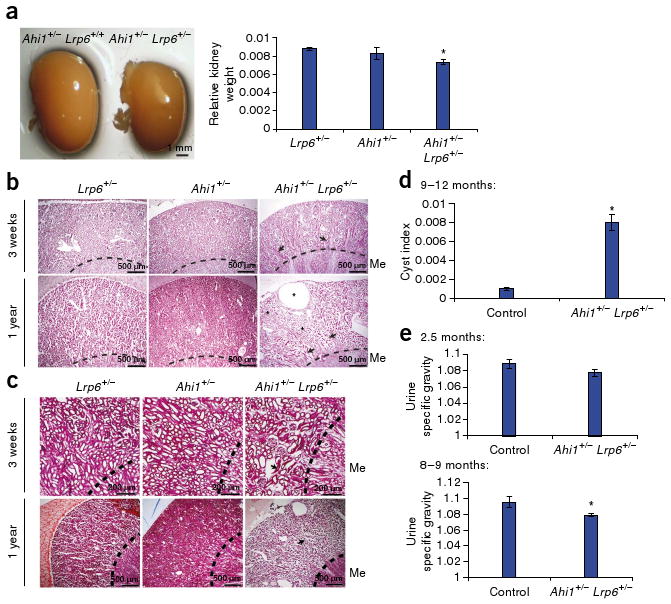
Ahi1 shows nonallelic noncomplementation with Lrp6. (a) Left, whole-mount image of Ahi1+/− kidney compared with Ahi1+/−;Lrp6+/− kidney. Right, the average kidney weight relative to total body weight (kidney/body weight ratio, arbitrary units). *P < 0.05, n = 3, Students's t test. (b,c) H&E (b) and Masson's trichrome (c) staining in Lrp6 and Ahi1 single heterozygotes compared with double heterozygotes. Arrows point to collapsed dysmorphic tubules within the cortex at 3 weeks, with a worsening of the phenotype at 1 year, at which a large cyst as well as tubule dilatation (*) are evident. (d) Average of cyst index measurements from three sections of each kidney of single-heterozygote control littermates (Ahi1+/− or Lrp6+/−) and double-heterozygote mutants (Ahi1+/−;Lrp6+/−) at 9–12 months of age (Student's t test, *P < 0.05, n = 3). (e) Average urine specific gravity at 2.5 months (top) and 8–9 months (bottom) of age. Both ages show decreased levels in Ahi1+/−;Lrp6+/− mutants compared to single-heterozygote littermates (Ahi1+/− or Lrp6+/−) with significance at 8–9 months (*P < 0.05, Student's t test, n = 3). Error bars in all histograms represent means ± s.e.m.
Jbn functions downstream of β-catenin stabilization
Although Jbn seems to act as a positive modulator of the canonical Wnt pathway, Ahi1−/− mice show a milder phenotype compared with the embryonic lethality and severe defects seen in classical Wnt mutants such as the Wnt3a-mutant mouse38. We therefore hypothesized that Jbn may have a modulatory role, rather than acting as an intrinsic Wnt pathway component. To test this hypothesis, we used an in vitro approach using the Super Topflash construct which expresses luciferase as a reporter of Wnt activity39,40. HEK293T cells transfected with this construct and Jbn alone did not show activation of the Wnt pathway, indicating that Jbn is not an activator of the pathway (Fig. 4a). However, transfection of Jbn into the cells potentiated the response to cotransfected disheveled homolog-1 (Dvl-1) (Fig. 4a) or treatment with Wnt3a (Supplementary Fig. 6a), suggesting that Jbn positively modulates the canonical Wnt pathway. Additionally, measurement of endogenous expression of the Wnt target cyclin D1 (ref. 41) revealed augmentation of the transcription response similar to that seen in the luciferase assay, whereas Jbn overexpression in the absence of Wnt had no effect on cyclin D1 (Fig. 4b). These results support the hypothesis that Jbn is a canonical Wnt pathway modulator.
Figure 4.
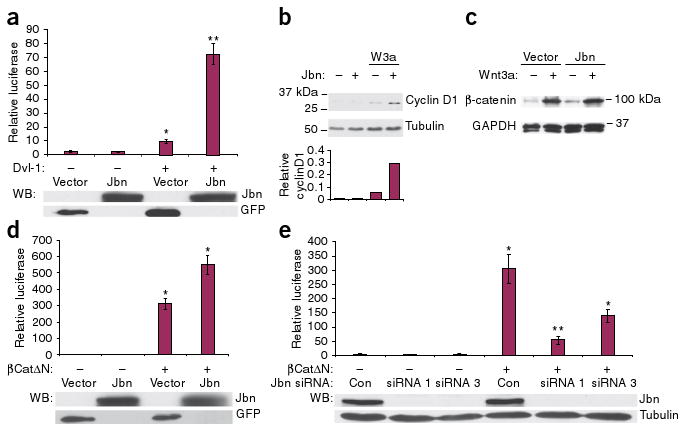
Jbn is a positive modulator of Wnt signaling downstream of β-catenin stabilization. (a) Top, induction of TCF/Lef by sevenfold by cotransfection of Jbn. *P < 0.001, **P < 5 × 10−7, n = 10 from four experiments, Student's t test. Values are relative to control untreated condition and are normalized for co-transfected β-gal. Western blot (WB) for construct expression from a representative luciferase is shown below each histogram. (b) Top, western blot for cyclin D1 with Wnt3a-conditioned medium treatment (W3a) and overexpression of Jbn. Bottom, quantification of cyclin D1 relative to α-tubulin (loading control). (c) Western blot of cytosolic extracts from 293T cells treated with Wnt3a-conditioned medium and expressing Jbn or empty vector. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is the loading control. (d) Luciferase activity relative to vector transfected alone in 293T cells transfected with βCatΔN and Jbn or GFP empty vector. *P < 0.0005, Student's t test, n = 24 from nine experiments. (e) Luciferase reporter activity in N2A cells transfected with Jbn-specific siRNA (Jbn siRNA) constructs and βCatΔN. *P < 0.05, **P < 0.001 Student's t test, n = 7 from four experiments. Values normalized for total protein concentration are expressed as relative to control untreated condition. Error bars in all experiments represent means ± s.e.m.
Wnt stimulation results in accumulation of cytosolic β-catenin due to disruption of the β-catenin destruction complex, a key step in downstream signaling42. To elucidate which step in the canonical Wnt pathway Jbn may modulate, we first tested whether Jbn overexpression had an effect on endogenous cytosolic β-catenin protein abundance. Treatment of 293T cells with Wnt3a-conditioned medium resulted in increased cytosolic β-catenin protein amounts, as expected (Fig. 4c). Overexpression of Jbn, however, did not potentiate this increase (Fig. 4c), suggesting that Jbn may function downstream in the pathway. To test this hypothesis, we used a constitutively active β-catenin lacking the amino terminus (β-catΔN), making it resistant to degradation43. We found that transfection of wild-type Jbn with β-catΔN enhanced the response to β-catΔN (Fig. 4d), implicating modulation downstream of activation of β-catenin.
To further test the hypothesis that Jbn acts downstream of β-catenin stabilization, we used small interfering RNA (siRNA) oligonucleotides targeted to mouse Jbn. Out of three siRNAs tested, siRNAs 1 and 3 substantially lowered endogenous Jbn expression in mouse N2A cells (Supplementary Fig. 6b); therefore, we used these two siRNAs for subsequent analyses. To test for a requirement for Jbn downstream of β-catenin activation, we transfected Jbn siRNAs with β-catΔN. Jbn knockdown by siRNA oligonucleotides led to a significant decrease in the response elicited by β-catΔN as compared to control siRNA cotransfection (Fig. 4e). These results support the hypothesis that Jbn modulates, and is required for, the Wnt signaling response downstream of β-catenin stabilization.
Jbn facilitates β-catenin nuclear translocation
We next used a coimmunoprecipitation approach to test the possibility that Jbn interacts with β-catenin to modulate its signaling effects. We subjected 293T cells transfected with GFP-tagged Jbn to immunoprecipitation with a GFP-specific antibody followed by immunoblotting for endogenous β-catenin. This method revealed an interaction between β-catenin and GFP-Jbn but not GFP empty vector (Fig. 5a). Treatment with Wnt3A led to an increase in the amount of β-catenin pulled down with Jbn, suggesting a specific interaction with the Wnt responsive pool of β-catenin (Fig. 5a). We next tested this interaction by reciprocal coimmunoprecipitation from wild-type mouse tissue. Endogenous Jbn specifically coimmunoprecipitated with β-catenin but not with GFP-specific negative control mouse IgG1 antibody, suggesting this interaction occurs reciprocally and in vivo (Fig. 5b).
Figure 5.
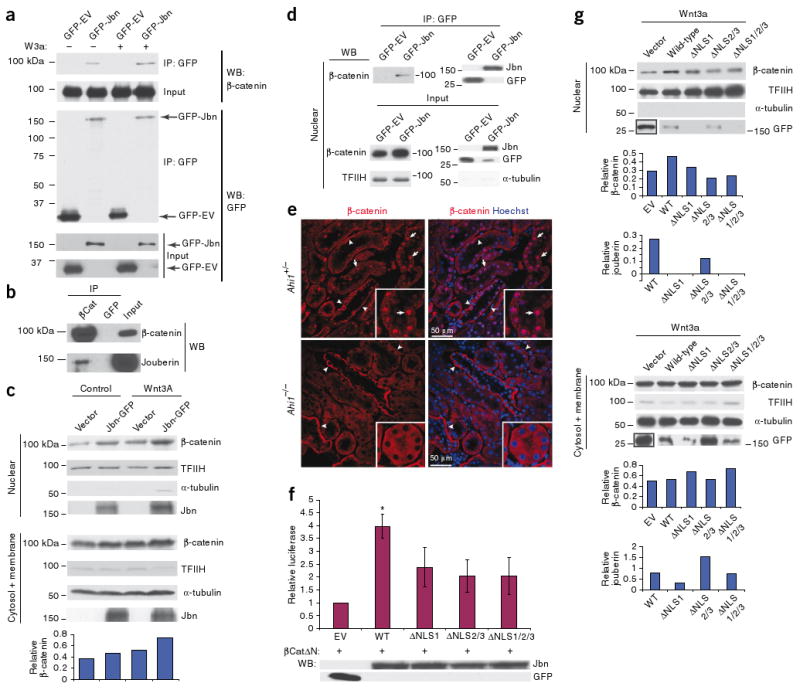
Jbn facilitates β-catenin nuclear accumulation. (a) Western blot for β-catenin after GFP-Jbn immunoprecipitation (IP) compared with vector control (GFP-EV) with and without Wnt3a treatment (W3a). Input is total cell lysate before IP. (b) IP for endogenous β-catenin from postnatal day 5 mouse whole-brain lysates, with western blotting for endogenous Jbn. (c) Western blot analysis of nuclear extraction from cells transfected with Jbn or vector, with or without Wnt3A treatment. Transcription factor II H (TFIIH) and tubulin are nuclear and cytosolic fractionation controls. Quantification of two repeats of this experiment is shown at the bottom β-catenin relative to TFIIH and controlling for cytosolic contamination by subtraction of tubulin). (d) IP from Cos-7 nuclear extracts for GFP-Jbn with GFP-specific antibody and western blotting for endogenous nuclear β-catenin. Input is nuclear lysate before the addition of GFP antibody. (e) β-catenin staining (red) of 1-year-old littermate kidneys revealing nuclear (Hoechst, blue) localization of β-catenin (arrows) that is absent in Ahi1-null kidney. Arrowheads denote basolateral localization. (f) Luciferase activity in 293T cells transfected with βCatΔN and NLS mutants compared to wild-type Jbn. *P < 0.05, n = 4 from four separate experiments, Student's t test. Values are relative to vector control and normalized for co-transfected β-galactosidase. Error bars represent means ± s.e.m. Western blot is from a representative luciferase assay. (g) Nuclear extraction and β-catenin western blotting from Cos-7 cells with overexpression of wild-type Jbn or NLS mutants. TFIIH and α-tubulin are controls for the nuclear extraction. Jbn and β-catenin levels are quantified relative to TFIIH levels for nuclear fraction or α-tubulin for cytosolic fraction.
Because Jbn functions downstream of cytosolic β-catenin stabilization and can associate with β-catenin while showing both nuclear and cytosolic localization, we hypothesized that Jbn may function to facilitate translocation and accumulation of β-catenin in the nucleus. To test this possibility, we first examined 293T cells overexpressing GFP-Jbn, which showed subtly higher levels of nuclear β-catenin when compared to neighboring untransfected cells (Supplementary Fig. 7a). We next performed nuclear extraction from Cos-7 cells transfected with GFP-Jbn followed by a western blot assay of nuclear β-catenin protein abundance to better visualize the effect on nuclear β-catenin. This assay revealed an enhancement of the Wnt3a-dependent nuclear β-catenin increase in the presence of Jbn overexpression (Fig. 5c). Notably, Jbn overexpression alone also led to higher levels of nuclear β-catenin (Fig. 5c). Luciferase reporter assay results (Supplementary Fig. 6a), however, indicate this is not sufficient for triggering a transcription response, probably owing to a requirement for other Wnt-induced transcription regulators. Notably, although Jbn was primarily present in the cytosolic fraction, it was also present in the nucleus, and this nuclear localization increased in a Wnt-dependent manner, along with β-catenin nuclear accumulation, suggesting it may translocate into the nucleus with β-catenin.
We therefore next examined whether Jbn and β-catenin interact in the nucleus by performing coimmunoprecipitation from nuclear extracts of Cos-7 cells, which revealed an interaction between endogenous nuclear β-catenin and GFP-Jbn (Fig. 5d). We then tested whether Jbn is required for β-catenin nuclear localization in vivo by examining endogenous β-catenin localization. In control kidneys, in addition to its more recognized basolateral localization, β-catenin was also clearly present in the nucleus of a subset of cortical tubules adjacent to the medulla (Fig. 5e), which costained for the proximal tubule marker lotus lectin (Supplementary Fig. 7b). A subset of Jbn protein similarly showed nuclear localization in addition to its primarily cytosolic and basal body staining (Supplementary Fig. 7c). Ahi1−/− kidneys, however, showed no evidence of nuclear β-catenin staining, whereas basolateral staining was intact (Fig. 5e), indicating that, in vivo, Jbn has a role in the nuclear localization of β-catenin. Given that Jbn enhances, and is required for, nuclear β-catenin accumulation, and given that Jbn's primary localization appears cytoplasmic with a nuclear subpopulation that is dynamically regulated by Wnt activation, these results suggest a role for Jbn in cytoplasmic-nuclear shuttling of β-catenin.
Because β-catenin does not contain nuclear localization signals (NLSs) we hypothesized that Jbn may facilitate translocation of activated β-catenin to the nucleus via three NLSs which we identified within the Jbn protein (with the PredictNLS program44). To test this possibility, we inactivated these NLSs by mutating key lysine residues, which is predicted to abolish NLS activity in NLS1 and in the overlapping region of NLS2 and NLS3 (Supplementary Fig. 8a). The three mutant Jbn proteins (ΔNLS1, ΔNLS2/3 and the combination of both mutations, ΔNLS1/2/3) showed comparable expression to wild-type Jbn, although ΔNLS1 was somewhat lower (Supplementary Fig. 8b), but all mutant constructs failed to localize to the nucleus when examined by fluorescence microscopy and showed significantly decreased Wnt signaling amplification compared with wild-type Jbn when transfected with Dvl-1 (Supplementary Fig. 8c,d) or β-catΔN (Fig. 5f). We then performed nuclear extraction with cells transfected with the mutant constructs and found less nuclear β-catenin localization compared with cells transfected with wild-type Jbn (Fig. 5g). We should note that although nuclear localization of ΔNLS2/3 was not completely abolished, it failed to potentiate β-catenin nuclear levels. However, the mutant lacking all NLS regions (ΔNLS1/2/3), which showed similar overall expression to that of wild-type Jbn and failed to enter the nucleus, also failed to potentiate β-catenin nuclear accumulation (Fig. 5g). These data support a mechanism whereby Jbn functions through interaction with cytosolic β-catenin to facilitate the its nuclear translocation, a function that requires all three NLS regions of Jbn.
Ahi1−/− mice show defective injury repair and Wnt response
We next sought to address how abrogated Wnt signaling might lead to the nephronophthisis phenotype in Ahi1−/− mice. Wnt activity has previously been reported as upregulated in mouse renal injury, lasting as long as 28 d after injury45, suggesting it may be involved in adult renal homeostatic injury repair. We therefore performed injury experiments using established protocols for either cisplatin or ischemia-reperfusion injury (IRI). We subjected control and Ahi1-null littermates (4-months-old) to intraperitoneal injection of cisplatin or saline or unilateral IRI. At this age, mutant kidneys do not yet have cysts, as indicated in mock-treated kidneys (Fig. 6a). However, 2.5– 4 weeks after injury, microcysts were evident in mutant kidneys, which was reminiscent of the pathology normally observed at one year of age, suggesting acceleration of the phenotype. Control kidneys, in contrast, showed signs of injury recovery without evidence of cysts. We quantified these findings by measuring cyst index (Fig. 6b).
Figure 6.
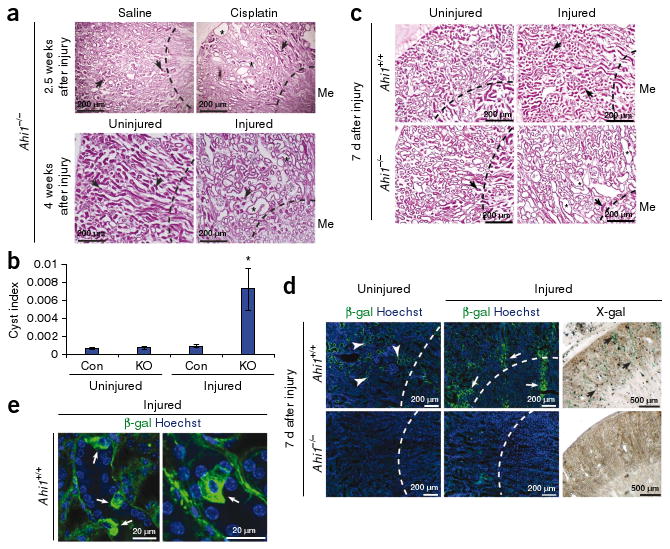
Ahi1−/− mice show defective recovery from renal injury. (a) H&E histology in 4-month-old injured Ahi1−/− kidneys (cisplatin administration or renal IRI (Injured) on the left kidney) compared with saline-treated or uninjured kidneys. Arrows indicate early signs of nephronophthisis, although cysts are not present. Asterisks depict cysts and tubule dilatation 2.5 weeks after cisplatin injection or 4 weeks after IRI of the left kidney (*). Dashed line indicates medullary boundary. (b) Average cyst index measurement from littermate control (Con) and Ahi1−/− (KO) uninjured and injured kidneys at 2.5–4 weeks after injury with either cisplatin or IRI (n = 3 kidneys each, *P < 0.05, Student's t test). (c) H&E histology of injured left kidney of littermate wild-type mice 7 d after IRI showing tubular obstruction and collapse (arrows). Ahi1−/− injured kidney instead shows tubular dilatation and microcysts (*) with evident glomerulosclerosis. Dashed line indicates medullary boundary. n = 3 mutant mice and n = 4 control mice. (d) β-galactosidase–specific antibody (green) and X-gal staining in control littermate uninjured right kidney and injured left kidney. Hoechst (blue) labels nuclei, and the dashed line indicates the medullary boundary. (e) Higher magnification of β-galactosidase–positive kidney tubules in control injured sections, revealing Wnt-responsive cells with fibroblast-like morphology.
To test for the possibility that Ahi1-null mice were more susceptible to the injury itself, we also performed a representative renal IRI in a pair of littermate control and Ahi1-null mice and examined pathology as well as serum chemistry 2 h after injury. Histology revealed comparable signs of injury (vacuolization and epithelial cell sloughing) in mice of both genotypes (Supplementary Fig. 9a), and serum chemistry revealed similarly elevated creatinine (control, 0.6 mg dl−1; Ahi1−/−, 0.5 mg dl−1), suggesting the renal insult was not more severe in Ahi1-null mice. These results support the hypothesis that renal injury repair, rather than susceptibility, is abnormal in Ahi1−/− mice, leading to cystogenesis.
To investigate Wnt signaling in this repair process, we performed IRI on Ahi1+/+ TOPGAL+ and Ahi1−/− TOPGAL+ kidneys and compared β-galactosidase Wnt reporter activity 5–7 d after injury. In the uninjured right kidney, morphology and Wnt activity appeared similar to that described above for the mutant and control mice, although there were mild defects in the wild-type contralateral kidney (Fig. 6c), possibly a result of indirect injury46. However, the wild-type injured left kidney showed clear morphological indications of injury 7 d after IRI (Fig. 6c), along with a striking upregulation of Wnt activity, as measured by X-gal and β-galactosidase–specific antibody staining (Fig. 6d). High magnification revealed a subpopulation of highly Wnt-responsive cells that showed nonpolarized morphology (Fig. 6e). Histological examination of Ahi1−/− injured kidney, in contrast, revealed tubular dilatation and glomerulosclerosis already at 7 d after injury (Fig. 6c) that lacked comparable Wnt reporter upregulation (Fig. 6d). We then performed quantification of Wnt activity by a β-galactosidase luminescence assay on whole kidney lysates from an independent set of injured littermates aged 3 months with no prior renal impairment (Supplementary Fig. 4a). This assay revealed upregulation of Wnt activity in the control injured kidney 5 d after IRI compared with the uninjured kidney (Supplementary Fig. 9b), whereas both Ahi1−/− kidneys showed decreased Wnt activity, which was especially evident in the injured kidney lysate. These results suggest a role for canonical Wnt signaling in recovery from injury, a function that is abrogated in Ahi1-mutant kidneys, leading to renal cyst pathology.
Discussion
Our findings provide what is to our knowledge the first indication that the canonical Wnt pathway is necessary for adult kidney homeostasis, and that abrogation of this signaling can lead to cystic kidney pathogenesis. Despite the ciliopathy nomenclature, the nephronophthisis phenotype that arises with loss of Jbn seems not to relate to structural ciliary defects. Instead, Jbn acts as a direct positive modulator of canonical Wnt signaling. Some ciliopathy proteins have been found to regulate the structure of the cilium (NPHP-1 and NPHP-4)47, yet several others (such as inversin and Glis2) seem to function primarily in signaling3,23. With recent evidence pointing to a negative regulatory role for the primary cilium itself in canonical Wnt signaling, we hypothesize that the renal pathology associated with ciliopathies is at least partly due to disruption of downstream signaling. This may occur either through an indirect effect of the cilium on these pathways or through direct regulation of the pathway. Whereas indirect regulation through disruption of the cilium might be expected to result in an increase in canonical Wnt activity, Jbn instead directly facilitates the pathway, as indicated by a lack of effect on the primary cilium and reduced endogenous Wnt signaling.
Like Jbn, several other ciliopathy proteins are modulators of canonical Wnt signaling, but, to date, the data suggest that they mainly function as negative regulators of the pathway. These seemingly contradictory results suggest a unique balance of Wnt–β-catenin regulation modulating renal development and homeostasis. Parallels can be discerned from renal tubule development in which disruption of canonical Wnt signaling in either direction leads to inhibition of terminal epithelial differentiation, suggesting a similar delicate balance of regulation48–50. Likewise, disruption of noncanonical Wnt signaling (that is, disruption of PCP) similarly leads to defects in formation of the polarized epithelium51, and abnormal PCP has also been implicated in cystic kidney disease pathogenesis52,53. Thus, differentiation of the renal tubular epithelium shows sensitivity to precise Wnt regulation similar to that seen in adult cystogenesis with loss of Jbn, suggesting similar mechanisms.
Given the parallels between renal tubular development and cystogenesis, the following question arises: how might these developmental processes be involved in pathogenesis of an adult-onset disease? One model suggests developmental processes can be reactivated in adult renal epithelial cells during basal cell turnover or in response to injuries to regenerate renal tubules54. Differentiated epithelial cells may in fact be capable of dedifferentiating in order to proliferate and repopulate damaged tubules with new epithelium31,55. Our findings suggest that precise Wnt signaling regulation, similar to that seen in developing tubules, may be vital to tubular epithelial cell renewal in the adult kidney, and that this signaling is particularly active after injury.
Similar to knockout of Ahi1 in mice, acute injury has been shown to trigger cystogenesis in other cystic kidney mouse models, whereas conditional ablation of Kif3a or Pkd1 leads to a slow accumulation of cysts in the absence of acute injury17,56,57. These late-onset phenotypes may reflect a gradual accumulation of mild damage, leading to vulnerability to cystogenesis when tubular regeneration is initiated. Thus, the homeostatic balance of Wnt activity in adult tissues may regulate the regenerative response to subtle injuries and basal cell turnover, a function whose loss leads to a mammalian ciliopathy58. These findings may reflect a general requirement for reactivation of developmental pathways in injury repair. And, given the vital role in signaling by the primary cilium during development, adult-onset ciliopathy may result if signaling by this cellular organ is abnormally regulated during these repair processes.
Methods
Methods and any associated references are available in the online version of the paper at http://www.nature.com/naturemedicine/.
Supplementary Material
Acknowledgments
We are grateful to members of the Gleeson lab for technical expertise and feedback and the Nigam lab for helpful kidney-related discussions and reagents, as well as B. Brinkman and the UCSD Neuroscience Microscopy Core. We also thank the K. Kaushansky, M. Karin, and P.L. Mellon labs, as well as E.L. Stone for technical expertise. We are grateful to S. Piccolo at the Departments of Histology, Microbiology and Medical Biotechnologies, University of Padua, for the BATGAL mice. We thank S. Pleasure at the Department of Neurology, University of California–San Francisco, for Lrp6-mutant mice. We also thank M.G. Rosenfeld at the School of Medicine, UCSD, for the β-catΔN construct and R.T. Moon at the Department of Pharmacology, University of Washington, for the Super Topflash construct. M.A.L. and C.M.L received support from the US National Institutes of Health–National Institute of General Medical Sciences–funded UCSD Genetics Training Program (T32 GM08666). This work was supported by the US National Institutes of Health and the Burroughs Wellcome Fund in Translational Research (J.G.G.). J.G.G. is an investigator with Howard Hughes Medical Institute.
Footnotes
Note: Supplementary information is available on the Nature Medicine website.
Author Contributions: M.A.L. designed the experimental approach, conducted the experiments and wrote the manuscript. J.G.G. supervised the project and experimental approach, interpreted data and contributed to manuscript preparation. C.M.L. designed and generated the Ahi1−/− mouse mutant and provided feedback. J.L.S. generated mutant constructs and assisted in microscopy. L.S. contributed to in vitro localization experiments. M.D. contributed to IRI experiments. S.K.N. provided feedback regarding renal characterization and manuscript preparation. K.W. provided feedback and reagents for in vitro Wnt assays.
Published online at http://www.nature.com/naturemedicine/.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
References
- 1.Harris PC. Molecular basis of polycystic kidney disease: PKD1, PKD2 and PKHD1. Curr Opin Nephrol Hypertens. 2002;11:309–314. doi: 10.1097/00041552-200205000-00007. [DOI] [PubMed] [Google Scholar]
- 2.Hildebrandt F, Zhou W. Nephronophthisis-associated ciliopathies. J Am Soc Nephrol. 2007;18:1855–1871. doi: 10.1681/ASN.2006121344. [DOI] [PubMed] [Google Scholar]
- 3.Simons M, et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet. 2005;37:537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergmann C, et al. Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber–like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am J Hum Genet. 2008;82:959–970. doi: 10.1016/j.ajhg.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saadi-Kheddouci S, et al. Early development of polycystic kidney disease in transgenic mice expressing an activated mutant of the β-catenin gene. Oncogene. 2001;20:5972–5981. doi: 10.1038/sj.onc.1204825. [DOI] [PubMed] [Google Scholar]
- 6.Qian CN, et al. Cystic renal neoplasia following conditional inactivation of apc in mouse renal tubular epithelium. J Biol Chem. 2005;280:3938–3945. doi: 10.1074/jbc.M410697200. [DOI] [PubMed] [Google Scholar]
- 7.Marose TD, Merkel CE, McMahon AP, Carroll TJ. β-catenin is necessary to keep cells of ureteric bud/Wolffian duct epithelium in a precursor state. Dev Biol. 2008;314:112–126. doi: 10.1016/j.ydbio.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC. An LDL-receptor–related protein mediates Wnt signalling in mice. Nature. 2000;407:535–538. doi: 10.1038/35035124. [DOI] [PubMed] [Google Scholar]
- 9.Ferland RJ, et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet. 2004;36:1008–1013. doi: 10.1038/ng1419. [DOI] [PubMed] [Google Scholar]
- 10.Dixon-Salazar T, et al. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet. 2004;75:979–987. doi: 10.1086/425985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Louie CM, Gleeson JG. Genetic basis of Joubert syndrome and related disorders of cerebellar development. Hum Mol Genet. 2005;14(Spec No. 2):R235–R242. doi: 10.1093/hmg/ddi264. [DOI] [PubMed] [Google Scholar]
- 12.Utsch B, et al. Identification of the first AHI1 gene mutations in nephronophthisis-associated Joubert syndrome. Pediatr Nephrol. 2006;21:32–35. doi: 10.1007/s00467-005-2054-y. [DOI] [PubMed] [Google Scholar]
- 13.Rauchman MI, Nigam SK, Delpire E, Gullans SR. An osmotically tolerant inner medullary collecting duct cell line from an SV40 transgenic mouse. Am J Physiol. 1993;265:F416–F424. doi: 10.1152/ajprenal.1993.265.3.F416. [DOI] [PubMed] [Google Scholar]
- 14.Eley L, et al. Jouberin localizes to collecting ducts and interacts with nephrocystin-1. Kidney Int. 2008;74:1139–1149. doi: 10.1038/ki.2008.377. [DOI] [PubMed] [Google Scholar]
- 15.Davison AM, et al. Oxford Textbook of Clinical Nephrology Section 16.3. Oxford University Press; Oxford: 2005. [Google Scholar]
- 16.Faraggiana T, Malchiodi F, Prado A, Churg J. Lectin-peroxidase conjugate reactivity in normal human kidney. J Histochem Cytochem. 1982;30:451–458. doi: 10.1177/30.5.7077075. [DOI] [PubMed] [Google Scholar]
- 17.Patel V, et al. Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet. 2008;17:1578–1590. doi: 10.1093/hmg/ddn045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Attanasio M, et al. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat Genet. 2007;39:1018–1024. doi: 10.1038/ng2072. [DOI] [PubMed] [Google Scholar]
- 19.Kim YS, et al. Kruppel-like zinc finger protein Glis2 is essential for the maintenance of normal renal functions. Mol Cell Biol. 2008;28:2358–2367. doi: 10.1128/MCB.01722-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krishnan R, Eley L, Sayer JA. Urinary concentration defects and mechanisms underlying nephronophthisis. Kidney Blood Press Res. 2008;31:152–162. doi: 10.1159/000129648. [DOI] [PubMed] [Google Scholar]
- 21.Parisi MA, et al. AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert syndrome. J Med Genet. 2006;43:334–339. doi: 10.1136/jmg.2005.036608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 23.Kim YS, Kang HS, Jetten AM. The Kruppel-like zinc finger protein Glis2 functions as a negative modulator of the Wnt/β-catenin signaling pathway. FEBS Lett. 2007;581:858–864. doi: 10.1016/j.febslet.2007.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang K, et al. PKD1 inhibits cancer cells migration and invasion via Wnt signaling pathway in vitro. Cell Biochem Funct. 2007;25:767–774. doi: 10.1002/cbf.1417. [DOI] [PubMed] [Google Scholar]
- 25.Kim E, et al. The polycystic kidney disease 1 gene product modulates Wnt signaling. J Biol Chem. 1999;274:4947–4953. doi: 10.1074/jbc.274.8.4947. [DOI] [PubMed] [Google Scholar]
- 26.Zheng R, et al. Polycystin-1 induced apoptosis and cell cycle arrest in G0/G1 phase in cancer cells. Cell Biol Int. 2008;32:427–435. doi: 10.1016/j.cellbi.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 27.Lal M, et al. Polycystin-1 C-terminal tail associates with β-catenin and inhibits canonical Wnt signaling. Hum Mol Genet. 2008;17:3105–3117. doi: 10.1093/hmg/ddn208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 1999;126:4557–4568. doi: 10.1242/dev.126.20.4557. [DOI] [PubMed] [Google Scholar]
- 29.Iglesias DM, et al. Canonical WNT signaling during kidney development. Am J Physiol Renal Physiol. 2007;293:F494–F500. doi: 10.1152/ajprenal.00416.2006. [DOI] [PubMed] [Google Scholar]
- 30.Weiss DJ, Liggitt D, Clark JG. Histochemical discrimination of endogenous mammalian β-galactosidase activity from that resulting from lac-Z gene expression. Histochem J. 1999;31:231–236. doi: 10.1023/a:1003642025421. [DOI] [PubMed] [Google Scholar]
- 31.Duffield JS, et al. Restoration of tubular epithelial cells during repair of the postischemic kidney occurs independently of bone marrow–derived stem cells. J Clin Invest. 2005;115:1743–1755. doi: 10.1172/JCI22593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maretto S, et al. Mapping Wnt/β-catenin signaling during mouse development and in colorectal tumors. Proc Natl Acad Sci USA. 2003;100:3299–3304. doi: 10.1073/pnas.0434590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Filali M, Cheng N, Abbott D, Leontiev V, Engelhardt JF. Wnt-3A/β-catenin signaling induces transcription from the LEF-1 promoter. J Biol Chem. 2002;277:33398–33410. doi: 10.1074/jbc.M107977200. [DOI] [PubMed] [Google Scholar]
- 34.Jho EH, et al. Wnt/β-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niida A, et al. DKK1, a negative regulator of Wnt signaling, is a target of the β-catenin/TCF pathway. Oncogene. 2004;23:8520–8526. doi: 10.1038/sj.onc.1207892. [DOI] [PubMed] [Google Scholar]
- 36.Hovanes K. β-catenin–sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat Genet. 2001;28:53–57. doi: 10.1038/ng0501-53. [DOI] [PubMed] [Google Scholar]
- 37.Li CM, et al. CTNNB1 mutations and overexpression of Wnt/β-catenin target genes in WT1-mutant Wilms' tumors. Am J Pathol. 2004;165:1943–1953. doi: 10.1016/s0002-9440(10)63246-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takada S, et al. Wnt-3a regulates somite and tailbud formation in the mouse embryo. Genes Dev. 1994;8:174–189. doi: 10.1101/gad.8.2.174. [DOI] [PubMed] [Google Scholar]
- 39.Korinek V, et al. Constitutive transcriptional activation by a β-catenin–Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 40.Kaykas A, et al. Mutant Frizzled 4 associated with vitreoretinopathy traps wild-type Frizzled in the endoplasmic reticulum by oligomerization. Nat Cell Biol. 2004;6:52–58. doi: 10.1038/ncb1081. [DOI] [PubMed] [Google Scholar]
- 41.Tetsu O, McCormick F. β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 42.Willert K, Nusse R. β-catenin: a key mediator of Wnt signaling. Curr Opin Genet Dev. 1998;8:95–102. doi: 10.1016/s0959-437x(98)80068-3. [DOI] [PubMed] [Google Scholar]
- 43.van Noort M, Meeldijk J, van der Zee R, Destree O, Clevers H. Wnt signaling controls the phosphorylation status of β-catenin. J Biol Chem. 2002;277:17901–17905. doi: 10.1074/jbc.M111635200. [DOI] [PubMed] [Google Scholar]
- 44.Cokol M, Nair R, Rost B. Finding nuclear localization signals. EMBO Rep. 2000;1:411–415. doi: 10.1093/embo-reports/kvd092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Surendran K, Schiavi S, Hruska KA. Wnt-dependent β-catenin signaling is activated after unilateral ureteral obstruction, and recombinant secreted frizzled-related protein 4 alters the progression of renal fibrosis. J Am Soc Nephrol. 2005;16:2373–2384. doi: 10.1681/ASN.2004110949. [DOI] [PubMed] [Google Scholar]
- 46.Meldrum KK, Meldrum DR, Meng X, Ao L, Harken AH. TNF-α–dependent bilateral renal injury is induced by unilateral renal ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2002;282:H540–H546. doi: 10.1152/ajpheart.00072.2001. [DOI] [PubMed] [Google Scholar]
- 47.Jauregui AR, Nguyen KC, Hall DH, Barr MM. The Caenorhabditis elegans nephrocystins act as global modifiers of cilium structure. J Cell Biol. 2008;180:973–988. doi: 10.1083/jcb.200707090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmidt-Ott KM, Barasch J. WNT/β-catenin signaling in nephron progenitors and their epithelial progeny. Kidney Int. 2008;74:1004–1008. doi: 10.1038/ki.2008.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuure S, Popsueva A, Jakobson M, Sainio K, Sariola H. Glycogen synthase kinase-3 inactivation and stabilization of β-catenin induce nephron differentiation in isolated mouse and rat kidney mesenchymes. J Am Soc Nephrol. 2007;18:1130–1139. doi: 10.1681/ASN.2006111206. [DOI] [PubMed] [Google Scholar]
- 50.Park JS, Valerius MT, McMahon AP. Wnt/β-catenin signaling regulates nephron induction during mouse kidney development. Development. 2007;134:2533–2539. doi: 10.1242/dev.006155. [DOI] [PubMed] [Google Scholar]
- 51.Osafune K, Takasato M, Kispert A, Asashima M, Nishinakamura R. Identification of multipotent progenitors in the embryonic mouse kidney by a novel colony-forming assay. Development. 2006;133:151–161. doi: 10.1242/dev.02174. [DOI] [PubMed] [Google Scholar]
- 52.Saburi S, et al. Loss of Fat4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nat Genet. 2008;40:1010–1015. doi: 10.1038/ng.179. [DOI] [PubMed] [Google Scholar]
- 53.Kishimoto N, Cao Y, Park A, Sun Z. Cystic kidney gene seahorse regulates cilia-mediated processes and Wnt pathways. Dev Cell. 2008;14:954–961. doi: 10.1016/j.devcel.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 54.Bonventre JV, Zuk A. Ischemic acute renal failure: an inflammatory disease? Kidney Int. 2004;66:480–485. doi: 10.1111/j.1523-1755.2004.761_2.x. [DOI] [PubMed] [Google Scholar]
- 55.Bonventre JV. Dedifferentiation and proliferation of surviving epithelial cells in acute renal failure. J Am Soc Nephrol. 2003;14 1:S55–S61. doi: 10.1097/01.asn.0000067652.51441.21. [DOI] [PubMed] [Google Scholar]
- 56.Davenport JR, et al. Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol. 2007;17:1586–1594. doi: 10.1016/j.cub.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med. 2007;13:1490–1495. doi: 10.1038/nm1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Calvet JP. Injury and development in polycystic kidney disease. Curr Opin Nephrol Hypertens. 1994;3:340–348. doi: 10.1097/00041552-199405000-00017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.