

Abstract
To alleviate the problems in the receptor-based design of metalloprotein ligands due to inadequacies in the force-field description of coordination bonds, a four-tier approach was devised. Representative ligand-metalloprotein interaction energies are obtained by subsequent application of (1) docking with metal-binding-guided selection of modes; (2) optimization of the ligand-metalloprotein complex geometry by combined quantum mechanics and molecular mechanics (QM/MM) methods; (3) conformational sampling of the complex with constrained metal bonds by force-field based molecular dynamics (MD); and (4) a single point QM/MM energy calculation for the time-averaged structures. The QM/MM interaction energies are, in a linear combination with the desolvation-characterizing changes in the solvent-accessible surface areas, correlated with experimental data. The approach was applied to structural correlation of published binding free energies of a diverse set of 28 hydroxamate inhibitors to zinc-dependent matrix metalloproteinase 9 (MMP-9). Inclusion of step 3 and step 4 significantly improved both correlation and prediction. The two descriptors explained 90% of variance in inhibition constants of all 28 inhibitors, ranging from 0.08 to 349 nM, with the average unassigned error of 0.318 log units. The structural and energetic information obtained from the time-averaged MD simulation results helped understand the differences in binding modes of related compounds.
Keywords: molecular dynamics - MD, QM/MM, docking, linear response approximation, binding affinity, free energy of binding, matrix metalloproteinases, metalloproteins
Introduction
Metalloproteins play important roles in physiological processes (hemoglobin, cytochrome oxidase, catalase and superoxide dismutase), the receptor binding of potential drugs (carbonic anhydrases, matrix metalloproteinases, thermolysin, leucine aminopeptidase, phospolipase C, carboxypeptidase A and B, adenosine and cytidine deaminase), and drug metabolism (cytochromes P450 and methane monooxygenase), to give just a few examples.1–3 In drug design, a description of the ligand interactions with transition metals poses a challenge due to the possibility of multi-dentate coordination bonding that is most appropriately treated at the quantum mechanical level.4–11 This study presents an approach to a receptor-based estimation of binding affinities of metalloprotein ligands that is more reliable than standard ensemble-based techniques at the expense of a modest increase in the computing time.
The fastest and usually least precise descriptions of coordination bonds are obtained using molecular mechanical approaches. Several techniques are available with varying levels of sophistication. The nonbonded approach12,13 uses optimized electrostatics and van der Waals terms,14,15 occasionally using dummy cations placed around the metal atom16,17 to enforce the correct coordination geometry. The geometry enforcement is more stringent in the bonded model18–20 that utilizes the bond terms including bond stretching, angle bending, and torsional terms. These approaches require a predefined valence of the coordinating metal, in contrast to the directional force field YETI that is more flexible in selection of appropriate valence.21,22 Further force-field enhancements include addition of polarizable bonds,23,24 directionality based on orbital hybridization,25 and ligand field stabilization energy.26,27 Unfortunately, the more sophisticated force fields are not readily available for a routine use in modeling of ligand-metal interactions.
Combined quantum mechanical and molecular mechanical (QM/MM) methods represent an economical approach to characterization of macromolecular processes that include changes in the covalent bond status.28–31 The atoms directly involved in the chemical steps are typically included in the QM region, whereas the rest of the system is treated at the MM level. Initially developed for gas-phase calculations,32 the QM/MM approach was first applied to enzyme systems by Warshel & Levitt.28 QM/MM methods combine the efficiency of MM allowing for free energy simulations of macromolecules, with the accuracy and a systematic improvement potential provided by QM.33 The methods have been implemented in combination with semi empirical and ab initio molecular orbital theory and with density functional theory (DFT). QM/MM-based molecular dynamics (MD) simulations have been used for modeling of enzymatic reactions,31,34,35 but are computationally intensive and currently impractical for the prediction of binding affinities of a series of metalloprotein ligands. Alternatively, a large system as a whole can be treated at the QM level by fragmenting, linearly scaling approaches;36 however, the QM/MM treatment seems to provide a better focus by applying the most sophisticated methods to the key chemical steps and the fast methods to the less important noncovalent processes.
Several categories of the methods for prediction of binding affinities to the receptors with known structures are available. Free Energy Perturbation (FEP),37 Thermodynamic Integration,38 and similar techniques39 are the most sophisticated tools for free energy calculations. Extensive sampling resulting in extreme demands on computational resources and limitation to close homologs currently preclude their routine use in drug design. Computational costs can be reduced by partitioning of the binding energy into individual contributions and, in further simplification, by replacing the ensembles of structures generated in molecular simulations by single structures.
In ensemble-based partitioning approaches, only two states needs to be considered: complexes and free interaction partners. The approaches in this category can be classified according to the use of adjustable parameters in the final relation between the binding affinity and the calculated free energy contributions. The MM-PBSA40,41 and MM-GBSA42 methods form the parameter-free category while the Linear Response (LR, a.k.a. Linear Interaction Energy) method43–45 and its extended version (ELR)46–48 represent the parametrized category. The contributions to the binding free energy are expressed as the differences Δ between the solvated ligand in the bound and free states in the ensemble averages (denoted by angle brackets) of respective quantities.
In ELR type methods, binding free energy ΔGb is calculated as the linear combination of the differences Δ in the van der Waals energies, electrostatic energies,43–45 and the solvent-accessible surface areas46–48 (SASA) obtained from MD or Monte Carlo simulations:
| (1) |
The adjustable parameters α, β, and γ contain the protein-solvent and solvent-solvent interactions.49–51 The van der Waals parameter α depends on the used force field, as was shown e.g. for thrombin.47,49,52 Its magnitude was analyzed with respect to hydrophobicity of the binding site.49 The coulombic scaling coefficient β varies with the ligand nature and ligand surroundings (protein or water),53–55 although its equality to ½ was assumed initially based on the linear response of the surroundings to electric fields.43–45 Similar method for treatment of hydration of more complex molecules56 required the use H-bond donor and acceptor counts, in addition to the quantities present in Eq. 1. Continuing studies of more diverse ligands may reveal the need for further empirical corrections.
Single-structure-based partitioning approaches, represented by VALIDATE,57 the Free Energy Force Field approach,58,59 COMBINE analysis,60 and a single-structure version of the LR method using continuum electrostatics,61 replace the ensemble averages by a single configuration, usually obtained by a direct geometry optimization of the receptor-ligand complex. Scoring functions are simplified single-structure-based partitioning approaches that are categorized as force field-based methods,62,63 empirical free energy scoring functions,64–66 and knowledge-based scoring functions.67–69 They are mainly used in high-throughput virtual screening, in connection with fast docking procedures.
To overcome the limitations of the aforementioned methods in prediction of binding affinities to metalloproteins due to the difficulties in handling the transition metal atoms, we have combined docking, QM/MM calculations, and force-field-based MD methods into a coherent approach. The approach is tested using inhibitors of matrix metalloproteinases (MMPs),70 which belong to the most-studied metalloenzymes. Development of MMP inhibitors is complicated by structural relatedness of the MMP family,71 in which some members assume normal physiological roles and others are pathological, depending upon given concentration or activity.
Results and Discussion
Interactions of metalloproteins with ligands are often difficult to describe effectively due to the limited availability of appropriate force fields. To predict binding affinities in these cases, we devised a four-tier procedure consisting of: (1) docking with the selection of poses based upon appropriate metal binding; (2) QM/MM optimization of the best docked geometries; (3) MD simulation with the metal binding group of the ligand confined in the geometry from Step 2; and (4) QM/MM single point interaction energy calculation based on the time-averaged structures from Step 3:
| (2) |
The QM/MM interaction energies are correlated in an ELR-type approach, along with SASA crudely parametrizing desolvation, with experimental affinities. The use of the energies of the time-averaged structures in place of the ensemble averages of energies was previously shown to provide equivalent results in a parametrized partitioning approach to binding affinity correlations72 and in protein pKa prediction.73 The approach was tested using published data70 on inhibition of 28 hydroxamate inhibitors of MMP-9 (Table 1).
Table 1.
Structures, inhibition constants,70 and the simulation results of the studied MMP-9 inhibitors.
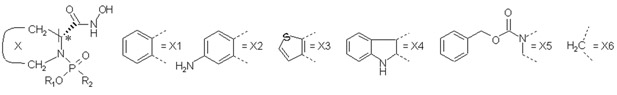 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No | X | R1 | R2 | log(1/Ki), Ki in M | −Δ<EvdW> a (kcal/mol) | −Δ<Eel> a (kcal/mol) | −Δ<EQM/MM> b (kcal/mol) | −Δ<SASA> (Å2) | |||
| exp. | calc. d | Min | MD | Min c | MD a | ||||||
| 1 | X1 | CH2CH3 |  |
8.297 | 8.035 | 111.93 | 189.63 | 1216.74 | 689.95 | 417.06 | 389.34 |
| 2 e | X1 | CH2CH3 |  |
6.457 | 6.728 | 86.41 | 192.93 | 1177.15 | 489.14 | 324.51 | 311.53 |
| 3 | X1 | CH3 |  |
8.693 | 8.819 | 107.04 | 174.78 | 1130.66 | 860.87 | 350.53 | 411.94 |
| 4 | X1 | 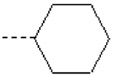 |
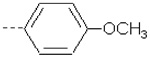 |
8.182 | 7.803 | 106.40 | 169.62 | 1105.70 | 721.32 | 425.77 | 343.59 |
| 5 | X1 | 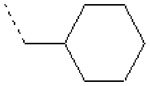 |
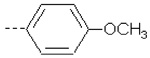 |
8.341 | 8.233 | 95.40 | 155.90 | 1140.95 | 520.52 | 437.80 | 496.27 |
| 6 | X1 | 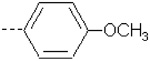 |
8.189 | 8.315 | 121.79 | 192.67 | 723.49 | 736.12 | 370.67 | 404.369 | |
| 7 | X1 | (CH2)2N(CH2CH3)2 | 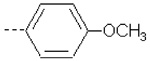 |
7.777 | 7.924 | 72.34 | 148.89 | 1107.33 | 646.02 | 455.69 | 395.52 |
| 8 | X1 | 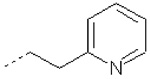 |
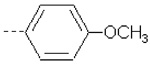 |
8.466 | 8.337 | 127.92 | 239.22 | 1090.74 | 533.07 | 460.21 | 504.04 |
| 9 | X1 | (CH2)2OCH2CH3 | 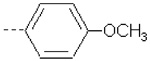 |
8.197 | 8.322 | 120.93 | 190.55 | 980.36 | 746.42 | 381.41 | 400.46 |
| 10 | X1 | CH2CH3 | 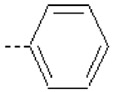 |
7.370 | 7.226 | 88.32 | 159.09 | 874.43 | 639.75 | 320.68 | 305.84 |
| 11 | X1 | CH2CH3 | 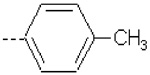 |
7.379 | 7.562 | 103.30 | 179.29 | 1016.72 | 696.22 | 353.45 | 323.61 |
| 12 | X1 | CH2CH3 | 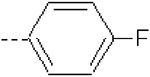 |
7.285 | 7.253 | 88.67 | 192.33 | 879.98 | 614.65 | 303.00 | 321.47 |
| 13 | X1 | CH2CH3 | 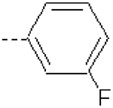 |
7.096 | 7.017 | 90.80 | 182.57 | 868.42 | 589.55 | 316.57 | 302.09 |
| 14 | X1 | CH2CH3 | 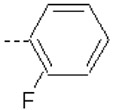 |
7.493 | 7.122 | 98.18 | 176.21 | 879.45 | 595.82 | 322.45 | 312.99 |
| 15 | X1 | CH2CH3 | 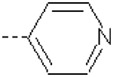 |
6.556 | 7.218 | 87.85 | 156.83 | 887.22 | 608.37 | 348.26 | 319.82 |
| 16 | X1 | CH2CH3 | 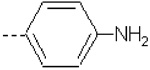 |
7.020 | 7.635 | 95.44 | 183.74 | 982.55 | 696.22 | 334.14 | 333.28 |
| 17 | X1 | CH2CH3 | 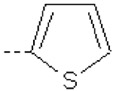 |
7.228 | 7.224 | 94.51 | 159.36 | 949.87 | 595.82 | 319.49 | 326.53 |
| 18 | X1 | CH2CH3 | 7.848 | 8.158 | 123.38 | 183.20 | 1307.85 | 551.90 | 459.85 | 471.31 | |
| 19 | X1 | CH2CH3 | 9.509 | 9.197 | 121.99 | 227.93 | 1127.44 | 940.95 | 429.04 | 423.94 | |
| 20 | X1 | CH2CH3 | 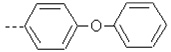 |
10.10 | 10.18 | 123.45 | 206.88 | 1121.86 | 834.28 | 451.32 | 604.77 |
| 21 | X1 | CH2CH3 | 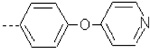 |
9.222 | 8.554 | 124.01 | 195.86 | 1058.55 | 740.15 | 456.49 | 434.23 |
| 22 | X1 | CH2CH3 | 10.00 | 9.977 | 137.04 | 224.55 | 1127.20 | 771.52 | 472.26 | 608.05 | |
| 23 | X1 | CH2CH3 | 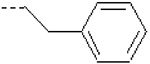 |
7.417 | 7.675 | 111.28 | 197.02 | 1037.06 | 771.52 | 389.28 | 302.73 |
| 24 | X2 | CH2CH3 | 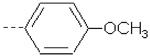 |
7.959 | 8.114 | 0.74 | 0.82 | 1023.59 | 727.60 | 384.47 | 381.87 |
| 25 | X3 | CH2CH3 | 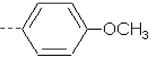 |
8.420 | 8.026 | 103.83 | 203.22 | 1013.16 | 683.67 | 361.72 | 391.05 |
| 26 | X4 | CH2CH3 | 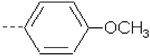 |
8.947 | 9.193 | 110.63 | 189.12 | 1009.13 | 771.52 | 345.50 | 504.02 |
| 27 | X5 | CH2CH3 | 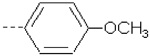 |
8.284 | 8.218 | 132.48 | 237.82 | 879.21 | 740.15 | 428.73 | 389.68 |
| 28 | X6 | CH2CH3 | 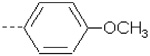 |
6.500 | 6.081 | 95.55 | 180.16 | 549.68 | 464.73 | 253.08 | 237.38 |
The LR terms for van der Waals and electrostatic energies and for the SASA change were obtained using MD simulation with explicit solvent.
The QM/MM energy terms calculated for optimized structures after QM/MM minimization and for time-averaged structures after MD simulation.
The SASA change for optimized structures after QM/MM minimization.
Calculated from Eq. 3 with optimized values of adjustable parameters for Step 4 (Table 2, last row).
Configuration of C-3 (marked with an asterisk) is S in 2 while in other compounds it is R.
Step 1 - Docking
Docking of the inhibitors to the MMP-9 structure taken from the Protein Data Bank74 (PDB file 1GKC) was performed using FlexX.64,75 The ranking of poses was based upon the distance between catalytic zinc and both hydroxamate oxygens in the interval 1.5 – 2.5 Å as the primary criterion and the FlexX score as the secondary criterion.76 Docking provided a greater variety of the initial binding modes than superposition of the ligands with the ligand in the PDB file of the complex.77 Since the MD simulations routinely do not sample conformations too different from the starting conformations, this step was instrumental in selecting the binding mode explaining the experimental data for several compounds. To reduce the QM/MM convergence time, for the top complexes of each ligand, the mobile region within 5 Å of the ligand superposition was briefly optimized by the conjugate gradient minimization using OPLS-AA force field.
Step 2 - QM/MM Geometry Optimization
The charges and inter-atomic distances for all 28 ligands (Table 1) do not differ much; therefore we present them as average values in Figure 1. The maximum standard deviations were 8.1 % for charges and 2.5 % for distances. Charge transfer from hydroxamate group to zinc is significant, albeit very similar for all studied compounds (Figure 1). For the free/bound hydroxamates, the average bond lengths were (in Å): C=0 1.236/1.284, C-N 1.355/1.308, N–O 1.401/1.364, and O–H 0.982/1.590, respectively. In the QM/MM geometry optimization of the complex, the proton from the hydroxamate OH group was transferred to the Glu402 oxygen but remained H-bonded to the parent oxygen. The average C=0, C-N and N-O bond lengths for the bound state obtained here (1.284, 1.308 and 1.364 Å) are closer to deprotonated Zn-coordinated hydroxamate (1.294, 1.317 and 1.378 Å) than to neutral hydroxamate with respective distances of 1.279, 1.337 and 1.416 Å.78 The distance between Glu402 oxygen and oxygen of the reverse hydroxamate (2.70 Å) in MMP-9 and hydroxamate oxygen in MMP-779 and MMP-880,81 (2.6–2.8 Å) complex are in close agreement with the distance found in Step 2 (2.604 Å). These facts suggest that bound hydroxamate structure is, in the presence of proton-accepting Glu402, closer to deprotonated state than to neutral form. Hydroxamates approach the binding site as neutral molecules (pKa ≥ 8.9 for the studied compounds)82 and require ionized Glu402 for full potency.83
Figure 1.

Average Mulliken charges and bond lengths (Å, in italics) for the complete set of 28 hydroxamate complexes (Table 1) after QM/MM optimization. The maximum standard deviations were 8.1 % for charges and 2.5 % for distances.
Two hydroxamate oxygens may bind to zinc in monodentate or bidentate configurations that result in four- or five-fold coordination, respectively. Among the structures of hydroxamates bound to MMPs in PDB, the bidentate zinc binding prevails, with trigonal bipyramidal (TB) configurations occurring about six times more frequently than the square-based pyramid (SP). After QM/MM optimization, the bidentate TB coordination was observed in the studied hydroxamate structures, as evidenced by the average geometries of all 28 complexes (Figure 1). The bond lengths of the two Zn-O bonds (Zn-O1 and Zn-O2) are 2.029±0.018 Å and 2.089±0.012 Å, respectively, which indicates that the two oxygens coordinate with zinc about equally. The average bond angles O1-Zn-N in His401, His405 and His411 in these complexes are 121.77±14.81°, 135.58±8.95°, and 82.06±1.65°, respectively (ideal values for TB: 120°, 120°, 90°; and for SP: 90°, 180°, 90°). The average bond angles O2-Zn-N in the same order are 101.55±1.89°, 95.07±2.13°, and 155.23±2.98°, respectively (both TB and SP: 90°, 90°, 180°). The average bond angle of O2-Zn-O1 is 79.66±0.61° , approaching the ideal value of 90° for both TB and SP configurations.
The QM/MM approach for the entire binding domain provided a more realistic picture than the studies using reduced systems5,10,78,84,85 that do not consider the protein surroundings of bound ligands. In this case, ionized Glu402 is crucial for a correct description of binding. The QM/MM approach handled the zinc-ligand charge transfer,36 bond length changes, polarization, and ionization25 upon binding to zinc, which would be difficult to describe by advanced force fields.21–27 Albeit very similar in the studied set, the electronic phenomena can be anticipated to play a more important role in a heterogeneous series of ligands.
Step 3 - MD Simulations
The QM/MM optimized complexes were subjected to the MD simulation with the constrained zinc-hydroxamate oxygens bond lengths and angles, to obtain conformational sampling for the rest of the complex. To reduce the computational expense, only one MD simulation, for the enzyme-inhibitor complex, was performed.41 The energy terms for the enzyme and inhibitor taken from the MD simulation for the complex were comparable to those obtained from running separate simulations for the enzyme and inhibitor, as checked for several inhibitors. Inspection of the trajectories revealed that the secondary and tertiary structure of the ligand-receptor complex remained stable during the entire 200-ps simulation period (data not shown). The influence of zinc bond constrains on movement of other ligand parts is illustrated in Figure 2. The chiral carbon (Table 1) that is only 2–3 bonds apart from the constrained hydroxamate oxygens is comparatively rigid. The ring nitrogen and phosphorus are separated by 1–2 more bonds from the constrained part and exhibit amplitudes comparable to those of distant binding site parts like the oxygen of Glu402 that is H-bonded to hydroxamate oxygen. The ensemble averages of the van der Waals and electrostatic energies that were calculated for the time-averaged structures obtained after 5 ps simulations are listed in Table 1. Longer simulation times provided similar samplings that did not improve the correlations using Eq. 1 and Eq. 3 and the results are not shown. The ΔSASA terms were lower for the time-averaged structures from Step 3 than for the minimized structures from Step 2 in 16 cases (Table 1). For 12 ligands, however, the ΔSASA terms increased, indicating that MD sampling found better binding modes than minimization. The largest increases were observed for ligands 3, 5, 6, 8, 20, 22, 25, and 26.
Figure 2.

The effect of constrained zinc binding on the MD simulation of ligand 20 (Table 1) in the MMP-9 active site. Distances between catalytic zinc and, from the top, phosphorus, ring nitrogen, and chiral carbon atoms (structures in Table 1) and between hydroxamate oxygen (O2) and Glu402 oxygen in 10 fs intervals throughout the 5-ps MD simulation.
Step 4 - Calculation of QM/MM Interaction Energies
For the time-averaged structures resulting from 5-ps MD simulations, single point QM/MM interaction energies were calculated according to Eq. 2 and are summarized in Table 1. The time-averaged structures were used to preserve conformational sampling obtained in Step 3 that should result in more realistic descriptions than using the single optimized structures.61 As can be expected, the QM/MM energy changes are much smaller for the time-averaged structures from Step 3 than for the minimized structures in Step 2. The QM/MM calculations properly treat the coordination bonds between zinc and the ligands. Therefore, the QM/MM interaction energies are expected to provide better energy estimates than the MM-based force field simulations.
Correlations with Inhibitory Activities
The QM/MM energies, along with SASA characterizing ligand desolvation, were correlated with experimental binding affinities using Eq. 3, in a way reminiscent of the ELR approach (Eq. 1):
| (3) |
For a comparison, the MD results were correlated with biological data using Eq. 1. The influence of the SASA’s polar and nonpolar components, as well as that of the constant term κ were examined. The resulting equations are not listed because they improved neither correlations nor predictions.
The fits of Eq. 1 and Eq. 3, with logKi on the left side, to the inhibition data for the 28 compounds for individual Steps are summarized in Table 2. Prediction of the MMP-9 inhibition constant based upon the linear correlation of the experimental data with the FlexX scores in Step 1 resulted in a poor correlation characterized by r2 = 0.044, with r being the correlation coefficient.
Table 2.
Correlations of inhibitory potencies with the energy and SASA terms obtained by different methods for 28 MMP-9 inhibitors. The logKi, Ki in M, values were used instead of ΔGb in Eq. 1 and Eq. 3.
| Method | Step | Eq. | a α×10−3 (mol/kcal) | b β×10−3 (mol/kcal) | c γ×10−3 (1/Å2) | κ | r2 | SD | F | LOO d | LSO e | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| q2 | RMSE | q2 | RMSE | ||||||||||
| FlexX f | 1 | - | 69.7±63.71 | - | - | −5.831±1.997 | 0.044 | 0.966 | 1.198 | −0.079 | 0.989 | −1.110 | 0.994 |
| QM/MM Minimization | 2 | 3 | −0.420±1.190 | - | 12.30±3.12 | −3.744±0.966 | 0.504 | 0.709 | 12.72 | 0.380 | 0.750 | −0.457 | 0.808 |
| QM/MM Minimization | 2 | 3 | - | - | 11.55±2.26 | −3.606±0.870 | 0.502 | 0.697 | 26.21 | 0.423 | 0.723 | −0.278 | 0.751 |
| MD | 3 | 1 | 11.25±10.50 | −4.090±6.010 | 8.380±1.240 | −4.315±0.551 | 0.764 | 0.499 | 25.96 | 0.566 | 0.627 | 0.180 | 0.662 |
| MD | 3 | 1 | 4.630±4.060 | - | 8.680±1.150 | −4.137±0.478 | 0.760 | 0.494 | 39.55 | 0.602 | 0.600 | 0.272 | 0.629 |
| MD | 3 | 1 | - | - | 9.230±1.105 | 4.399±0.422 | 0.747 | 0.496 | 76.91 | 0.717 | 0.506 | 0.442 | 0.531 |
| QM/MM | 4 | 3 | 3.592±0.580 | - | 7.543±0.727 | −2.623±0.394 | 0.900 | 0.318 | 112.8 | 0.879 | 0.331 | 0.769 | 0.319 |
Characterizes electrostatic energy ( Eq. 1 only).
Leave-one-out cross-validation.
Leave-several-out cross-validation: random selection of 6-member test set, repeated 200 times.
The correlation between logKi and the FlexX scores, with the slope α and the intercept κ.
The QM/MM energies of the ligand-enzyme complexes with optimized geometries, obtained in Step 2, resulted in a slightly improved statistics (r2 = 0.504, Table 2). The correlation is dominated by the SASA term. Inclusion of the QM/MM energy for the optimized geometry did not improve the correlation. The pertinent regression parameter α has a negative sign and an inflated error, possibly due to a moderate cross-correlation between the QM/MM energy and the SASA term (r2=0.460).
The use of the energy terms from the MD conformational sampling in Step 3 pushed the correlation to r2=0.764 (Table 2). Compounds 3, 7, 20, 22, and 26 (Table 1) that saw the greatest description improvement in Step 3 (0.6 – 1.1 log units), exhibit a significant change in the conformation of the bound ligand upon MD sampling. The change in the binding conformation is accompanied by a large increase of ΔSASA, except compound 7. Descriptions for other compounds with ΔSASA increased upon MD sampling (5, 6, 8, and 25, Table 1) improved by 0.1 – 0.3 log units. For the van der Waals coefficient α, the error is almost equal to the parameter estimate, while for the coulombic coefficient β, the sign of the parameter estimate is negative and the error term larger than the parameter estimate. This misbehavior may be caused by the collinearity problem of the van der Waals and electrostatic terms (r2=0.712 – 0.839 for different simulation times). The low value of the van der Waals coefficient α might indicate a low hydrophobicity of the MMP 9 binding site, if the results of the original analysis49 are also valid for correlations with the explicit SASA term. Cross-correlation of van der Waals and SASA terms was very weak (r2=0.174).
The best model for Step 4 contained just the single point QM/MM energy for the time-averaged structures and the SASA term and was obtained using the data from a 5-ps MD simulation. This treatment was most beneficial for compounds 18 and 19, the residuals of which decreased from ~1.0 in Step 3 to ~0.3 log units in Step 4, and compounds 5 and 8, with residuals improving from ~ 0.6 to ~0.1 log units. The correlation for all 28 compounds is characterized by r2 = 0.900 and the standard deviation SD = 0.318 reflecting a good agreement between actual and calculated values (Table 2). For each parameter, the probability >F ratio was <0.0001, implying that the likelihood of a random occurrence of a significant parameter is negligible. The cross-correlation between the QM/MM energy and SASA is very weak as indicated by the r2 value of 0.140. The dominance of the SASA terms, clearly seen in Table 2, is probably reflecting the effect of burial of the inhibitor in the binding site. This phenomenon was described previously in the analysis of binding energies of several ligand-protein complexes.86 A plot of experimental activity as a linear combination of contributions from QM/MM energy and SASA is shown in Figure 3. The quality of correlations in Step 4 remained at about the same level with the increase in the MD simulation time for obtaining the time-averaged structures. Consequently, the simulation time of 5 ps seems to be sufficient for the binding energy analyses in the studied case, which is characteristic by constrained geometry of the zinc binding group in the complex and rigid protein structure outside the 5-Å region around the ligand superposition.
Figure 3.

Experimental inhibition constants Ki (M) of hydroxamates (Table 1) vs MMP-9 as a linear combination of the change in the SASA (Å2) caused by binding and the QM/MM interaction energy (kcal/mol) for the time-averaged structures obtained by MD simulation.
The adjustable parameter κ in Eq. 3 yields an attractive term of about −2.623 log units (Table 2), providing a base value for the inhibitors that is then modulated by the QM/MM interaction and SASA terms. The values of the QM/MM terms (Table 1) are negative and the associated positive coefficient (Table 2) implies that a strong interaction between the inhibitor and the binding site is important for inhibition. The SASA terms (Table 1) are negative, implying burial of the surface area upon binding. The associated parameter γ (Table 2) is positive so that the removal of mostly hydrophobic surface area from the contact with water upon binding promotes the binding, which simply reflects the hydrophobic effect.87 The obtained values of γ (Table 2: 0.00754-0.011 Å−2; multiplied by R×T×ln10 = 1.419 kcal/mol to account for the change of the dependent variable from free energy to log Ki as described in part Methods/Data Set) are in the same range as the slopes of the linear dependencies of solvation free energies on SASA: 0.007 kcal/(mol×Å2) for alkanes,88 and 0.01689 or 0.020 kcal/(mol×Å2)46 for various compounds.
The robustness of the regression equations and their predictive abilities were probed by cross-validation. The leave-one-out (LOO) procedure and especially the leave-several-out (LSO) procedure with a random selection of 6-member test set that was repeated 200 times provided a thorough evaluation. The predictive root mean squared error (RMSE) for Eq 3 obtained for the 5 ps MD simulation time is the lowest among all correlations. The RMSE values using LOO (0.331) and LSO (0.319) were comparable to that of the RMSE of the whole data set (0.315). Inclusion of all Steps in the correlation was warranted by the improvement in descriptive and predictive ability. The quality of correlations for individual Steps is documented in Figure 4.
Figure 4.

Correlations between experimental and calculated inhibition potencies of hydroxamates vs. MMP-9 as obtained by FlexX docking with the zinc binding based selection of modes in Step 1 (green), QM/MM minimization in Step 2 (blue), MD simulation with constrained zinc bonds in Step 3 (red), and by QM/MM energy calculations for the time-averaged structures from MD simulation in Step 4 (black). All correlation results are summarized in Table 2.
The correlation described by Eq. 3 with the optimized parameters given in Table 2 is much better than our previous ELR results77 obtained from MD simulations with nonbonded zinc-ligand interactions. The predictive ability of the ELR model for all 28 compounds was characterized by RMSE from LSO cross-validation between 0.584 and 1.173, depending upon the simulation time (Table 6, model A in ref. 77). Comparison of these values with equivalent LSO RMSE values in the right-most column of Table 2 shows that current correlations were significantly better only for Step 4 (RMSE 0.319), where single-point QM/MM energies of the time-averaged structures were used. The results from MD simulations employing the same time-averaged structures (Step 3), although run with improved starting geometries of hydroxamate groups from the QM/MM minimization in Step 2, did not show any major progress: the best RMSE was 0.531 that is only slightly better than 0.584 resulting from a less constrained MD simulation. The QM/MM energy from the minimization procedure (Step 2) ended with the best RMSE of 0.751 that is not much better than RMSE of 0.785 that was obtained previously with a force-field minimization. Apparently, conformational sampling embodied in the time-averaged structures (Step 3) and a good description of the zinc coordination bonds (Step 4) are jointly required for a good correlation with experimental inhibitory potencies and none of these procedures alone is making a major breakthrough.
Binding Trends
Eq. 3 and underlying structural information on bound ligands present a straightforward framework for understanding the trends in the observed activities. For a given series of hydroxamate derivatives in Table 1, the ranges for the contributions from QM/MM interactions and burial of solvent accessible surface area are 451.81 kcal/mol and 305.97 Å2, respectively. The SASA term contributes more to the computed activities than the QM/MM term. Representative binding modes of compound 22 (Table 1) obtained after FlexX docking, minimization, and MD simulation are illustrated in Figure 5. The differences between minimized structures and time-averaged structures after MD sampling in Step 3 were sometimes larger than those shown in Figure 5, especially for compounds 3, 7, 20, and 26 (Table 1). The S1 subsite is structurally defined by Asp185, Gly186, Leu187, Leu188, Ala189, His190, Ala191, and Phe192, while the S2’ subsite is defined by Ala417, Leu418, Met419, Tyr420, Pro421, Met422, and Tyr423. The P=O group of all the ligands except 2, 3, 13, 16, 20, and 25 forms hydrogen bonds with the backbone NH of Leu188. In general, the R1 substituents occupy the pocket lined with residues of the S1 subsite while R2 substituents occupy the S2’ subsite (Figure 6). Even compounds with longer R2 substituents (compounds 18–22) leave an empty pocket in the S2’ subsite. Since these are the most potent compounds in the given series, further lengthening of the R2 substituents may be helpful in designing more potent inhibitors.
Figure 5.

The binding modes of compound 22 (Table 1) in active site of MMP-9 obtained from FlexX docking (green), QM/MM optimization (blue), and MD simulation (red). The R1 and R2 substituents bind in S1 and S2’ subsites, respectively. The time-averaged protein surface after MD simulation (Step 3) is shown in white for clarity. The imidazole rings of His401, His405, and His411, as well as catalytic zinc were included in creating the surface but are also shown in corresponding colors. The surface of the protein was z-clipped for better view.
Figure 6.

The binding modes of compounds with longer R1 (compounds 4 – 9) and R2 (18 – 22) substituents (Table 1) shown in ball and stick mode. Compounds with smaller R1 and R2 substituents follow a similar pattern. The catalytic zinc is represented as sphere. The empty pocket of the S2’ subsite, created with the SiteID module of Sybyl,96 is represented by transparent green space fill. The residues of the S2’ and S1 subsites as well as residues surrounding the empty pocket for compound 4 are shown in atom color in the capped stick mode. All structures are time-averaged structures from MD simulation (Step 3).
The Effect of Stereoisomerism
Compound 1 is a potent inhibitor (Ki = 5.05 nM) while compound 2 exhibits only moderate activity (Ki = 349 nM). The compounds are structurally similar except the configuration on C-3 where 1 exhibits the R-configuration while in 2 it is the S-configuration. The importance of the R-configuration at the α carbon is well documented.90–94 Binding modes after MD simulation (Step 3) of the two inhibitors are shown in Figure 7. The zinc-binding group is tightly bound to zinc in both the compounds but the rest of the ligand is flipped by 180°. In case of 1, several hydrogen bonds can be discerned: between the oxygen atom of the ethyl ester group and the main chain NH of Leu188; between the hydroxamate OH and oxygen of Glu402 (and also an intramolecular H-bond with the phosphonamide oxygen). In contrast, compound 2 forms only one hydrogen bond with the Glu402 (Figure 7). The H-bond pattern is reflected in computed QM/MM interaction terms (Table 1) that are equal to −698.95 kcal/mol for 1 and −489.14 kcal/mol for 2. Additional benefit for 1 comes from the burial of more SASA (−389.34 Å2) than for 2 (−311.53 Å2).
Figure 7.

Stereo view of the binding modes and key interactions of isomers 1 (atom color) and 2 (purple) in MMP-9 active site (time-averaged structures after the Step 3). Compound 1 forms hydrogen bonds with Leu188 and Glu402, while compound 2 engages in hydrogen bond with Glu402 only. The hydrogen bonds are shown in dashed lines (magenta). Structures of isomers are shown in Table 1.
The Role of 4-Methoxyphenyl in R2 Position
Substitution of 4-methoxyphenyl in the R2 position in 1 with pyridine ring in 15 resulted in a severe loss of activity. The fact can be explained by the lower QM/MM energy values as well as a lower burial of SASA in 15 as compared to 1 (Table 1). Compounds 10 – 12 (Ki = 42.7, 41.8, and 51.9 nM, respectively) are structural analogs of compound 1 but exhibit lower activity as compared to 1 (Ki = 5.05 nM). The 4-methoxyphenyl group in R2 position of 1 is replaced with phenyl (10), 4-methylphenyl (11), and 4-fluorophenyl (12) substituents. Compounds 10 – 12 exhibit less burial of SASA and similar (11) or lower QM/MM energy values (10 and 12), as compared to 1 (Table 1).
Interactions vs. SASA Burial
Compound 19 (R2=Ph-Ph) differs from 20 (R2=Ph-O-Ph) in the interlinking oxygen atom and exhibits a lower activity. The ether linkage in 20 is expected to be better solvated in the unbound state of the ligand than in the bound state. However, this factor is not encoded in the overall ΔSASA value: greater ΔSASA (−604.77 Å2) of 20 than 19 (−423.94 Å2) merely reflects the size difference as both ligands bind in a similar way. Although the QM/MM energy term is more favorable for 19 (−940.95 kcal/mol) than for 20 (−834.28 kcal/mol), this contribution is negated by the dominance of the SASA term and consequently higher activity for compound 20.
Compounds 20 and 21 differ in isosteric R2 substituents (Ph-O-Ph and Ph-O-C6H5N, respectively). The QM/MM as well as SASA terms are more favorable for compound 20 as compared to compound 21 (Table 1). For compound 20, MD simulation in Step 3 found a new time-averaged binding mode resulting in a substantial increase in ΔSASA that explains higher activity of 20 as compared to 21. In this mode, the P=O group of 20 forms H-bond interaction with backbone NH of Ala189 that was not observed after QM/MM minimization in Step 2.
MD simulation also found a new time-averaged binding mode for compound 26, resulting in a significant increase in ΔSASA and improvement in description of activity after Step 3 as compared to Step 2. A closer look at the structural changes shows that the new mode also exhibits a different H-bonding interaction pattern: the P=0 group forms H-bond with backbone NH of Leu188, while after Step 2 the NH group in X4 was H-bonded with the backbone C=0 group of Gly186.
Methods
Data Set
Published inhibitory potencies, characterized by the inhibition constants Ki at 37 °C, of a series of 28 hydroxamate derivatives70 towards MMP-9 were used (Table 1). The inhibition constants are the inverse values of the association constants K. The left sides of Eq. 1 and Eq. 3 are the binding free energies ΔGb = − R×T×lnK = R×T×ln10×logKi. The logKi values were used as dependent variables, in order to work with the dependent variables that are most common in medicinal chemistry. The optimized values of adjustable parameters α, β, γ, and κ (Table 2) can be recalculated for the ΔGb as independent variable by multiplication with R×T×ln10 = 1.419 kcal/mol.
Coordinates of the MMP-9 catalytic domain were taken from the recently reported x-ray crystal structure of the MMP-9 in the complex with N-formyl-N-hydroxy-2-(2-methylpropyl)-β-alanyl-N,3-dimethyl-L-valinamide, a ‘reverse hydroxamate’ inhibitor, as deposited in the PDB (file 1GKC).95
Construction of Initial Inhibitor/Enzyme Complexes
Three-dimensional structures of ligands were constructed using the SYBYL6.9 suite of programs96 running under Irix 6.5. Full geometry optimization and charges were calculated using DFT/B3LYP-6–31G** approach.97 The inhibitors were docked into the active site of MMP-9 using the FlexX program that considers ligand conformational flexibility by an incremental fragment placing technique.64,75 For each ligand, the best conformation in the active site was selected from the top 30 poses generated by FlexX using the distances in the interval 1.5 – 2.5 Å between both hydroxamate oxygens and the catalytic zinc atom as the primary criterion and the FlexX ranking as the secondary criterion.76 Step 1 was completed by a brief MM geometry optimization using the OPLS-AA force field with a distance-dependent dielectrics and conjugate gradient algorithm with a convergence criterion of 0.001 kcal/(mol×Å).
QM/MM Calculations98 were used for three purposes: optimization of the initial geometries of the inhibitor-enzyme complexes and estimation of their interaction energies (Step 2), as well as estimation of the interaction energies for the time-averaged structures (Step 4) obtained by MD simulation (Step 3). The QM region consisted of side chains of His405 and His411, the backbone atoms and side chains of His401 and Glu402, the entire inhibitor, and the zinc ion. The backbone atoms were included to obtain valid QM/MM cuts. The rest of the protein was treated with MM. The protein and water outside 5 Å of the ligand superposition after Step 1 were frozen.
The interface between QM and MM regions is mediated by frozen orbitals.99 The QM and MM regions interact via two mechanisms: electrostatic interactions between MM point charges and the QM wave function, and van der Waals interactions between QM and MM atoms. For the MM and QM parts of the QM/MM calculations, OPLS-AA force field100 and DFT functional B3LYP101 were deployed, respectively. All charges in the MM region were treated using the OPLS-AA force field. The 6–31G* basis set was used in the interface region between the QM and MM regions. The LAV3P** basis set was employed for geometry optimization: for Zn, S and P atoms, this means the Los Alamos effective core potential (ECP)102,103 with all the s functions and the last p and d gaussian uncontracted; for the remaining atoms, it implies 6–31G** basis set. The maximum number of iterations was set to 100 cycles and all calculations converged before reaching this limit: the root mean squared change in density matrix elements was less than the criterion of 5.0×10−6. B3LYP provides as good or better geometries and energies as those from correlated ab initio methods for the first-row transition metal complexes.5 We therefore selected B3LYP to optimize the structures of the complexes in Step 2. In Step 4, the interaction energies were calculated by subtracting the QM/MM energy of the ligand and the receptor from the QM/MM energy of the complex for the time-averaged structures (Eq. 2).
Molecular Dynamics Simulations were performed using SYBYL 6.9 under isothermal/isobaric (NPT) conditions with Tripos force field.104 The QM/MM-optimized bonds between the zinc-binding group (hydroxamate oxygens) of inhibitors, zinc and nitrogens of His401, His405, and His411, slightly different for each ligand, were restrained with a harmonic potential (the force constant 200, power 2) throughout the simulation. The Mulliken charges105 resulting from the QM/MM optimization were used for the QM region (ligand, catalytic zinc, coordinating histidine and glutamate residues). For the rest of the protein and water, Gasteiger-Hückel charges106 were used. For each system, only one set of simulations was performed with the ligand bound to the protein with a cap comprising two layers of water (TIP3P) molecules,104 surrounding the complex. The water molecules were then minimized for 10,000 cycles using conjugate gradient minimization keeping the protein and ligand atom fixed to its initial positions. The outer portion of the water cap was farther than 5 Å from the ligand superposition after Step 2 and was assigned to the frozen region. Subsequently MD simulation was performed for 20 ps for the mobile water molecules, keeping the protein, ligand, and crystal water and frozen water fixed. This solvent equilibration phase was performed in order for the solvent molecule to readjust to the potential field of the ligand-receptor complex.
After the 15-ps heating phase, when the temperature of the system was raised from 0 to 300 K, the equilibration run was performed for 100 ps. Finally, the production phase was carried out at 300 K for 200 ps. The time step of the simulations was 1 fs with a cutoff of 12 Ǻ for nonbonded interactions. The nonbonded pairs were updated every 25 fs. All residues within 5 Ǻ of any atom in the ligand superposition after Step 2 were allowed to move freely and the remaining part of the protein and water was kept frozen. This setting was identical for all ligands. The time-averaged structures, obtained from the readings in 100 fs intervals, of the complete mobile region were collected at appropriate times. These structures were briefly minimized, to relieve the worst conflicts, using the Tripos force field with a distance-dependent dielectrics and the Powell conjugate gradient algorithm with a convergence criterion of 0.001 kcal/(mol×Å). The minimization produces structures with standard bond length and angles, with the dihedrals representing the ensemble. The time-averaged structures were shown to provide similar LIE correlations as ensemble averages.72,73 The single-point QM/MM energy calculations on the time-averaged structures were used to estimate the zinc binding energies. The polar, non-polar and total solvent accessible surface area (SASA) terms were calculated using the ProsSat option in the Homology module of the Insight II modeling package.107
Regression and Cross-Validation
The least-square fits108 were based on Eq 1 and Eq 3 with the constant term. The robustness of the regression equations and their predictive abilities were probed by cross-validation. For this purpose, the fits to the potency data are generated leaving out one or more inhibitors from the calibration process. The resulting equation for each fit is used to predict the potencies of the omitted compounds. We used the leave-one-out (LOO) approach and the leave-several-out (LSO) approach, where 6 inhibitors were randomly omitted and the process was repeated 200 times. The correlations of the LOO and LSO predictions with the actual potencies were characterized by the root mean square errors (RMSE).
Conclusions
A computational approach combining docking, QM/MM calculations, and MD simulations was developed for prediction of binding affinities of ligands to metalloproteins. The use of QM/MM energies in the ELR-type correlations was facilitated by the use of time-averaged structures from MD simulations. The application of the approach to the MMP-9 inhibition by 28 hydroxamates resulted in an excellent correlation (r2 = 0.900) between experimental and calculated values for all tested compounds that exhibit ~4000-fold difference in binding affinity, with the inhibition constants Ki ranging from 0.08 to 349 nM. Prediction ability of the correlation is characterized by RMSE ~ 0.3 for the logKi values, as compared to RMSE > 0.6 if the QM/MM term is not used. The examination of the energetic and structural results provides a basis for understanding activity differences of individual inhibitors and for their rational design. The proposed approach improves the descriptive and predictive abilities for metalloprotein ligand affinity prediction as compared to the LR approach at the expense of about a four-fold increase in the computational time.
ACKNOWLEDGMENT
This work was supported in part by the NIH NCRR grants 1P20RR15566 and 1P20RR16471, as well as by the access to resources of the Computational Chemistry and Biology Network and the Center for High Performance Computing, both at the North Dakota State University.
REFERENCES
- 1.Lipscomb WN, Straeter N. Recent advances in zinc enzymology. Chem. Rev. 1996;96:2375–2433. doi: 10.1021/cr950042j. [DOI] [PubMed] [Google Scholar]
- 2.Messerschmidt A, Huber R, Poulos T, Wieghardt K. Handbook of Metalloproteins. John Wiley & Sons; 2001. [Google Scholar]
- 3.Messerschmidt A, Bode O, Cygler M. Handbook of Metalloproteins. John Wiley & Sons; 2004. [Google Scholar]
- 4.Zheng YJ, Merz KM., Jr Mechanism of the human carbonic anhydrase II-catalyzed hydration of carbon dioxide. J. Am. Chem. Soc. 1992;114:10498–10507. [Google Scholar]
- 5.Ryde U. Carboxylate binding modes in zinc proteins: A theoretical study. Biophys. J. 1999;77:2777–2787. doi: 10.1016/S0006-3495(99)77110-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dudev T, Lim C. Metal binding in proteins: The effect of the dielectric medium. J. Phys. Chem. B. 2000;104:3692–3694. [Google Scholar]
- 7.Rogalewicz F, Ohanessian G, Gresh N. Interaction of neutral and zwitterionic glycine with Zn2+ in gas phase: Ab initio and SIBFA molecular mechanics calculations. J. Comput. Chem. 2000;21:963–973. [Google Scholar]
- 8.Rulisek L, Havlas Z. Theoretical studies of metal ion selectivity. 1. DFT calculations of interaction energies of amino acid side chains with selected transition metal ions (Co2+, Ni2+, Cu2+, Zn2+, Cd2+, and Hg2+) J. Am. Chem. Soc. 2000;122:10428–10439. [Google Scholar]
- 9.Deerfield DW, Carter CW, Pedersen LG. Models for protein-zinc ion binding sites. II. The catalytic sites. Int. J. Quant. Chem. 2001;83:150–165. [Google Scholar]
- 10.Remko M, Garaj V. Thermodynamics of binding of Zn2+ to carbonic anhydrase inhibitors. Mol. Phys. 2003;101:2357–2368. [Google Scholar]
- 11.Koca J, Zhan CG, Rittenhouse RC, Ornstein RL. Coordination number of zinc ions in the phosphotriesterase active site by molecular dynamics and quantum mechanics. J. Comput. Chem. 2003;24:368–378. doi: 10.1002/jcc.10217. [DOI] [PubMed] [Google Scholar]
- 12.Stote RH, Karplus M. Zinc binding in proteins and solution: A simple but accurate nonbonded representation. Proteins. 1995;23:12–31. doi: 10.1002/prot.340230104. [DOI] [PubMed] [Google Scholar]
- 13.Hu X, Shelver WH. Docking studies of matrix metalloproteinase inhibitors: zinc parameter optimization to improve the binding free energy prediction. J. Mol. Graph. Model. 2003;22:115–126. doi: 10.1016/S1093-3263(03)00153-0. [DOI] [PubMed] [Google Scholar]
- 14.Donini OA, Kollman PA. Calculation and prediction of binding free energies for the matrix metalloproteinases. J. Med. Chem. 2000;43:4180–4188. doi: 10.1021/jm000040d. [DOI] [PubMed] [Google Scholar]
- 15.Hou TJ, Guo SL, Xu XJ. Predictions of binding of a diverse set of ligands to gelatinase-A by a combination of molecular dynamics and continuum solvent models. J. Phys. Chem. B. 2002;106:5527–5535. [Google Scholar]
- 16.Pang Y, Xu K, Yazal J, Prendergast FG. Successful molecular dynamics simulation of the zinc-bound farnesyltransferase using the cationic dummy atom approach. Protein Sci. 2000;9:1857–1865. [PMC free article] [PubMed] [Google Scholar]
- 17.Pang YP. Novel zinc protein molecular dynamics simulations: Steps toward antiangiogenesis for cancer treatment. J. Mol. Model. 1999;5:196–202. [Google Scholar]
- 18.Hoops SC, Anderson KW, Merz KM. Force field design for metalloproteins. J. Am. Chem. Soc. 1991;113:8262–8270. [Google Scholar]
- 19.Hou TJ, Zhang W, Xu XJ. Binding affinities for a series of selective inhibitors of gelatinase-A using molecular dynamics with a linear interaction energy approach. J. Phys. Chem. B. 2001;105:5304–5315. [Google Scholar]
- 20.Toba S, Damodaran KV, Merz KM., Jr Binding preferences of hydroxamate inhibitors of the matrix metalloproteinase human fibroblast collagenase. J. Med. Chem. 1999;42:1225–1234. doi: 10.1021/jm980577f. [DOI] [PubMed] [Google Scholar]
- 21.Vedani A. YETI: An interactive molecular mechanics program for small-molecule protein complexes. J. Comput. Chem. 1988;9:269–280. [Google Scholar]
- 22.Vedani A, Huhta DW. A new force field for modeling metalloproteins. J. Am. Chem. Soc. 1990;112:4759–4767. [Google Scholar]
- 23.Piquemal JP, Williams-Hubbard B, Fey N, Deeth RJ, Gresh N, Giessner-Prettre C. Inclusion of the ligand field contribution in a polarizable molecular mechanics: SIBFA-LF. J. Comput. Chem. 2003;24:1963–1970. doi: 10.1002/jcc.10354. [DOI] [PubMed] [Google Scholar]
- 24.Sternberg U, Koch FT, Brauer M, Kunert M, Anders E. Molecular mechanics for zinc complexes with fluctuating atomic charges. J. Mol. Model. 2001;7:54–64. [Google Scholar]
- 25.El-Yazal J, Pang YP. Novel stable configurations and tautomers of the neutral and deprotonated hydroxamic acids predicted from high-level ab initio calculations. J. Phys. Chem. A. 1999;103:8346–8350. [Google Scholar]
- 26.Burton VJ, Deeth RJ, Kemp CM, Gilbert PJ. Molecular mechanics for coordination complexes: The impact of adding d-electron stabilization energies. J. Am. Chem. Soc. 1995;117:8407–8415. [Google Scholar]
- 27.Deeth RJ. The ligand field molecular mechanics model and the stereoelectronic effects of d and s electrons. Coord. Chem. Rev. 2001;212:11–34. [Google Scholar]
- 28.Warshel A, Levitt M. Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976;103:227–249. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 29.Singh UC, Kollmann PA. A combined ab initio quantum mechanical and molecular mechanical method for carrying out simulations on complex molecular systems: applications to the CH3Cl + Cl- exchange reaction and gas phase protonation of polyethers. J. Comput. Chem. 1986;7:718–730. [Google Scholar]
- 30.Tapia O, Lluch JM, Cardenas R, Andres J. Theoretical study of solvation effects on chemical reactions. A combined quantum chemical/Monte Carlo study of the Meyer-Schuster reaction mechanism in water. J. Am. Chem. Soc. 1989;111:829–835. [Google Scholar]
- 31.Field MJ, Bash PA, Karplus M. A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J. Comput. Chem. 1990;11:700–733. [Google Scholar]
- 32.Warshel A, Karplus M. Calculation of ground and excited state potential surfaces of conjugated molecules. I. Formulation and parametrization. J. Am. Chem. Soc. 1972;94:5612–5625. [Google Scholar]
- 33.Gao J, Furlani TR. Simulating solvent effects in organic chemistry: combining quantum and molecular mechanics. IEEE Comput. Sci. Eng. 1995;2:24–33. [Google Scholar]
- 34.Mulholland AJ. The QM/MM approach to enzymatic reactions. Theor. Comput.Chem. 2001;9:597–653. [Google Scholar]
- 35.Aqvist J, Warshel A. Simulation of enzyme reactions using valence bond force fields and other hybrid quantum/classical approaches. Chem. Rev. 1993;93:2523–2544. [Google Scholar]
- 36.Raha K, Merz KM., Jr A Quantum mechanics-based scoring function: Study of zinc ion-mediated ligand binding. J. Am. Chem. Soc. 2004;125:1020–1021. doi: 10.1021/ja038496i. [DOI] [PubMed] [Google Scholar]
- 37.Kollman P. Free energy calculations: Applications to chemical and biochemical phenomena. Chem. Rev. 1993;93:2395–2417. [Google Scholar]
- 38.van Gunsteren WF. Methods for calculation of free energies and binding constants: success and problems. In: van Gunsteren WF, Weiner PK, editors. Computer Simulation of Biomolecular Systems. Leiden: ESCOM; 1989. pp. 27–59. [Google Scholar]
- 39.Radmer RJ, Kollman PA. Free energy calculation methods: A theoretical and empirical comparison of numerical errors and a new method for qualitative estimates of free energy changes. J. Comput. Chem. 2003;18:902–919. [Google Scholar]
- 40.Srinivasan J, Cheatham TE, III, Cieplak P, Kollman PA, Case DA. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate-DNA helices. J. Am. Chem. Soc. 1998;120:9401–9409. [Google Scholar]
- 41.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 42.Rizzo RC, Toba S, Kuntz ID. A molecular basis for the selectivity of thiadiazole urea inhibitors with stromelysin-1 and gelatinase-A from generalized Born molecular dynamics simulations. J. Med. Chem. 2004;47:3065–3074. doi: 10.1021/jm030570k. [DOI] [PubMed] [Google Scholar]
- 43.Aqvist J, Medina C, Samuelsson JE. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994;7:385–391. doi: 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- 44.Hansson T, Aqvist J. Estimation of binding free energies for HIV proteinase inhibitors by molecular dynamics simulations. Protein Eng. 1995;8:1137–1144. doi: 10.1093/protein/8.11.1137. [DOI] [PubMed] [Google Scholar]
- 45.Aqvist J. Calculation of absolute binding free energies for charged ligands and effects of long-range electrostatic interactions. J. Comput. Chem. 1996;17:1587–1597. [Google Scholar]
- 46.Carlson HA, Jorgensen WL. An Extended Linear Response method for determining free energies of hydration. J. Phys. Chem. 1995;99:10667–10673. [Google Scholar]
- 47.Jones-Hertzog DK, Jorgensen WL. Binding affinities for sulfonamide inhibitors with human thrombin using Monte Carlo simulations with a Linear Response method. J. Med. Chem. 1997;40:1539–1549. doi: 10.1021/jm960684e. [DOI] [PubMed] [Google Scholar]
- 48.Lamb ML, Tirado-Rives J, Jorgensen WL. Estimation of the binding affinities of FKBP12 inhibitors using a linear response method. Bioorg. Med. Chem. 1999;7:851–860. doi: 10.1016/s0968-0896(99)00015-2. [DOI] [PubMed] [Google Scholar]
- 49.Wang W, Wang J, Kollman PA. What determines the van der Waals coefficient beta in the LIE (linear interaction energy) method to estimate binding free energies using molecular dynamics simulations? Proteins. 1999;34:395–402. [PubMed] [Google Scholar]
- 50.Sham YY, Chu ZT, Tao H, Warshel A. Examining methods for calculations of binding free energies: LRA, LIE, PDLD-LRA, and PDLD/S-LRA calculations of ligands binding to an HIV protease. Proteins. 2000;39:393–407. [PubMed] [Google Scholar]
- 51.Almlof M, Brandsdal BO, Aqvist J. Binding affinity prediction with different force fields: examination of the linear interaction energy method. J. Comput. Chem. 2004;25:1242–1254. doi: 10.1002/jcc.20047. [DOI] [PubMed] [Google Scholar]
- 52.Ljungberg KB, Marelius J, Musil D, Svensson P, Norden B, Åqvist J. Computational modelling of inhibitor binding to human thrombin. Eur. J. Pharm. Sci. 2001;12:441–446. doi: 10.1016/s0928-0987(00)00185-8. [DOI] [PubMed] [Google Scholar]
- 53.Hansson T, Marelius J, Aqvist J. Ligand binding affinity prediction by linear interaction energy methods. J. Comput. Aided Mol. Des. 1998;12:27–35. doi: 10.1023/a:1007930623000. [DOI] [PubMed] [Google Scholar]
- 54.Åqvist J, Marelius J. The linear interaction energy method for predicting ligand binding free energies. Comb. Chem. High Throughput Screen. 2001;4:613–626. doi: 10.2174/1386207013330661. [DOI] [PubMed] [Google Scholar]
- 55.Åqvist J, Luzhkov VB, Brandsdal BO. Ligand binding affinities from MD simulations. Acc. Chem. Res. 2002;35:358–365. doi: 10.1021/ar010014p. [DOI] [PubMed] [Google Scholar]
- 56.Duffy EM, Jorgensen WL. Prediction of properties from simulations: Free energies of solvation in hexadecane, octanol, and water. J. Am. Chem. Soc. 2000;122:2878–2888. [Google Scholar]
- 57.Head RD, Smythe ML, Oprea TI, Waller CL, Green SM, Marshall GR. VALIDATE: A new method for the receptor-based prediction of binding affinities of novel ligands. J. Am. Chem. Soc. 1996;118:3959–3969. [Google Scholar]
- 58.Tokarski JS, Hopfinger AJ. Prediction of ligand-receptor binding thermodynamics by free energy force field (FEFF) 3D-QSAR analysis: Application to a set of peptidometic renin inhibitors. J. Chem. Inf. Comp. Sci. 1997;37:792–811. doi: 10.1021/ci970006g. [DOI] [PubMed] [Google Scholar]
- 59.Venkatarangan P, Hopfinger AJ. Prediction of ligand-receptor binding thermodynamics by free energy force field three-dimensional quantitative structure-activity relationship analysis: Applications to a set of glucose analogue inhibitors of glycogen phosphorylase. J. Med. Chem. 1999;42:2169–2179. doi: 10.1021/jm980515p. [DOI] [PubMed] [Google Scholar]
- 60.Ortiz AR, Pisabarro MT, Gago F, Wade RC. Prediction of drug binding affinities by comparative binding energy analysis. J. Med. Chem. 1995;38:2681–2691. doi: 10.1021/jm00014a020. [DOI] [PubMed] [Google Scholar]
- 61.Huang D, Caflisch A. Efficient evaluation of binding free energy using continuum electrostatics solvation. J. Med. Chem. 2004;47:5791–5797. doi: 10.1021/jm049726m. [DOI] [PubMed] [Google Scholar]
- 62.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 63.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 64.Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 65.Bohm HJ. Prediction of binding constants of protein ligands: a fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J. Comput. Aided Mol. Des. 1998;12:309–323. doi: 10.1023/a:1007999920146. [DOI] [PubMed] [Google Scholar]
- 66.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 67.Muegge I, Martin YC. A general and fast scoring function for protein-ligand interactions: A simplified potential approach. J. Med. Chem. 1999;42:791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 68.Gohlke H, Hendlich M, Klebe G. Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 2000;295:337–356. doi: 10.1006/jmbi.1999.3371. [DOI] [PubMed] [Google Scholar]
- 69.Ishchenko AV, Shakhnovich EI. SMall Molecule Growth 2001 (SMoG2001): an improved knowledge-based scoring function for protein-ligand interactions. J. Med. Chem. 2002;45:2770–2780. doi: 10.1021/jm0105833. [DOI] [PubMed] [Google Scholar]
- 70.Sawa M, Kiyoi T, Kurokawa K, Kumihara H, Yamamoto M, Miyasaka T, Ito Y, Hirayama R, Inoue T, Kirii Y, Nishiwaki E, Ohmoto H, Maeda Y, Ishibushi E, Inoue Y, Yoshino K, Kondo H. New type of metalloproteinase inhibitor: Design and synthesis of new phosphonamide-based hydroxamic acids. J. Med. Chem. 2002;45:919–929. doi: 10.1021/jm0103211. [DOI] [PubMed] [Google Scholar]
- 71.Lukacova V, Zhang Y, Mackov M, Baricic P, Raha S, Calvo JA, Balaz S. Similarity of binding sites of human matrix metalloproteinases. J. Biol. Chem. 2004;279:14194–14200. doi: 10.1074/jbc.M313474200. [DOI] [PubMed] [Google Scholar]
- 72.Zoete V, Michielin O, Karplus M. Protein-ligand binding free energy estimation using molecular mechanics and continuum electrostatics. Application to HIV-1 protease inhibitors. J. Comput. Aided Mol. Des. 2003;17:861–880. doi: 10.1023/b:jcam.0000021882.99270.4c. [DOI] [PubMed] [Google Scholar]
- 73.Van Vlijmen HWT, Schaefer M, Karplus M. Improving the accuracy of protein pKa calculations: conformational averaging versus the average structure. Proteins. 1998;33:145–158. doi: 10.1002/(sici)1097-0134(19981101)33:2<145::aid-prot1>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 74.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kramer B, Rarey M, Lengauer T. Evaluation of the FlexX incremental construction algorithm for protein-ligand docking. Proteins. 1999;37:228–241. doi: 10.1002/(sici)1097-0134(19991101)37:2<228::aid-prot8>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 76.Hu X, Balaz S, Shelver WH. A practical approach to docking of zinc metalloproteinase inhibitors. J. Mol. Graphics. Model. 2004;22:293–397. doi: 10.1016/j.jmgm.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 77.Khandelwal A, Lukacova V, Kroll DM, Comez D, Raha S, Balaz S. Simulation-based predictions of binding affinities of matrix metalloproteinase inhibitors. QSAR Comb. Sci. 2004;23:754–766. [Google Scholar]
- 78.El-Yazal J, Pang YP. Proton dissociation energies of zinc-coordinated hydroxamic acids and their relative affinities for zinc: Insight into design of inhibitors of zinc-containing proteinases. J. Phys. Chem. B. 2000;104:6499–6504. [Google Scholar]
- 79.Browner MF, Smith WW, Castelhano AL. Matrilysin-inhibitor complexes: Common themes among metalloproteases. Biochemistry. 1995;34:6602–6610. doi: 10.1021/bi00020a004. [DOI] [PubMed] [Google Scholar]
- 80.Grams F, Crimmin M, Hinnes L, Huxley P, Pieper M, Tschesche H, Bode W. Structure determination and analysis of human neutrophil collagenase complexed with a hydroxamate inhibitor. Biochemistry. 1995;34:14012–14020. doi: 10.1021/bi00043a007. [DOI] [PubMed] [Google Scholar]
- 81.Grams F, Reinemer P, Powers JC, Kleine T, Pieper M, Tschesche H, Huber R, Bode W. X-ray structures of human neutrophil collagenase complexed with peptide hydroxamate and peptide thiol inhibitors - implications for substrate binding and rational drug design. Eur. J. Biochem. 1995;228:830–841. doi: 10.1111/j.1432-1033.1995.tb20329.x. [DOI] [PubMed] [Google Scholar]
- 82.Advanced Chemistry Development. Toronto, ON, Canada: 2001. ACD/pKa DB 4.59. [Google Scholar]
- 83.Johnson LL, Pavlovsky AG, Johnson AR, Janowicz JA, Man CF, Ortwine DF, Purchase CF, II, White AD, Hupe DJ. A rationalization of the acidic pH dependence for stromelysin-1 (matrix metalloproteinase-3) catalysis and inhibition. J. Biol. Chem. 2000;275:11026–11033. doi: 10.1074/jbc.275.15.11026. [DOI] [PubMed] [Google Scholar]
- 84.Smiesko M, Remko M. Coordination and thermodynamics of stable Zn(II) complexes in the gas phase. J. Biomol. Struct. Dyn. 2003;20:759–770. doi: 10.1080/07391102.2003.10506893. [DOI] [PubMed] [Google Scholar]
- 85.Linder DP, Rodgers KR. A theoretical study of imidazole- and thiol-based zinc binding groups relevant to inhibition of metzincins. J. Phys. Chem. B. 2004;108:13839–13849. [Google Scholar]
- 86.Luque I, Freire E. Structural parameterization of the binding enthalpy of small ligands. Proteins. 2002;49:181–190. doi: 10.1002/prot.10208. [DOI] [PubMed] [Google Scholar]
- 87.Katz RA, Skalka AM. The retroviral enzymes. Ann. Rev. Biochem. 1994;63:133–173. doi: 10.1146/annurev.bi.63.070194.001025. [DOI] [PubMed] [Google Scholar]
- 88.Still WC, Tempczyk A, Hawley RC. Hendrickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990;112:6127. [Google Scholar]
- 89.Eisenberg D, McLachlan AD. Solvation energy in protein folding and binding. Nature. 1986;319:199–203. doi: 10.1038/319199a0. [DOI] [PubMed] [Google Scholar]
- 90.Whittaker M, Floyd CD, Brown P, Gearing AJH. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem. Rev. 1999;99:2735–2776. doi: 10.1021/cr9804543. [DOI] [PubMed] [Google Scholar]
- 91.Beckett RP, Whittaker M. Matrix Metalloproteinase Inhibitors. Expert Opin. Ther. Pat. 1998:259–282. [Google Scholar]
- 92.Jacobson IC, Reddy PG, Wasserman ZR, Hardman KD, Covington MB, Arner EC, Copeland RA, Decicco CP, Magolda RL. Structure-based design and synthesis of a series of hydroxamic acids with a quaternary-hydroxy group in P1 as inhibitors of matrix metalloproteinases. Bioorg. Med. Chem. Lett. 1998;8:837–842. doi: 10.1016/s0960-894x(98)00125-5. [DOI] [PubMed] [Google Scholar]
- 93.Tamura Y, Watanabe F, Nakatani T, Yasui K, Fuji M, Komurasaki T, Tsuzuki H, Maekawa R, Yoshioka T, Kawada K, Sugita K, Ohtani M. Highly selective and orally active inhibitors of type IV collagenase (MMP-9 and MMP-2): N-sulfonylamino acid derivatives. J. Med. Chem. 1998;41:640–649. doi: 10.1021/jm9707582. [DOI] [PubMed] [Google Scholar]
- 94.O'Brien PM, Ortwine DF, Pavlovsky AG, Picard JA, Sliskovic DR, Roth BD, Dyer RD, Johnson LL, Man CF, Hallak H. Structure-activity relationships and pharmacokinetic analysis for a series of potent, systemically available biphenylsulfonamide matrix metalloproteinase inhibitors. J. Med. Chem. 2000;43:156–166. doi: 10.1021/jm9903141. [DOI] [PubMed] [Google Scholar]
- 95.Rowsell S, Hawtin P, Minshull CA, Jepson H, Brockbank SMV, Barratt DG, Slater AM, McPheat WL, Waterson D, Henney AM, Pauptit RA. Crystal structure of human MMP9 in complex with a reverse hydroxamate inhibitor. J. Mol. Biol. 2002;319:173–181. doi: 10.1016/S0022-2836(02)00262-0. [DOI] [PubMed] [Google Scholar]
- 96.Sybyl 6.91. St. Louis, MO: Tripos Inc.; [Google Scholar]
- 97.Jaguar 5.0. Portland, OR: Schrödinger Inc.; [Google Scholar]
- 98.QSite 2.5. Portland, OR: Schrödinger Inc.; [Google Scholar]
- 99.Murphy RB, Philipp DM, Friesner RA. A mixed quantum mechanics/molecular mechanics (QM/MM) method for large-scale modeling of chemistry in protein environments. J. Comput. Chem. 2000;21:1442–1457. [Google Scholar]
- 100.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. [Google Scholar]
- 101.Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- 102.Hay PJ, Wadt WR. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms scandium to mercury. J. Chem. Phys. 1985;82:270–283. [Google Scholar]
- 103.Wadt WR, Hay PJ. Ab initio effective core potentials for molecular calculations. Potentials for main group elements sodium to bismuth. J. Chem. Phys. 1985;82:284–298. [Google Scholar]
- 104.Clark M, Cramer RDI, van Op den Bosch N. Validation of the general purpose Tripos 5.2 force field. J. Comput. Chem. 1989;10:982–1012. [Google Scholar]
- 105.Mulliken RS. Electronic population analysis on LCAO-MO [linear combination of atomic orbital-molecular orbital] molecular wave functions. I. J. Chem. Phys. 1955;23:1833–1840. [Google Scholar]
- 106.Force field manual. St Louis, MO: Tripos Inc.; 2003. SYBYL 6.9; p. 164. [Google Scholar]
- 107.Homology module. San Diego, CA: Accelrys, Inc.; Insight II. [Google Scholar]
- 108.Origin 6.0. Northampton, MA: Microcal Software, Inc.; [Google Scholar]