

Abstract
Common fragile sites are loci that preferentially form gaps and breaks on metaphase chromosomes when DNA synthesis is perturbed, particularly after treatment with the DNA polymerase inhibitor, aphidicolin. We and others have identified several cell cycle checkpoint and DNA repair proteins that influence common fragile site stability. However, the initial events underlying fragile site breakage remain poorly understood. We demonstrate here that aphidicolin-induced gaps and breaks at fragile sites are prevented when cells are co-treated with low concentrations of the topoisomerase I inhibitor, camptothecin. This reduction in breakage is accompanied by a reduction in aphidicolin-induced RPA foci, CHK1 and RPA2 phosphorylation, and PCNA monoubiquitination, indicative of reduced levels of single stranded DNA. Furthermore, camptothecin reduces spontaneous fragile site breakage seen in cells lacking ATR, even in the absence of aphidicolin. These data from cultured human cells demonstrate that topoisomerase I activity is required for DNA common fragile site breaks and suggest that polymerase-helicase uncoupling is a key initial event in this process.
1. Introduction
Common fragile sites (CFSs) are loci that demonstrate reproducible, non-random gaps and breaks on metaphase chromosomes when cells are grown under conditions that partially perturb replication, particularly in the presence of low doses of the polymerase inhibitor, aphidicolin (APH) or following folate stress [1,2]. CFSs are large, with breakage occurring over a broad region ranging from hundreds of kilobases to over a megabase. FRA3B at 3p14.2 stands out as the most fragile site in the human genome and can be induced to form gaps or breaks in the majority of treated cells. Other highly expressed CFSs in cultured lymphocytes include those at 16q23 (FRA16D), 6q26 (FRA6E), 7q32.3 (FRA7H), and Xp22.3 (FRAXB). These and at least eight other CFSs have been molecularly characterized [3–14]. They share a number of characteristics that may contribute to their instability, such as long stretches of AT-rich sequence, including AT-repeats, late replication, and their presence within very large genes (Reviewed in [15,16]). In addition, it has been shown that histone hypoacetylation is able to reduce the incidence of CFS breakage after APH treatment [17]. CFSs are normally stable in somatic cells in vivo but are often associated with chromosome rearrangements in tumor cells, particularly large, submicroscopic deletions or copy number alterations (Reviewed in [18]), and we have recently shown that APH induces similar deletions at FRA3B and elsewhere in the genome in cultured cells [19,20]. CFSs may be among the earliest loci in the genome to be deleted during tumorigenesis in association with replication stress [21–23]. The presence of putative tumor suppressor genes at some CFSs, such as FHIT at FRA3B [24] and WWOX at FRA16D [25](Reviewed in [26]), suggests that CFS instability may lead to a selective growth advantage via inactivation of these genes in some cancers, while other CFS deletions may be neutral but act as signatures of replication stress.
We and others have identified a number of cell cycle checkpoint and DNA repair proteins that are important in maintaining CFS stability, including ATR, BRCA1, CHK1, FANCD2, RAD51, DNA-PKcs, Ligase IV, HUS1, and SMC1 [27–31]. The fact that CFS breakage occurs following modest levels of replication inhibition and that it is regulated by these checkpoint and repair pathways has led to the use of CFS breakage as a cytological assay in studies of the DNA damage response to replication stress.
While considerable progress has been made in identifying the cellular pathways required for maintenance of CFS stability, little is known about the mechanisms involved in the initial breakage events. CFS regions complete replication late in the cell cycle and contain AT-rich sequences that have the potential to form secondary structures that could further impede replication [5,32,33]. Current models for CFS breakage suggest that polymerase stalling and perhaps fork collapse caused by APH and certain other forms of replication stress lead to incomplete replication at these sites that can result in DNA double strand breaks [1].
Topoisomerase I (TopoI) unwinds positive supercoils in DNA created by the replicative helicase during replication [34](reviewed in [35]). TopoI acts by transiently cleaving one strand of duplex DNA, unwinding the DNA, and religating the cleavage site. It has been shown that TopoI is part of the GINS-MCM replication complex and is recruited to replication origins, after which it moves with the replication fork[36]. Camptothecin (CPT) is a powerful chemotherapeutic agent used to treat multiple types of cancer (Reviewed in [37]). CPT inhibits TopoI, its only known target, by blocking religation of the DNA, resulting in a dose-dependent reduction in replication [38–41]. At high doses, CPT results in large amounts of DNA double strand breaks, which are thought to be formed when the replication machinery proceeds to the site of the stabilized, TopoI-induced, single stranded nick [42]. At these high doses, this replication-dependant damage can be prevented by co-treating cells with high doses of APH, which stall the replication forks before they can collide with the stabilized TopoI cleavage complex [43–45]. Because of its critical role in replication dynamics, we hypothesize that TopoI plays a role in CFS instability.
To test this hypothesis, we treated normal human cells with APH and varying doses of CPT to partially inhibit TopoI and evaluated them for CFS breaks on metaphase chromosomes as well as for evidence of S-phase or G2/M checkpoint activation. Our results show that treatment with low doses of CPT almost completely prevents APH-induced breaks at CFSs. We also demonstrate that betulinic acid, a TopoI inhibitor that prevents DNA cleavage also prevents APH-induced CFS breaks. In addition, CPT reduces the spontaneous CFS breakage that occurs in ATR-deficient cells. This reduction in CFS gaps and breaks on metaphase chromosomes is accompanied by a decrease in activation of CHK1, PCNA, and RPA2, consistent with reduced amounts of single stranded DNA (ssDNA) at stalled replication forks. These results from cultured human cells indicate that TopoI activity is required for CFS breakage, and are consistent with in vitro models of polymerase-helicase uncoupling and suggest that this uncoupling is an initial key event in CFS instability after replication perturbation.
2. Materials and Methods
2.1 Cell Culture and Fragile Site Analysis
Normal UML-49 lymphoblastoid cells were grown in RPMI medium (Invitrogen) supplemented with 15% FBS. Primary blood lymphocytes were cultured immediately after venipuncture using conventional techniques. Aliquots of blood were transferred to RPMI (Invitrogen) supplemented with 10% FBS. Lymphocytes were stimulated with phytohemagglutinin (Sigma) for three days prior to experimental treatments. All cells were grown at 37°C in a humidified atmosphere containing 5% CO2.
Common fragile sites were induced by exposure of cells to 0.2 μM to 0.4 μM APH for 24 hours prior to harvest, in the presence or absence of camptothecin (CPT, Sigma) or betulinic acid (BA, Sigma). Cells were harvested for chromosome preparation using standard conditions of 45 minutes of colcemid treatment (50 ng/ml) followed by an 18 minute incubation in 0.075 M KCl at 37°C and multiple changes of Carnoy fixative (3:1 methanol: glacial acetic acid). Cells were dropped onto slides and baked for one hour at 60°C before Giemsa banding or FISH protocols were carried out. Chromosome preparations and G-banding were done by standard methods.
YAC and BAC probes that map to fragile site regions were used for FISH analysis, following standard protocols [46]. YAC 850A6 was used to detect FRA3B and BAC 264L1 (RP-11) was used to detect FRA16D. Probes were labeled with biotin-14-dATP or digoxigenin-11-dUTP using a BioNick Translation Kit (Invitrogen). FISH signals were visualized by incubation with fluorescein isothiocyanate (FITC)-conjugated avidin-DCS and fluorescein-conjugated anti-avidin IgG (Vector Laboratories). Chromosomes were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, Vector Laboratories). Since the FISH probes are smaller than CFSs, chromosome gaps or breaks with a FISH signal immediately proximal, distal, or crossing the break were scored as an induced CFS. FISH results were analyzed with a Zeiss Axioscope epifluorescence microscope and Quips PathVysion imaging software (Vysis Inc).
2.2 Immunocytochemistry
For the detection of RPA2 nuclear foci, cells were fixed in 4% w/v paraformaldehyde at 4°C followed by permeabilization in 0.3% v/v Triton X-100 in PBS. Fixed cells were incubated with anti-RPA2 antibody (RPA34-19, Calbiochem) in 5% v/v goat serum, 0.1% v/v NP-40, in PBS for 2 h, washed three times in PBS and incubated for 1 h at room temperature with a mouse-specific fluorescein-conjugated secondary antibody (Molecular Probes). Cells were counterstained and mounted in vectashield plus 406-diamidine-2-phenylindole dihydrochloride (DAPI) (Vector Laboratories). Cells were visualized and images acquired using a Zeiss Axioscope epifluorescence microscope with Quips PathVysion imaging software (Vysis Inc.).
2.3 ATR depletion
HCT116 ATRflox/− cells, which allow for cre-lox mediated removal of ATR, were obtained from Dr. Stephen J. Elledge (Harvard Medical School, Boston, MA) and maintained in DMEM medium + 10% FBS + 200 μg/ml G418. Expression of cre recombinase in these cells was accomplished through infection with adenovirus AdCre1, which was obtained from Dr. Frank Graham (McMaster University, Hamilton, Ontario, Canada) as previously described [27,47].
2.4 Western Blots
Cell lysates were prepared by resuspending cell pellets in SDS-lysis buffer followed by sonication. NuPAGE 4–12% Bis-Tris, 10% Bis-Tris, or 3–8% Tris-acetate gels were used to resolve proteins. Whole-cell lysate (25 ug) was loaded per lane. Gels were transferred to PVDF membrane (Millipore Inc.) using a Trans-Blot SD Semi-Dry Transfer system (Bio-Rad Laboratories). Antibody hybridization and chemiluminescence detection were performed according to standard protocols. CHK1 protein was detected with sc-8408 (Santa-Cruz Biotechnology Inc.), CHK2 protein was detected with ab8108 (Novus Biologicals), RPA2 protein was detected with RPA34-19 (EMD), CHK1 phosphorylation on Ser317 was detected with Phosho-Chk1 (Ser317) (Cell Signaling), CHK2 phosphorylation on Thr68 was detected with Phospho-Chk2 (Thr68) (Cell Signaling), and RPA2 phosphorylation on Ser4/8 was detected with BL647 (Bethyl Laboratories, Inc.). PCNA protein was detected with sc-56 (Santa-Cruz Biotechnology Inc.). ATR was detected with a rabbit anti-ATR antibody (Abcam, Inc.). HRP-conjugated anti-mouse and anti-rabbit antibodies were obtained from Amersham Biosciences, Piscataway, NJ, USA. Protein bands were quantitated using Scion Image software (Scion Corp.).
2.5 Cell Cycle Profiles
UML-49 lymphoblasts were grown for 24 hours in medium containing 0.4uM APH, 30nM CPT, or both. After a 15 min treatment with 30 μM BrdU, cells were harvested, resuspended in PBS, fixed with 70% ethanol at −20°C, treated with RNase A (Roche Applied Science) and stained with propidium iodide (Sigma). Flow cytometric analysis to determine cell cycle profiles were performed using a Coulter Elite ESP Cell Sorter.
2.6 Statistical Analysis
Total gaps and breaks data were analyzed using the Student’s t-Test for equal or unequal variance. Variance for each data set was determined using the sample variance F-test. Fisher’s exact test (two sided) was used for analysis of specific fragile site induction data.
3. Results
3.1 CPT reduces APH-induced common fragile site breaks
To determine if TopoI plays a role in CFS stability, we treated normal human UML-49 lymphoblastoid cells with 0.4 μM APH for 24 hours, a standard dose that induces CFS breaks but still allows cell division, plus varying doses of the TopoI inhibitor, CPT. Since CFSs are detected on metaphase chromosomes from dividing cells, replication cannot be completely inhibited experimentally. As a result, we set out to only partially inhibit TopoI, thus allowing replication and cell divisions to continue, albeit at lower levels. To achieve partial TopoI inhibition, very low concentrations of CPT were used, ranging from 3 nM to 30 nM. Two independent experiments were performed and scored for chromosomal gaps and breaks.
When treated with 0.4 μM APH alone, UML-49 lymphoblastoid cells showed an average of 2.3 gaps and breaks per cell, compared with a mean of 0.05 in untreated cells (Figure 1A). When cells were treated with increasing doses of CPT alone, a modest number of gaps and breaks was observed in a dose dependent manner, with a maximum of 0.61 per cell at 15 nM CPT, consistent with reports showing that CPT induces DNA breaks [42]. At doses higher than 30 nM, replication was completely arrested and no metaphases were observed. However, when cells were treated with CPT in addition to APH, a decrease in gaps and breaks was observed, relative to cells treated with APH alone. This reduction in total gaps and breaks was dependent on the dose of CPT, with a maximum reduction seen in cells treated with 30 nM CPT. These cells were characterized by an average of only 0.48 gaps and breaks per cell, a five-fold, statistically significant (p < 0.001) decrease compared to APH treatment alone.
Figure 1.

Low doses of CPT reduce the frequency of APH-induced common fragile site breaks. (A) Average total chromosomal gaps and breaks per cell in UML-49 cells after 18 h in the presence (gray) or absence (white) of 0.4 μM APH and 0 to 30 nM CPT; n= 80 metaphases for each data set except for cells treated with only 30 nM CPT (asterisk), in which case n=8 due to very few metaphases being present for scoring. Error bars indicate the 95% confidence interval. (B) Frequency (%) of gaps and breaks at specific fragile sites FRA3B and FRA16D in UML-49 cells after 18 h in the presence or absence of 0.4 μM APH and 0 to 30 nM CPT; n= 96 sites examined. Fragile sites were identified by G-banding. Frequency of fragile-site induction is presented as the percentage of chromosome 3 or 16 homologs with breaks at FRA3B (white) or FRA16D (gray), respectively. Error bars indicate the 95% confidence interval. (C) Average total chromosomal gaps and breaks per cell in LC-1 cells after 18 h in the presence (gray) or absence (white) of 0.3 μM APH and 0 or 60 nM CPT; n= 80 metaphases for each data set. Error bars indicate the 95% confidence interval. (D) Frequency (%) of gaps and breaks at specific fragile sites FRA3B and FRA16D in LC-1 cells after 18 h in the presence or absence of 0.3 μM APH and 0 or 60 nM CPT; n= 100 sites examined. Fragile sites were identified by G-banding. Frequency of fragile-site induction is presented as the percentage of chromosome 3 or 16 homologs with breaks at FRA3B (white) or FRA16D (gray), respectively. Error bars indicate the 95% confidence interval.
To verify that the reduction in total gaps and breaks by CPT co-treatment corresponds to a reduction in APH-induced CFS breaks, metaphases from APH- and CPT-treated UML-49 cells were G-banded and the location of each break scored from two independent experiments. Results are shown for breaks at FRA3B (3p14) and FRA16D (16q23), the two most frequently-broken CFSs in the genome (Figure 1B). APH-induced breakage at these two CFSs was reduced in a dose-dependent manner by co-treatment with CPT, consistent with the results seen with total gaps and breaks. In cells co-treated with 0.4 μM APH and 30 nM CPT, FRA3B and FRA16D breaks were reduced 8-fold (p < 0.001) and 6-fold (p < 0.001), respectively, compared to cells treated with 0.4 μM APH alone. This treatment reduced FRA3B and FRA16D breakage to levels not significantly different from those seen in untreated cells (p=0.50 and 0.06, respectively).
These experiments were repeated, in duplicate, in primary human blood lymphocytes (LC-1) using 0.3 μM APH and 60 nM CPT with similar results. APH and CPT doses varied from those used with UML-49 due to differences in drug response between cell types. In lymphoblastoid cells, replication was completely arrested and no metaphases were observed at doses higher than 60 nM. A 4-fold reduction in APH-induced total gaps and breaks on G-banded chromosomes was observed when these cells were treated with both APH and CPT, as compared to APH-treatment alone (p < 0.001; Figure 1C). Consistent with this result, LC-1 cells treated with APH and CPT showed a ten-fold reduction in breaks at FRA3B (p < 0.001), compared to APH-treatment alone, and FRA16D breaks were reduced to zero (p < 0.001; Figure 1D).
All sites of chromosome gaps and breaks seen in UML-49 lymphoblasts treated with 0.4 μM APH, 30 nM CPT, or both are listed in Table 1. Ninety-three percent of all gaps and breaks induced by APH were found at the 32 CFSs that account for 93% of all gaps and breaks in normal human lymphocytes [16], with the FRA3B and FRA16D sites showing the highest incidence of breaks. While a slight increase in total gaps and breaks was seen in cells treated with low dose CPT alone, as compared to untreated cells, the location of these breaks did not correlate with CFSs. Instead, they appeared to be random, indicating that CPT alone is not an effective inducer of CFS breaks. Co-treatment of cells with APH and CPT did not completely eliminate breaks at CFSs, although almost half the low numbers of breaks seen under these conditions were at apparently random sites, compared to 93% at CFSs in APH treated cells. In addition, breaks at the most frequently observed CFSs, FRA3B and FRA16D, were almost completely eliminated in co-treated cells.
Table 1.
List of all chromosome bands and associated fragile sites with breaks in UML-49 cells after treatment with 0.4 μM APH, 30 nM CPT, or both.
| G band | APH (0.4uM)a | CPT (30nM)a | APH + CPTa | Associated Fragile Site |
|---|---|---|---|---|
| 3p14.2 | 24/96 | 1/96 | 2/96 | FRA3Bc |
| 16q23.2 | 32/96 | 0/96 | 5/96 | FRA16Dc |
| 7q31.2 | 5/96 | 0/96 | 3/96 | FRA7Gc |
| 7q32.3 | 0/96 | 0/96 | 1/96 | FRA7Hc |
| Xp22.3 | 1/96 | 0/96 | 0/96 | FRAXBc |
| 1p36.1-36.3 | 0/96 | 0/96 | 1/96 | FRA1A |
| 1p22.1-22.3 | 1/96 | 0/96 | 0/96 | FRA1D |
| 2q21.3 | 1/96 | 0/96 | 0/96 | FRA2F |
| 2q31 | 0/96 | 0/96 | 1/96 | FRA2Gc |
| 2q37.3 | 1/96 | 0/96 | 0/96 | FRA2J |
| 3q25.1-25.3 | 1/96 | 0/96 | 0/96 | FRA3D |
| 4q31.1 | 0/96 | 0/96 | 1/96 | FRA4C |
| 6q13 | 1/96 | 0/96 | 0/96 | FRA6D |
| 6q21 | 0/96 | 1/96 | 0/96 | FRA6Fc |
| 8q22.1 | 0/96 | 1/96 | 0/96 | FRA8B |
| 10q25.2 | 0/96 | 1/96 | 0/96 | FRA10E |
| 10q11.2 | 0/96 | 0/96 | 1/96 | FRA10G |
| 11p15.1 | 0/96 | 0/96 | 1/96 | FRA11C |
| 13q32 | 1/96 | 0/96 | 0/96 | FRA13D |
| 19q13.1-13.3 | 0/96 | 1/96 | 0/96 | FRA19A |
| 1p13.1-13.3 | 0/96 | 0/96 | 1/96 | - |
| 1q31 | 0/96 | 1/96 | 0/96 | - |
| 2p23 | 0/96 | 1/96 | 0/96 | - |
| 2q23 | 0/96 | 0/96 | 1/96 | - |
| 2q31 | 0/96 | 0/96 | 1/96 | - |
| 3p25.1-25.3 | 0/96 | 1/96 | 0/96 | - |
| 4q31.1-31.3 | 0/96 | 1/96 | 0/96 | - |
| 5q33.1-33.3 | 0/96 | 0/96 | 1/96 | - |
| 7p11.2 | 0/96 | 1/96 | 0/96 | - |
| 8q23 | 0/96 | 1/96 | 0/96 | - |
| 10p11.2 | 0/96 | 0/96 | 1/96 | - |
| 10q11.2 | 0/96 | 0/96 | 1/96 | - |
| 12p12.1-12.3 | 0/96 | 0/96 | 1/96 | - |
| 13q14.1-14.3 | 0/96 | 1/96 | 0/96 | - |
| Total # of breaks | 68 | 12 | 23 | |
| % of breaks at fragile sitesb | 93% | 25% | 57% | |
| % of breaks at non-fragile sites | 0% | 75% | 48% | |
Number of breaks at indicated band seen in 96 chromosomes.
Percentage of total breaks occurring at the 32 fragile sites that account for 93% of all gaps and breaks in human lymphocytes.
Fragile sites whose breakpoints have been defined by FISH.
3.2 CPT treatment during DNA replication is required for reduction of fragile site breaks
We hypothesized that TopoI inhibition must occur during replication perturbation by APH in order to reduce CFS induction. To test this hypothesis, UML-49 cells were treated for 18 hours with 0.4 μM APH and 30 nM CPT added 1, 2, 6, and 18 hours prior to cell harvest. We found that cells treated with CPT for shorter durations than APH showed levels of gaps and breaks similar to those seen with APH alone (Figure 2A). The only samples in which total gaps and breaks were significantly reduced as compared to cells treated with APH alone were those that were concurrently treated with CPT, with a small effect seen at 6 hours (p = 0.01) and the full effect seen with co-treatment for the entire 18 hour time course (p < 0.001; Figure 2A). Similar results were obtained when these cells were evaluated for breakage at the specific CFSs, FRA3B and FRA16D (Figure 2B). The same experiments were carried out using primary lymphocytes (LC-1), with similar results (Figure 2C and 2D). These results indicate that CPT treatment during DNA replication is required for reduction of APH-induced CFS breaks, suggesting that CPT acts to prevent the formation of CFS breaks, rather than acting on a downstream event.
Figure 2.

Concurrent CPT treatment is required to reduce the frequency of APH-induced common fragile site breaks. (A) Average total chromosomal gaps and breaks per cell in UML-49 cells after 18 h in the presence of 0.4 μM APH and 1 to 18 hours in the presence of 30 nM CPT; n= 42–60 metaphases for each data set. Error bars indicate the 95% confidence interval. (B) Frequency (%) of gaps and breaks at specific fragile sites FRA3B and FRA16D in UML-49 cells after 18 h in the presence of 0.4 μM APH and 1 to 18 hours in the presence of 30 nM CPT; n= 80 sites examined. Fragile sites were identified by G-banding. Frequency of fragile-site induction is presented as the percentage of chromosome 3 or 16 homologs with breaks at FRA3B (white) or FRA16D (gray), respectively. Error bars indicate the 95% confidence interval. (C) Average total chromosomal gaps and breaks per cell in LC-1 cells after 18 h in the presence of 0.3 μM APH and 1 to 18 hours in the presence of 60 nM CPT; n= 42–60 metaphases for each data set. Error bars indicate the 95% confidence interval. (D) Frequency (%) of gaps and breaks at specific fragile sites FRA3B and FRA16D in LC-1 cells after 18 h in the presence of 0.4 μM APH and 1 to 18 hours in the presence of 60 nM CPT; n= 100 sites examined. Fragile sites were identified by G-banding. Frequency of fragile-site induction is presented as the percentage of chromosome 3 or 16 homologs with breaks at FRA3B (white) or FRA16D (gray), respectively. Error bars indicate the 95% confidence interval.
3.3 Betulinic acid treatment reduces CFS breaks in APH-treated cells
Since CPT stabilizes the TopoI cleavage complex, leading to stabilized DNA breaks, it is possible that the induction of DNA breaks is a key step in the prevention of APH-induced CFS breakage by CPT, for example, by inducing cell cycle checkpoints or stimulating DNA repair. Betulinic acid is a TopoI inhibitor that acts by preventing the interaction of TopoI with DNA, blocking the formation of the cleavage complex [48,49]. As a result, betulinic acid inhibits TopoI activity without inducing DNA breaks. To determine if the TopoI-DNA cleavage complex, and associated DNA breakage is required for reduction of CFS breaks, we treated normal human UML-49 lymphoblastoid cells with 0.4 μM APH plus varying doses of betulinic acid for 24 hours in two independent experiments. As with CPT, very low concentrations of betulinic acid were used in order to only partially inhibit TopoI activity.
Cells treated with 0.4 μM APH alone showed an average of 1.0 breaks per metaphase, compared to a mean of 0.04 in untreated cells (Figure 3A). As was the case with CPT cotreatment, when APH-treated cells were cotreated with betulinic acid, a dose-dependent reduction in chromosome breaks was observed, with a 2.5-fold reduction at 0.3 μM betulinic acid to 0.4 breaks per cell. This difference is statistically significant (p < 0.001). No change in the baseline chromosome breakage was observed at any dose of betulinic acid, consistent with this drug’s lack of DNA damage induction.
Figure 3.

Low doses of betulinic acid reduce the frequency of APH-induced common fragile site breaks. (A) Average total chromosomal gaps and breaks per cell in UML-49 cells after 18 h in the presence (gray) or absence (white) of 0.4 μM APH and 0 to 3 μM betulinic acid (BA); n= 50 metaphases for each data set. Error bars indicate the 95% confidence interval. (B) Frequency (%) of gaps and breaks at specific fragile sites FRA3B and FRA16D in UML-49 cells after 18 h in the presence or absence of 0.4 μM APH and 0 to 3 μM betulinic acid (BA); n= 92–105 sites examined. Fragile sites were identified by FISH. Frequency of fragile-site induction is presented as the percentage of chromosome 3 or 16 homologs with breaks at FRA3B (white) or FRA16D (gray), respectively. Error bars indicate the 95% confidence interval.
To verify that the reduction in APH-induced total chromosome breaks after betulinic acid treatment corresponds to a reduction in CFS breakage, we evaluated metaphases from cells treated with 0.4 μM APH alone or APH plus 0.3 μM betulinic acid for breakage specifically at FRA3B and FRA16D by FISH (Figure 3B). As with total gaps and breaks, breaks at these specific CFSs were reduced after betulinic acid treatment. APH-induced breakage at FRA3B and FRA16D were reduced 3- to 4-fold, a statistically significant reduction (FRA3B p=0.02; FRA16D p < 0.001).
It should be noted that betulinic acid also inhibits topoisomerase IIα by preventing its binding to DNA [48]. However, ICRF-193, which inhibits TopoII in a manner analogous to betulinic acid, did not have an effect on CFS breakage in the presence or absence of APH (data not shown), suggesting that betulinic acid is not reducing CFS breaks via its effect on TopoII.
These experiments indicate that the reduction in APH-induced CFS breakage after CPT treatment is not the result of the stabilization of the TopoI cleavage complex, but rather the result of the inhibition of TopoI-dependent unwinding of supercoiled DNA during replication.
3.4 APH-induced RPA2 phosphorylation and nuclear focus formation are reduced in the presence of CPT
RPA is normally associated with replication forks, but forms visible foci at sites of ssDNA after stalled replication and DNA damage in an ATR-dependent manner [30,50,51]. RPA2 nuclear focus formation and hyperphosphorylation is stimulated by APH-mediated replication arrest [30,52]. To determine if CPT is able to reduce the formation of APH-induced RPA2 nuclear foci, HeLa cells were grown for 24 hours in the presence of 4 μM APH, 300 nM CPT, or both and evaluated for RPA2 nuclear foci (Figure 4A and 4B). The low doses of APH used in our chromosome breakage studies did not induce detectable levels of RPA2 phosphorylation or focus formation due to limitations of the assays used for detection. Therefore, we used higher doses of APH and CPT in these experiments than in chromosome breakage experiments. However, it is important to note that the relative amounts of APH and CPT (a ~13:1 molar ratio) that maximally reduced CFS breaks also reduce phosphorylation and focus formation, suggesting that the same mechanisms are invoked in all of these results.
Figure 4.

CPT reduces APH-dependent RPA2 nuclear focus formation and RPA2 phosphorylation. (A) Examples of immunocytochemistry performed with a RPA2-specific antibody in HeLa cells following 24 hour treatment with 4.0μM APH, 300nM CPT, or both. Nuclei are stained with DAPI. (B) Quantitation of the number of nuclei with >5 nuclear RPA2 foci after drug treatments (n > 200 nuclei per treatment). (C) Western blots showing the induction by APH of RPA2 phosphorylation as evidenced by a gel shift (panel 1) and by detection of phosphorylation on serines 4 and 8 (panel 2). Addition of CPT reduces the APH-depended phosphorylation of RPA2 in a dose-dependent manner. Tubulin is included as a loading control. Concentrations of APH and CPT, added for 24 hours, are indicated. A 10 μM CPT treatment for 2 hours was included as a positive control. (D) Quantitation of RPA2 phosphorylation in (C), normalized to tubulin.
Only 3.4% of untreated cells were characterized by RPA2 nuclear foci, similar to previous studies [30,51]. In contrast, upon APH or CPT treatment, 21% and 36% of cells, respectively, had focal RPA2 staining. When cells were grown in the presence of both drugs, only 9.5% of nuclei contained RPA2 foci, a significant reduction compared to APH treated cells (p = 0.001) (Figure 4A and B). These experiments were also conducted using nonimmortalized, primary human fibroblasts, with similar results (Supplemental Figure 1).
We next examined RPA2 phosphorylation via western blot analysis in UML-49 cells following exposure to APH and/or CPT (Figure 4C). RPA2 is a target of ATR and DNA-PK, and its phosphorylation reflects ATR-activation [45,51,53,54]. Consistent with previous results [30], APH induces RPA2 phosphorylation, as evidenced by a gel shift, as well as by detection of a band with an antibody specific to RPA2 phosphorylated on serines 4 and 8 (Figure 4C, Lane 5). Similarly, CPT was also found to phosphorylate RPA2, consistent with the literature [45,51,55] (Figure 4C, Lanes 2–4). However, when APH and CPT were added together, at the same stoichiometry used to reduce APH-induced CFS breaks, RPA2 phosphorylation was attenuated (Figure 4C, Lane 8). These data suggest that the two drugs combined result in a reduction of ssDNA, and thus a reduction of ATR activation, compared to treatment with either drug alone.
3.5 APH-induced phosphorylation of CHK1, but not CHK2, is reduced in the presence of low dose CPT
Treatment of cells with high concentrations of CPT has previously been demonstrated to activate the ATR-dependent DNA damage cell cycle checkpoint, as detected by phosphorylation of CHK1 [56,57]. Our RPA2 phosphorylation results suggest that CPT cotreatment may downregulate ATR after APH. Because ATR and CHK1 have previously been shown to play a critical role in suppressing CFS breakage [27,29], we examined this further. To test whether CPT has an effect on APH-induced ATR activation, UML-49 cells were treated with 0.4 μM APH and various doses of CPT, and levels of CHK1 and CHK2 phosphorylation were examined by western blot analysis. In agreement with previous observations [29], 0.4 μM APH resulted in a 11-fold increase in CHK1 phosphorylation on residue S317, compared to untreated controls (Figure 5A). Addition of CPT reduced the APH-induced CHK1 phosphorylation in a dose-dependent manner, with phosphorylation levels partially reduced at 30 nM CPT, and the lowest levels of phosphorylation seen at 150 nM CPT (Figures 5A and 5B). In addition, 30 nM CPT treatment alone was not sufficient to induce high levels of CHK1 phosphorylation. In contrast, addition of CPT to APH-treated UML-49 cells had only minor effects on phosphorylation of CHK2 on residue T68 (Figures 5A and 5B), consistent with ATM activation due to double strand break formation. These results show a decrease, rather than an increase, in CHK1-phosphorylation after combined APH/CPT treatment as compared to APH alone that is consistent with decreased chromosome breaks and suggest that CPT is attenuating ATR activation by APH.
Figure 5.

CPT reduces APH-induced phosphorylation of CHK1, but not CHK2. (A) Western blots showing the induction by APH of CHK1 and CHK2 phosphorylation on residues S317 and T68, respectively. Addition of CPT reduces the APH-dependent phosphorylation of CHK1, but not CHK2. Tubulin is included as a loading control. Concentrations of APH and CPT, added for 24 hours, are indicated. A 10 μM CPT treatment for 2 hours was included as a positive control. (B) Quantitation of CHK1 S317 and CHK2 T68 phosphorylation in (A), normalized to CHK1 and CHK2, respectively.
3.6 ATR-deficient cells treated with CPT have a reduced incidence of spontaneous total and CFS breaks
The RPA and CHK1 phosphorylation results described above suggest that CPT cotreatment is reducing APH-dependent ATR activation. Cells deficient in ATR show an elevated frequency of spontaneous chromosome gaps and breaks at CFSs, without APH treatment, indicating that there is a baseline level of replication stress occurring at these loci [27]. To determine if TopoI inhibition affects the spontaneous CFS breakage seen in cells with reduced ATR, we treated ATR-deficient cells with several doses of CPT and analyzed them for total gaps and breaks as well as for breaks at FRA3B and FRA16D. Cre-lox mediated removal of ATR from HCT116 ATRflox/− cells was performed and reduction in protein expression was confirmed by western blot (Figure 6A). As previously observed [27], loss of ATR in the absence of APH results in an increase in spontaneous chromosomal gaps and breaks, including gaps and breaks at CFSs (Figure 6B). When these cells are treated with 1 pM, 10 pM, or 100 pM CPT, a reduction in total gaps and breaks was seen. At higher doses of CPT, an increase in random breakage occurred in ATR-deficient cells, likely because ATR is required for checkpoint induction after CPT-induced damage [43,45,51,55–58]. The greatest reduction in breakage was seen with 1pM CPT, in which a five-fold, significant (p < 0.001) decrease is seen relative to cells without CPT (Figure 6B). This trend was also seen when breakage at FRA3B and FRA16D was evaluated by FISH. Ten percent of FRA3B sites had a gap or break in ATR-deficient cells while cells cotreated with 1pM CPT showed no breaks at FRA3B, a significant (p = 0.01) reduction (Figure 6C). While breaks at FRA16D were also reduced, the frequency of spontaneous breakage at FRA16D in ATR-deficient cells was too low in these experiments (~5%) to determine significance. These results support the hypothesis that CPT is affecting the dynamics of DNA unwinding by TopoI at CFSs.
Figure 6.
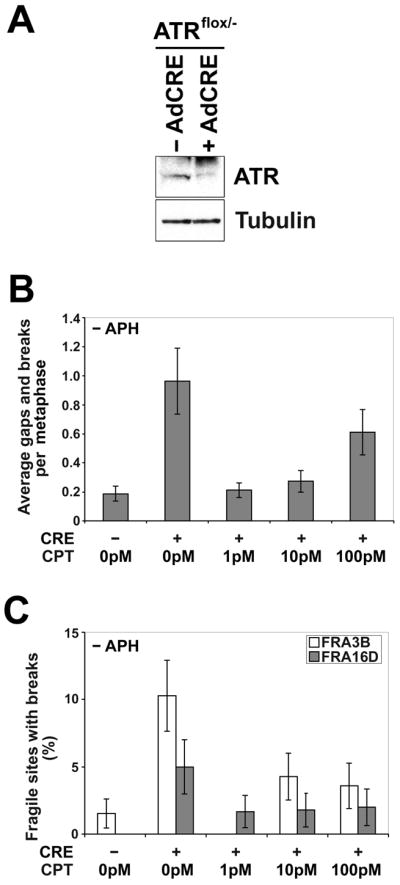
Low doses of CPT reduce the frequency of spontaneous common fragile site breaks in ATR-deficient cells. (A) Western blot showing depletion of ATR after ATRflox/− cells are infected with adenovirus AdCre1. (B) Average spontaneous total chromosomal gaps and breaks per cell in ATRflox/− cells after 18 h in the presence of 0 to 100 pM CPT; n= 80 metaphases for each data set. Error bars indicate the 95% confidence interval. (C) Frequency (%) of spontaneous gaps and breaks at specific fragile sites FRA3B and FRA16D in ATRflox/− cells after 18 h in the presence of 0 to 100 pM CPT; n=51–75 sites examined. Fragile sites were identified by FISH. Frequency of fragile-site induction is presented as the percentage of chromosome 3 or 16 homologs with breaks at FRA3B (white) or FRA16D (gray), respectively. Error bars indicate the 95% confidence interval.
3.7 CPT does not alter the cell cycle profile of APH-treated cells
To investigate if the reduction in CFS breakage after CPT treatment is due to changes in cell cycle progression, a cell cycle profile of UML-49 cells was determined by flow cytometry after treatment with APH and CPT. As expected, treatment with 0.4 μM APH results in an increase in the proportion of cells in S phase with a concomitant reduction in the number of cells in G1, indicating that cells continue to enter S phase, but proceed through it slowly (Figure 7A–B). In contrast, when cells are treated with 30 nM CPT alone, there is an increase in the percentage of cells in G2, with a slight reduction of cells in G1 and S. In particular, there is a decrease in the number of cells in early-S. This change in the cell cycle profile can be explained by the fact that CPT-induced DNA damage results in checkpoint activation and G2 arrest [59]. However, when cells are treated with both APH and CPT, the CPT-induced accumulation of G2 cells is prevented. Instead, the cell cycle profile closely resembles that of APH treatment alone, including the presence of cells in early S, indicating that combined treatment with both drugs does not induce a stronger checkpoint response than does APH alone.
Figure 7.

CPT treatment does not alter the cell cycle profile of APH-treated cells. (A) Cell cycle profiles of UML-49 treated with the indicated doses of APH and CPT. Cells were pulsed for 15 minutes with 30 μM BrdU and stained with propidium idodide. To the right of each FACs analysis is a representative histogram showing cell cycle profile. (B) Quantitation of the proportion of cells in the G1, S, and G2 phases of the cell cycle. Addition of drugs is indicated under each group of data. Bars represent the mean value of two experiments. Error bars indicate standard deviation. (C) Mitotic index after treatment with the indicated doses of APH and CPT, as measured by counting metaphase spreads on slides; n = 1980–2034. (D) Quantitation of the number of dead cells as seen in the lower left quadrant of the cell cycle profiles in (A). Addition of drugs is indicated under each group of data. Bars represent the mean value of two experiments. Error bars indicate standard deviation.
We also examined the mitotic index of cells treated with APH and CPT (Figure 7C). We found that cells treated with 0.4 μM APH show a slight decrease in mitotic index compared to untreated cells. In contrast, cells treated with 30 nM CPT demonstrate a decrease in mitotic index compared to untreated cells. Cells treated with both APH and CPT have a mitotic index that is intermediate between either drug alone, indicating that cotreatment of these two drugs has less of a cell cycle effect than CPT treatment alone.
There is an increase in the number of dead cells seen in the lower left quadrant of the cell cycle profiles in drug treated UML-49 cells compared to untreated controls (Figure 7A and D). Cell populations treated with 0.4 μM APH, 30 nM CPT, or both demonstrated a roughly twofold increase in such dead cells, raising the possibility that treated cells are undergoing increased levels of apoptosis. We found that 30 nM CPT treatment for 24 hours in the presence or absence of 0.4 μM APH induces PARP cleavage, an indicator of apoptosis (Data not shown). APH treatment alone did not induce apoptosis. While it is possible that CPT-induced apoptosis of cells expressing CFSs is responsible for the observed decrease in CFS breakage, we think it is unlikely for several reasons. First, if this were the case, it would be expected that cells treated with both APH and CPT would have a much higher incidence of cell death than cells treated with APH alone, which was not observed (Figure 7D). In addition, CPT is a poor inducer of apoptosis in the presence of CHK1 at 20nM, a comparable dose to ours [60].
3.8 APH-induced monoubiquitination of PCNA is reduced in the presence of low dose CPT
PCNA is monoubiquitinated in response to ssDNA under conditions of replication stress, including that induced by low doses of APH [61–64]. It has been shown that uncoupling of the replicative polymerase and helicase activities is required for the APH-induced monoubiquitination of PCNA by the Rad18 E3 ligase [61], and that this monoubiquitination is independent of ATR-mediated checkpoint activation [61,65]. It is believed that PCNA monoubiquitination is initiated by the accumulation of ssDNA and RPA2 at stalled replication forks [61,65]. To determine if CPT can reduce APH-induced PCNA monoubiquitination, UML-49 cells were treated with 0.4 μM APH and various doses of CPT, and levels of PCNA monoubiquitination were examined by western blot analysis. We found that treatment with 0.4 μM APH induced PCNA monoubiquitination, compared to untreated controls (Figure 8), consistent with an increase in ssDNA. Addition of CPT decreased the APH-induced PCNA monoubiquitination in a dose-dependent manner, with levels reduced to approximately untreated levels at 30 and 150 nM CPT (Figure 8). In addition, it was found that 30 nM CPT treatment alone was not sufficient to induce PCNA monoubiquitination. These results suggest that co-treatment with CPT reduces the amount of ssDNA induced by APH, thus preventing PCNA monoubiquitination.
Figure 8.

CPT reduces APH-induced monoubiquitination of PCNA. (A) Western blots showing the monoubiquitination of PCNA after low-dose APH treatment. Addition of CPT reduces PCNA monoubiquitination in a dose-dependent manner. Tubulin is included as a loading control. Concentrations of APH and CPT, added for 24 hours, are indicated. (B) Quantitation of PCNA monoubiquitination in (A), normalized to PCNA.
4. Discussion
We have shown that low doses of CPT or betulinic acid, specific inhibitors of TopoI, result in a dose-dependent reduction of gaps and breaks at CFSs induced by APH treatment or ATR-deficiency, almost to levels seen in untreated controls. The reduction in CFS chromosome breakage by CPT-mediated TopoI inhibition is accompanied by a decrease in APH-induced CHK1 phosphorylation, PCNA monoubiquitination, RPA2 phosphorylation and RPA2 nuclear focus formation, indicating decreased amounts of ssDNA. These results demonstrate that TopoI unwinding of supercoiled DNA is necessary for fragility at CFSs.
CPT reversibly binds to the TopoI cleavage complex, physically blocking religation of the nicked DNA [38–40]. At high doses of CPT, stabilized cleavage complexes cause toxicity by inducing irreversible DNA breaks during replication [42]. It has been shown that co-treatment of cells with high doses of APH reduces the toxicity of higher doses of CPT than used here by completely blocking DNA replication via inhibition of DNA polymerase activity, thus preventing the conversion of TopoI cleavage complexes into DNA double strand breaks by stopping the replication fork from catching up to the nicked DNA [43–45]. At these high doses, these chemicals also block cell cycle progression. At low doses, CPT and betulinic acid should only partially inhibit TopoI, with individual TopoI molecules being transiently inhibited, resulting in a reduction in the movement rate of the replication complex. Thus, at high doses, APH decreases CPT-induced random DNA breakage and at low doses, CPT and betulinic acid decrease APH-induced CFS breaks, but through different mechanisms. Treatment of cells with nanomolar doses of CPT still appears to result in modest amounts of DNA breaks, as seen on metaphase chromosomes in our experiments (Figure 1A and C). However, these CPT-induced breaks are found primarily at chromosome bands lacking CFSs, and appear to be random, suggesting that CPT alone has no specificity for CFSs or other loci (Table 1).
It is known that inhibition of TopoI with high dose CPT results in strong, ATR-dependent S-phase checkpoint activation and RPA2 phosphorylation, with inhibition of DNA synthesis persisting for hours after CPT removal [43,45,51,55–58]. Therefore, it is conceivable that low dose APH and CPT co-treatment reduces CFS breaks by more effectively inducing DNA damage and activation of cell cycle checkpoints than low-dose APH alone, thus allowing cells to repair the gaps and breaks at CFSs or preventing cells with CFS breaks from entering metaphase. If this were the case, betulinic acid would not be expected to reduce CFS breakage as it does not induce DNA breaks. In addition, treatment with both APH and CPT should result in higher checkpoint activation than either drug alone. We examined this possibility in a number of ways and several lines of evidence discount the possibility that CPT is reducing APH-induced CFS breakage through a checkpoint effect, including CHK1, RPA2, and CHK2 phosphorylation, RPA2 focus formation, the cell cycle profile of treated cells, and the reduction of APH-induced breakage by betulinic acid. Our results argue that low-dose CPT reduces, rather than increases checkpoint responses, and therefore that CPT is reducing the initial damage events.
We have previously shown that CHK1 is an essential component in the checkpoint pathways that govern CFS breakage on metaphase chromosomes [29]. When we co-treated cells with APH and CPT, CHK1 phosphorylation was reduced to levels similar to those seen in untreated cells (Figure 5). Similarly, low-dose CPT reduced, rather than increased, the amount of APH-induced RPA2 phosphorylation and focus formation (Figure 4), suggesting reduced ATR activation due to a reduction of ssDNA. Furthermore, we found that CPT reduces CFS breakage in ATR-deficient cells not treated with APH (Figure 6). We also examined the cell cycle profiles of cells treated with APH and/or CPT. Cells co-treated with APH and CPT did not show a change in cell cycle profile compared to cells treated only with APH (Figure 7). Since CPT does not reduce the polymerase inhibition caused by APH, it is not expected that cotreatment would restore the cell cycle profile to that seen in untreated cells.
It is possible that the reduction in APH-induced CFSs after CPT treatment is the result of a global reduction of DNA replication rates. The reduction in RPA foci and phosphorylation is consistent with reduced replication in general. Such a slowing, whether checkpoint dependent or independent, could provide additional time for repair of breaks. However, the fact that there is no change in the cell cycle profile between cells treated with APH alone or with APH and CPT suggests that this is not the case.
In addition to being involved in replication, TopoI plays a role in transcriptional elongation [35]. It is conceivable that this function of TopoI could play a role in the chromosome breakage events described here if, for example, transcription of associated genes is necessary for chromosome breakage at CFSs. While we cannot rule out this possibility, there is no direct evidence supporting a requirement for transcription in CFS breakage. Furthermore, cotreatment of cells with APH and DRB, an inhibitor of transcription [66], had no affect on APH-induced chromosome breakage (Supplemental Figure 2).
While early studies demonstrated DNA polymerase inhibition by APH in vitro [67–69], recent studies have shown its effects on DNA replication to be more complex. Walter and Newport [70], Pacek and Walter [71] and Byun et al. [57] have shown that APH treatment also results in the functional uncoupling of the replicative polymerase and helicase complexes in vitro. When Xenopus cell extracts were treated with high doses of APH to arrest DNA synthesis, the helicase complex continued to unwind DNA ahead of the replication fork, resulting in long stretches of ssDNA. Recent work has indicated that this uncoupling is both physical as well as functional, at least in vitro [72], and occurs in mammalian cells as well [73]. This uncoupling can be reduced by direct inhibition of helicase or treatment with CPT [70].
It is intriguing that low dose CPT reduces the APH-induced monoubiquitination of PCNA (Figure 8). PCNA is monoubiquitinated under conditions of replication stress, including that induced by low doses of APH [61–64]. Chang et al. [61] have shown that the uncoupling of polymerase and helicase activities is required for PCNA monoubiquitination in a checkpoint-independent manner, which likely results from the accumulation of ssDNA at stalled replication forks. The fact that CPT reduces APH-induced PCNA monoubiquitination suggests that the drug is reducing the amount of polymerase-helicase uncoupling in these cells. This result suggests the possibility that CPT is having its effect on reducing APH-induced fragile site breakage through a decrease in ssDNA formation via a reduction in polymerase-helicase uncoupling.
While it has been demonstrated that polymerase-helicase uncoupling in vitro leads to ATR-dependent checkpoint activation [57], the effect of ATR deficiency on this uncoupling is unknown. We have previously shown that ATR serves a critical role in maintaining CFS stability, even in the absence of any APH-induced replication stress [27]. Our observation that CPT treatment reduces the spontaneous CFS breaks seen in ATR-deficient cells suggests that polymerase-helicase uncoupling occurs not only in response to overt replication stress (e.g. APH treatment) but also can occur during normal, unperturbed DNA replication and that ATR-deficiency leads to an increase in this uncoupling. This baseline uncoupling appears to occur preferentially at CFSs, probably due to the difficulty in replicating these regions, and suggests a possible biological function for the inherent instability at these evolutionarily-conserved CFSs. Not only does polymerase-helicase uncoupling lead to ATR-dependent checkpoint activation, it appears to be a necessary event for such activation [57]. The fact that CFSs are particularly sensitive to polymerase-helicase uncoupling may indicate that CFS regions act as sensors for cell cycle progression, inducing checkpoint activation while damage is limited to a small number of sites.
Our results, together with earlier findings from in vitro studies of APH-induced polymerase-helicase uncoupling [57,70,71] suggest a model of polymerase-helicase uncoupling as an initial event leading to CFS breakage (Figure 9). According to this model, in a normal cell the helicase/TopoI complex progresses along the template chromosome, unwinding the DNA ahead of it. Immediately behind this complex, the DNA polymerase complex replicates the unwound DNA, leaving the amount of ssDNA at a minimum. When cells are treated with low dose APH, the polymerases slow or pause, leaving the helicase/TopoI complex to continue unwinding DNA ahead of it and resulting in long stretches of ssDNA that can activate the ATR-dependent checkpoint. This uncoupling may affect leading and lagging strand synthesis to different degrees, consistent with models in which lagging strand synthesis is more readily disrupted than leading strand synthesis [74–76]. These ssDNA regions may form secondary structures, such as hairpins or cruciforms at AT-rich sequences associated with CFSs, which can perturb replication further as the polymerases encounter them. While the majority of these perturbations are likely detected and resolved by the checkpoint and repair machinery, some escape and present themselves as gaps and breaks on metaphase chromosomes, particularly at CFSs. When the helicase/TopoI complex is partially inhibited with doses of CPT like those used in this study, this complex is also slowed. While DNA replication will be somewhat slowed globally as a result of these combined drug treatments, the restored balance between polymerase and helicase/TopoI progression rates will limit the formation of the ssDNA stretches and allow replication to finish with minimal difficulty. The data presented here, particularly the reduction in PCNA monoubiquitination after CPT and APH cotreatment, support this hypothesis.
Figure 9.

Model of the effects of low-dose CPT on APH-induced common fragile site breaks. (A) During normal DNA replication, the replicative polymerases, helicase, and TopoI all proceed along the DNA in a coupled fashion, keeping the amount of unwound ssDNA between the polymerases and the helicase to a minimum. (B) When replication occurs in the presence of APH, polymerase activity slows while the helicase/TopoI complex continues to unwind template DNA ahead of the replication fork. The resulting stretches of ssDNA may be more likely to form secondary structures, such as hairpins, that further perturb polymerase activity. These structures might occur on the leading strand, lagging strand, or both. While fork stalling at these sites will induce checkpoint activity, some will escape and appear on metaphases as chromosome breaks. Common fragile sites may be more likely to form such secondary structures, making them more likely to form breaks. (C) When cells are treated with both APH and CPT, both the polymerases and the helicase/TopoI complex are slowed. While replication proceeds more slowly under these conditions, the formation of large stretches of ssDNA and secondary structures is prevented, resulting in a reduction of common fragile site breaks.
These studies show that normal TopoI function is required for APH-induced CFS expression. It is interesting to note that we are treating cells with nanomolar concentrations of CPT, which are orders of magnitude lower than peak serum levels seen when CPT is used as a chemotherapeutic agent (Reviewed in [37]). CFSs provide an assay for studying subtle, low dose drug effects on replication dynamics, rather than the typical approach in which replication is completely disrupted. Our results described here provide new mechanistic insight into the replication dynamics at work at CFSs, the initial events required for CFS induction, the mechanisms of chromosomal damage following replication stress in mammalian cells, and the role of ATR in maintaining DNA replication fork stability at CFSs during normal replication.
Supplementary Material
Acknowledgments
We thank Christine Canman, Mats Ljungman, Niall Howlett, John Moran, and David Ferguson for helpful discussions. This work was supported by NIH grant CA43222 to T.W.G.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Glover TW, Arlt MF, Casper AM, Durkin SG. Mechanisms of common fragile site instability. Human molecular genetics. 2005;14(Spec No 2):R197–205. doi: 10.1093/hmg/ddi265. [DOI] [PubMed] [Google Scholar]
- 2.El Achkar E, Gerbault-Seureau M, Muleris M, Dutrillaux B, Debatisse M. Premature condensation induces breaks at the interface of early and late replicating chromosome bands bearing common fragile sites. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:18069–18074. doi: 10.1073/pnas.0506497102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohta M, Inoue H, Cotticelli MG, Kastury K, Baffa R, Palazzo J, Siprashvili Z, Mori M, McCue P, Druck T, Croce CM, Huebner K. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell. 1996;84:587–597. doi: 10.1016/s0092-8674(00)81034-x. [DOI] [PubMed] [Google Scholar]
- 4.Huang H, Qian J, Proffit J, Wilber K, Jenkins R, Smith DI. FRA7G extends over a broad region: coincidence of human endogenous retroviral sequences (HERV-H) and small polydispersed circular DNAs (spcDNA) and fragile sites. Oncogene. 1998;16:2311–2319. doi: 10.1038/sj.onc.1200202. [DOI] [PubMed] [Google Scholar]
- 5.Mishmar D, Rahat A, Scherer SW, Nyakatura G, Hinzmann B, Kohwi Y, Mandel-Gutfroind Y, Lee JR, Drescher B, Sas DE, Margalit H, Platzer M, Weiss A, Tsui LC, Rosenthal A, Kerem B. Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:8141–8146. doi: 10.1073/pnas.95.14.8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bednarek AK, Laflin KJ, Daniel RL, Liao Q, Hawkins KA, Aldaz CM. WWOX, a novel WW domain-containing protein mapping to human chromosome 16q23.3-24.1, a region frequently affected in breast cancer. Cancer research. 2000;60:2140–2145. [PubMed] [Google Scholar]
- 7.Ried K, Finnis M, Hobson L, Mangelsdorf M, Dayan S, Nancarrow JK, Woollatt E, Kremmidiotis G, Gardner A, Venter D, Baker E, Richards RI. Common chromosomal fragile site FRA16D sequence: identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Human molecular genetics. 2000;9:1651–1663. doi: 10.1093/hmg/9.11.1651. [DOI] [PubMed] [Google Scholar]
- 8.Arlt MF, Miller DE, Beer DG, Glover TW. Molecular characterization of FRAXB and comparative common fragile site instability in cancer cells. Genes, chromosomes & cancer. 2002;33:82–92. doi: 10.1002/gcc.10000. [DOI] [PubMed] [Google Scholar]
- 9.Morelli C, Karayianni E, Magnanini C, Mungall AJ, Thorland E, Negrini M, Smith DI, Barbanti-Brodano G. Cloning and characterization of the common fragile site FRA6F harboring a replicative senescence gene and frequently deleted in human tumors. Oncogene. 2002;21:7266–7276. doi: 10.1038/sj.onc.1205573. [DOI] [PubMed] [Google Scholar]
- 10.Callahan G, Denison SR, Phillips LA, Shridhar V, Smith DI. Characterization of the common fragile site FRA9E and its potential role in ovarian cancer. Oncogene. 2003;22:590–601. doi: 10.1038/sj.onc.1206171. [DOI] [PubMed] [Google Scholar]
- 11.Denison SR, Wang F, Becker NA, Schule B, Kock N, Phillips LA, Klein C, Smith DI. Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene. 2003;22:8370–8378. doi: 10.1038/sj.onc.1207072. [DOI] [PubMed] [Google Scholar]
- 12.Limongi MZ, Pelliccia F, Rocchi A. Characterization of the human common fragile site FRA2G. Genomics. 2003;81:93–97. doi: 10.1016/s0888-7543(03)00007-7. [DOI] [PubMed] [Google Scholar]
- 13.Rozier L, El-Achkar E, Apiou F, Debatisse M. Characterization of a conserved aphidicolin-sensitive common fragile site at human 4q22 and mouse 6C1: possible association with an inherited disease and cancer. Oncogene. 2004;23:6872–6880. doi: 10.1038/sj.onc.1207809. [DOI] [PubMed] [Google Scholar]
- 14.Savelyeva L, Sagulenko E, Schmitt JG, Schwab M. The neurobeachin gene spans the common fragile site FRA13A. Human genetics. 2006;118:551–558. doi: 10.1007/s00439-005-0083-z. [DOI] [PubMed] [Google Scholar]
- 15.Durkin SG, Glover TW. Chromosome Fragile Sites. Annu Rev Genet. 2007 doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- 16.Glover TW, Berger C, Coyle J, Echo B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Human genetics. 1984;67:136–142. doi: 10.1007/BF00272988. [DOI] [PubMed] [Google Scholar]
- 17.Jiang Y, Lucas I, Young DJ, Davis EM, Karrison T, Rest JS, Le Beau MM. Common fragile sites are characterized by histone hypoacetylation. Human molecular genetics. 2009;18:4501–4512. doi: 10.1093/hmg/ddp410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arlt MF, Durkin SG, Ragland RL, Glover TW. Common fragile sites as targets for chromosome rearrangements. DNA repair. 2006;5:1126–1135. doi: 10.1016/j.dnarep.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 19.Durkin SG, Ragland RL, Arlt MF, Mulle JG, Warren ST, Glover TW. Replication stress induces tumor-like microdeletions in FHIT/FRA3B. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:246–251. doi: 10.1073/pnas.0708097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arlt MF, Mulle JG, Schaibley VM, Ragland RL, Durkin SG, Warren ST, Glover TW. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. American journal of human genetics. 2009;84:339–350. doi: 10.1016/j.ajhg.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 22.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr, Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 23.Michael D, Beer DG, Wilke CW, Miller DE, Glover TW. Frequent deletions of FHIT and FRA3B in Barrett’s metaplasia and esophageal adenocarcinomas. Oncogene. 1997;15:1653–1659. doi: 10.1038/sj.onc.1201330. [DOI] [PubMed] [Google Scholar]
- 24.Huebner K, Hadaczek P, Siprashvili Z, Druck T, Croce CM. The FHIT gene, a multiple tumor suppressor gene encompassing the carcinogen sensitive chromosome fragile site, FRA3B. Biochimica et biophysica acta. 1997;1332:M65–70. doi: 10.1016/s0304-419x(97)00009-7. [DOI] [PubMed] [Google Scholar]
- 25.Bednarek AK, Keck-Waggoner CL, Daniel RL, Laflin KJ, Bergsagel PL, Kiguchi K, Brenner AJ, Aldaz CM. WWOX, the FRA16D gene, behaves as a suppressor of tumor growth. Cancer research. 2001;61:8068–8073. [PubMed] [Google Scholar]
- 26.O’Keefe LV, Richards RI. Common chromosomal fragile sites and cancer: focus on FRA16D. Cancer Lett. 2006;232:37–47. doi: 10.1016/j.canlet.2005.07.041. [DOI] [PubMed] [Google Scholar]
- 27.Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell. 2002;111:779–789. doi: 10.1016/s0092-8674(02)01113-3. [DOI] [PubMed] [Google Scholar]
- 28.Arlt MF, Xu B, Durkin SG, Casper AM, Kastan MB, Glover TW. BRCA1 is required for common-fragile-site stability via its G2/M checkpoint function. Molecular and cellular biology. 2004;24:6701–6709. doi: 10.1128/MCB.24.15.6701-6709.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Durkin SG, Arlt MF, Howlett NG, Glover TW. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene. 2006;25:4381–4388. doi: 10.1038/sj.onc.1209466. [DOI] [PubMed] [Google Scholar]
- 30.Howlett NG, Taniguchi T, Durkin SG, D’Andrea AD, Glover TW. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Human molecular genetics. 2005;14:693–701. doi: 10.1093/hmg/ddi065. [DOI] [PubMed] [Google Scholar]
- 31.Schwartz M, Zlotorynski E, Goldberg M, Ozeri E, Rahat A, le Sage C, Chen BP, Chen DJ, Agami R, Kerem B. Homologous recombination and nonhomologous end-joining repair pathways regulate fragile site stability. Genes & development. 2005;19:2715–2726. doi: 10.1101/gad.340905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hellman A, Rahat A, Scherer SW, Darvasi A, Tsui LC, Kerem B. Replication delay along FRA7H, a common fragile site on human chromosome 7, leads to chromosomal instability. Molecular and cellular biology. 2000;20:4420–4427. doi: 10.1128/mcb.20.12.4420-4427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palakodeti A, Han Y, Jiang Y, Le Beau MM. The role of late/slow replication of the FRA16D in common fragile site induction. Genes, chromosomes & cancer. 2004;39:71–76. doi: 10.1002/gcc.10290. [DOI] [PubMed] [Google Scholar]
- 34.Stivers JT, Harris TK, Mildvan AS. Vaccinia DNA topoisomerase I: evidence supporting a free rotation mechanism for DNA supercoil relaxation. Biochemistry. 1997;36:5212–5222. doi: 10.1021/bi962880t. [DOI] [PubMed] [Google Scholar]
- 35.Thomas CJ, Rahier NJ, Hecht SM. Camptothecin: current perspectives. Bioorganic & medicinal chemistry. 2004;12:1585–1604. doi: 10.1016/j.bmc.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 36.Gambus A, Jones RC, Sanchez-Diaz A, Kanemaki M, van Deursen F, Edmondson RD, Labib K. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nature cell biology. 2006;8:358–366. doi: 10.1038/ncb1382. [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Carbonero R, Supko JG. Current perspectives on the clinical experience, pharmacology, and continued development of the camptothecins. Clin Cancer Res. 2002;8:641–661. [PubMed] [Google Scholar]
- 38.Coll JM, Hickey RJ, Wei Y, Malkas LH. The human cell multiprotein DNA replication complex (MRC): the effect of camptothecin on its ability to support in vitro DNA synthesis. Cancer chemotherapy and pharmacology. 1996;39:97–102. doi: 10.1007/s002800050543. [DOI] [PubMed] [Google Scholar]
- 39.Hsiang YH, Liu LF. Identification of mammalian DNA topoisomerase I as an intracellular target of the anticancer drug camptothecin. Cancer research. 1988;48:1722–1726. [PubMed] [Google Scholar]
- 40.Pommier Y, Pourquier P, Fan Y, Strumberg D. Mechanism of action of eukaryotic DNA topoisomerase I and drugs targeted to the enzyme. Biochimica et biophysica acta. 1998;1400:83–105. doi: 10.1016/s0167-4781(98)00129-8. [DOI] [PubMed] [Google Scholar]
- 41.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nature reviews. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 42.Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, Pommier Y. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Molecular and cellular biology. 2000;20:3977–3987. doi: 10.1128/mcb.20.11.3977-3987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holm C, Covey JM, Kerrigan D, Pommier Y. Differential requirement of DNA replication for the cytotoxicity of DNA topoisomerase I and II inhibitors in Chinese hamster DC3F cells. Cancer research. 1989;49:6365–6368. [PubMed] [Google Scholar]
- 44.O’Connor PM, Nieves-Neira W, Kerrigan D, Bertrand R, Goldman J, Kohn KW, Pommier Y. S-phase population analysis does not correlate with the cytotoxicity of camptothecin and 10,11-methylenedioxycamptothecin in human colon carcinoma HT-29 cells. Cancer communications. 1991;3:233–240. doi: 10.3727/095535491820873083. [DOI] [PubMed] [Google Scholar]
- 45.Shao RG, Cao CX, Zhang H, Kohn KW, Wold MS, Pommier Y. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. The EMBO journal. 1999;18:1397–1406. doi: 10.1093/emboj/18.5.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilke CM, Guo SW, Hall BK, Boldog F, Gemmill RM, Chandrasekharappa SC, Barcroft CL, Drabkin HA, Glover TW. Multicolor FISH mapping of YAC clones in 3p14 and identification of a YAC spanning both FRA3B and the t(3;8) associated with hereditary renal cell carcinoma. Genomics. 1994;22:319–326. doi: 10.1006/geno.1994.1390. [DOI] [PubMed] [Google Scholar]
- 47.Anton M, Graham FL. Site-specific recombination mediated by an adenovirus vector expressing the Cre recombinase protein: a molecular switch for control of gene expression. Journal of virology. 1995;69:4600–4606. doi: 10.1128/jvi.69.8.4600-4606.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Syrovets T, Buchele B, Gedig E, Slupsky JR, Simmet T. Acetyl-boswellic acids are novel catalytic inhibitors of human topoisomerases I and IIalpha. Molecular pharmacology. 2000;58:71–81. doi: 10.1124/mol.58.1.71. [DOI] [PubMed] [Google Scholar]
- 49.Ganguly A, Das B, Roy A, Sen N, Dasgupta SB, Mukhopadhayay S, Majumder HK. Betulinic acid, a catalytic inhibitor of topoisomerase I, inhibits reactive oxygen species-mediated apoptotic topoisomerase I-DNA cleavable complex formation in prostate cancer cells but does not affect the process of cell death. Cancer research. 2007;67:11848–11858. doi: 10.1158/0008-5472.CAN-07-1615. [DOI] [PubMed] [Google Scholar]
- 50.Manthey KC, Opiyo S, Glanzer JG, Dimitrova D, Elliott J, Oakley GG. NBS1 mediates ATR-dependent RPA hyperphosphorylation following replication-fork stall and collapse. Journal of cell science. 2007;120:4221–4229. doi: 10.1242/jcs.004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sakasai R, Shinohe K, Ichijima Y, Okita N, Shibata A, Asahina K, Teraoka H. Differential involvement of phosphatidylinositol 3-kinase-related protein kinases in hyperphosphorylation of replication protein A2 in response to replication-mediated DNA double-strand breaks. Genes Cells. 2006;11:237–246. doi: 10.1111/j.1365-2443.2006.00942.x. [DOI] [PubMed] [Google Scholar]
- 52.Nuss JE, Patrick SM, Oakley GG, Alter GM, Robison JG, Dixon K, Turchi JJ. DNA damage induced hyperphosphorylation of replication protein A. 1. Identification of novel sites of phosphorylation in response to DNA damage. Biochemistry. 2005;44:8428–8437. doi: 10.1021/bi0480584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anantha RW, Vassin VM, Borowiec JA. Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. The Journal of biological chemistry. 2007;282:35910–35923. doi: 10.1074/jbc.M704645200. [DOI] [PubMed] [Google Scholar]
- 54.Olson E, Nievera CJ, Klimovich V, Fanning E, Wu X. RPA2 is a direct downstream target for ATR to regulate the S-phase checkpoint. The Journal of biological chemistry. 2006;281:39517–39533. doi: 10.1074/jbc.M605121200. [DOI] [PubMed] [Google Scholar]
- 55.Wu X, Yang Z, Liu Y, Zou Y. Preferential localization of hyperphosphorylated replication protein A to double-strand break repair and checkpoint complexes upon DNA damage. The Biochemical journal. 2005;391:473–480. doi: 10.1042/BJ20050379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Capasso H, Palermo C, Wan S, Rao H, John UP, O’Connell MJ, Walworth NC. Phosphorylation activates Chk1 and is required for checkpoint-mediated cell cycle arrest. Journal of cell science. 2002;115:4555–4564. doi: 10.1242/jcs.00133. [DOI] [PubMed] [Google Scholar]
- 57.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes & development. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Horwitz SB, Chang CK, Grollman AP. Studies on camptothecin. I. Effects of nucleic acid and protein synthesis. Molecular pharmacology. 1971;7:632–644. [PubMed] [Google Scholar]
- 59.Tsao YP, D’Arpa P, Liu LF. The involvement of active DNA synthesis in camptothecin-induced G2 arrest: altered regulation of p34cdc2/cyclin B. Cancer research. 1992;52:1823–1829. [PubMed] [Google Scholar]
- 60.Rodriguez R, Meuth M. Chk1 and p21 cooperate to prevent apoptosis during DNA replication fork stress. Molecular biology of the cell. 2006;17:402–412. doi: 10.1091/mbc.E05-07-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chang DJ, Lupardus PJ, Cimprich KA. Monoubiquitination of proliferating cell nuclear antigen induced by stalled replication requires uncoupling of DNA polymerase and mini-chromosome maintenance helicase activities. The Journal of biological chemistry. 2006;281:32081–32088. doi: 10.1074/jbc.M606799200. [DOI] [PubMed] [Google Scholar]
- 62.Stelter P, Ulrich HD. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 2003;425:188–191. doi: 10.1038/nature01965. [DOI] [PubMed] [Google Scholar]
- 63.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 64.Leach CA, Michael WM. Ubiquitin/SUMO modification of PCNA promotes replication fork progression in Xenopus laevis egg extracts. The Journal of cell biology. 2005;171:947–954. doi: 10.1083/jcb.200508100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Niimi A, Brown S, Sabbioneda S, Kannouche PL, Scott A, Yasui A, Green CM, Lehmann AR. Regulation of proliferating cell nuclear antigen ubiquitination in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:16125–16130. doi: 10.1073/pnas.0802727105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dubois MF, Nguyen VT, Bellier S, Bensaude O. Inhibitors of transcription such as 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole and isoquinoline sulfonamide derivatives (H-8 and H-7) promote dephosphorylation of the carboxyl-terminal domain of RNA polymerase II largest subunit. The Journal of biological chemistry. 1994;269:13331–13336. [PubMed] [Google Scholar]
- 67.Ikegami S, Taguchi T, Ohashi M, Oguro M, Nagano H, Mano Y. Aphidicolin prevents mitotic cell division by interfering with the activity of DNA polymerase-alpha. Nature. 1978;275:458–460. doi: 10.1038/275458a0. [DOI] [PubMed] [Google Scholar]
- 68.Lee MY, Tan CK, Downey KM, So AG. Structural and functional properties of calf thymus DNA polymerase delta. Progress in nucleic acid research and molecular biology. 1981;26:83–96. doi: 10.1016/s0079-6603(08)60396-7. [DOI] [PubMed] [Google Scholar]
- 69.Goscin LP, Byrnes JJ. DNA polymerase delta: one polypeptide, two activities. Biochemistry. 1982;21:2513–2518. doi: 10.1021/bi00539a034. [DOI] [PubMed] [Google Scholar]
- 70.Walter J, Newport J. Initiation of eukaryotic DNA replication: origin unwinding and sequential chromatin association of Cdc45, RPA, and DNA polymerase alpha. Molecular cell. 2000;5:617–627. doi: 10.1016/s1097-2765(00)80241-5. [DOI] [PubMed] [Google Scholar]
- 71.Pacek M, Walter JC. A requirement for MCM7 and Cdc45 in chromosome unwinding during eukaryotic DNA replication. The EMBO journal. 2004;23:3667–3676. doi: 10.1038/sj.emboj.7600369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pacek M, Tutter AV, Kubota Y, Takisawa H, Walter JC. Localization of MCM2-7, Cdc45, and GINS to the site of DNA unwinding during eukaryotic DNA replication. Molecular cell. 2006;21:581–587. doi: 10.1016/j.molcel.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 73.Gorisch SM, Sporbert A, Stear JH, Grunewald I, Nowak D, Warbrick E, Leonhardt H, Cardoso MC. Uncoupling the replication machinery: replication fork progression in the absence of processive DNA synthesis. Cell cycle. 2008;7:1983–1990. doi: 10.4161/cc.7.13.6094. [DOI] [PubMed] [Google Scholar]
- 74.Svoboda DL, Vos JM. Differential replication of a single, UV-induced lesion in the leading or lagging strand by a human cell extract: fork uncoupling or gap formation. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:11975–11979. doi: 10.1073/pnas.92.26.11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McInerney P, O’Donnell M. Functional uncoupling of twin polymerases: mechanism of polymerase dissociation from a lagging-strand block. The Journal of biological chemistry. 2004;279:21543–21551. doi: 10.1074/jbc.M401649200. [DOI] [PubMed] [Google Scholar]
- 76.Lopes M, Foiani M, Sogo JM. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Molecular cell. 2006;21:15–27. doi: 10.1016/j.molcel.2005.11.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.