

Abstract
MMPs and TACE (ADAM-17) assume independent, parallel or opposite pathological roles in cancer, arthritis, and several other diseases. For therapeutic purposes, selective inhibition of individual MMPs and TACE is required in most cases due to distinct roles in diseases and the need to preserve activities in normal states. Toward this goal, we compared force-field interaction energies of five ubiquitous inhibitor atoms with flexible binding sites of 24 known human MMPs and TACE. The results indicate that MMPs 1−3, 10, 11, 13, 16, and 17 have at least one subsite very similar to TACE. S3 subsite is the best target for development of specific TACE inhibitors. Specific binding to TACE compared to most MMPs is promoted by placing a negatively charged ligand part at the bottom of S2 subsite, at the entrance of S1’ subsite, or the part of S3’ subsite that is close to catalytic zinc. Numerous other clues, consistent with available experimental data, are provided for design of selective inhibitors.
Keywords: matrix metalloproteinase - MMP, tumor necrosis factor-α converting enzyme – TACE, ADAM-17, binding site, similarity, selectivity, ligand design, drug design
Introduction
The members of the metzincin superfamily of zinc-dependent endopeptidases exhibit similar structural topology of the hexa-peptide catalytic sites due to the need to accommodate the peptide backbone and the presence of conserved zinc-binding motif HEbxHxbGbxHz (b indicates a bulky hydrophobic amino acid, x is a variable amino acid, and z is a family specific amino acid). The motif comprises three His residues that bind zinc and Glu forming H-bond to the water molecule coordinated by zinc. Three families belonging to the metzincin superfamily are expressed in humans: MMPs, disintegrins-metalloproteinases (ADAMs and ADAM-TS) and astacins.1 Metzincins, synthesized as proenzymes, are mostly secreted and anchored to extracellular matrix (ECM) or to the cell surface. This compartmentalization limits their activity radius to specific substrates within the pericellular space.2 After proteolytic activation, metzincins cleave ECM components and other proteins, including cytokines, bioactive peptides, receptors, and ligands.3 Membrane-anchored metzincins exhibit sheddase activities, which are most notably manifested in activation of the membrane-bound proforms of bioactive peptides. Proteolytic activities of metzincins are controlled by endogenous tissue inhibitors and, in plasma, by α2-macroglobulin. Despite shape similarity in the binding sites,4 all three human metzincin families have specific roles under both normal and pathological conditions.
The current 24-member MMP family exhibits cell-type dependent expression levels that are generally low, yet prone to a rapid increase when tissues undergo remodeling, such as in inflammation, wound healing, development, and cancer. Due to the potential for both adhesion and proteolysis, the ADAM family, presently comprising 34 members, is implicated in processes such as sperm-egg fusion, cell-cell adhesion, ectodomain shedding, myoblast fusion, and development.5 Astacins include bone morphogenetic protein-1, as well as meprins α and β.1 Meprins are found in mammalian epithelial cells in the kidneys and the intestine.6 In contrast to other metzincins, the latent homooligomeric meprin complexes can move through extracellular spaces and only become activated at sites where relevant proteases are present.7
Several MMPs and TACE (ADAM-17) are best-characterized human metzincins. Pathological roles of MMPs and TACE, due to either (i) increased or (ii) decreased activities, can be classified as (a) independent, (b) parallel, and (c) opposite. Category i.a is represented by participation of TACE in diseases such as diabetes,8 HIV cachexia,9 and sepsis.10 Deficiency of MMP-14 causing craniofacial dysmorphism, arthritis, osteopenia, dwarfism, and fibrosis of soft tissues11 can be assigned to category ii.a. Category i.b includes inflammatory diseases, in which TACE is implicated with MMPs 1 and 3 in rheumatoid arthritis,12 and with MMPs 8 and 9 in bacterial meningitis.13,14 In category c, Alzheimer's disease can be mentioned, with the activation of TACE showing a positive effect15 and that of MMP-2 being implicated in the neuropathology of the disease.16
The roles of MMPs and TACE in cancer are most complex and span all categories. MMPs are synthesized by tumor cells and frequently also by surrounding stromal cells.17 The ECM-degrading abilities of MMPs 1, 2, 3, 7, 9, 11, and 14 are important for tumor invasion into surrounding connective tissues, crossing blood vessel walls, and metastasis to distant organs.18 Cleavage of a variety of nonmatrix substrates forms the basis for a wide range of other roles of MMPs in the processes such as cell death, cell proliferation, malignant conversion, tumor promotion, and tumor-associated angiogenesis.19 For instance, an increase in activities of MMPs 7 and 9 suppresses tumor progression by generation of Angiostatin from plasminogen in plasma and subsequent reduction in tumor vascularization.20 TACE causes tumorigenesis in nude mice by shedding transforming growth factor α (TGF-α) from its transmembrane form.21 Soluble TGF-α activates the receptor for the epidermal growth factor, a tyrosine kinase family implicated in the development of tumors of epithelial origin. Inhibition of sheddase activities of ADAMs is considered the primary cause of side effects of some cancerostatic MMP inhibitors.22
For therapeutic use, selective inhibition of individual MMPs and TACE is required in most cases, although parallel inhibition of certain MMPs and TACE might occasionally be beneficial. Selectivity is needed because of distinct roles of the enzymes in a disease and, also, to preserve their functions in normal states.23 This task is complicated by shape similarity of binding sites of TACE and MMPs even though: (i) the sequence identity of the catalytic domains of TACE and MMPs is very low,24 only 12−15%; (ii) the polypeptide chain of TACE catalytic domain is significantly longer; (iii) unlike MMPs, TACE is stable in the absence of calcium;25 and (iv) some differences between MMP-2 and TACE in binding a selective MMP inhibitor have been demonstrated.26 The usual targeting of one subsite (mostly S1’ subsite that is often considered the MMP specificity pocket) may lead to limited selectivity toward TACE vs a small group of MMPs that fades away when a broader set of MMPs is analyzed.27
This study aims at a detailed comparison of binding sites of TACE and MMPs using force-field interaction energies of atomic probes in binding sites, which are treated as partially flexible to account for induced fit. To assess the similarities in individual parts of the binding sites, linear regression analysis (LRA) was preferred to previously used Principal Component Analysis,28-30 since the latter requires discarding of the repulsive energies that are an important factor for selectivity. The most pronounced differences between the binding sites were analyzed to estimate the properties of functional groups conferring selectivity.
Results and Discussion
The hexapeptide binding site of TACE was compared with those of 24 human MMPs in terms of their force-field interaction energies with probes representing the atoms most frequently occurring in inhibitors and substrates.
Structures and Superposition
Catalytic domains of 24 human MMPs were represented by experimental structures and comparative models as previously described.31 For TACE, the Protein Data Bank32 file 1BKC was used. The residues of five subsites (S1, S1’, S2, S2’, and S3’) in TACE were identified based on the positions of catalytic zinc and coordinating His residues (His405, His409, His415), the bound ligand, and the peptide backbone structure in this part of the binding site (Figure 1). S2 subsite33 was identified as the loop between His409 and His415 residues that are coordinating catalytic zinc (Figure 1A). The third coordinating His405 residue and adjacent conserved Glu406 residue are at the entrance into S1’ subsite, which extends along the α-helix from the Glu406 and His405 residues down to Leu401 residue forming the bottom of S1’ subsite. The bottom part of S1’ subsite was identified from the relative positions of Leu401 in TACE and Tyr214 forming the bottom33 of S1’ subsite in MMP-7 (PDB file 1MMQ). Bound ligand is positioned between the catalytic zinc coordinated by His residues and a characteristic series of β-sheets. The β-sheet on the surface including Leu348 is forming one side of S3’ subsite, and extends34 to S1 subsite (Figure 1C). The stretch of residues outlining the other side of S3’ subsite, including Ala439, lies on the opposite side of the bound ligand and continues to S2’ subsite34 (Figure 1D). The opposing side chains of Ala439 and Leu348 are separating S3’ subsite from S1’ subsite.25 The structure of entire catalytic domain with highlighted residues forming the five subsites is shown in Figure 1E.
Figure 1.

Identification of subsites in the catalytic domain of TACE (PDB file 1BKC). A-D: bound ligand is shown in magenta, the identified residues outlining individual subsites are shown in red, catalytic zinc is shown as a blue sphere. A: S2 subsite is defined by residues between His409 and His415 coordinating catalytic zinc, B: S1’ subsite is defined by an α-helix positioned around the third His405coordinating catalytic zinc, C: S1 and one side of S3’ subsites are outlined by the β-sheet along the bound ligand, D: the residues outlining the other side of S3’ continuing into the S2’ subsite. E: the entire catalytic domain with highlighted residues forming the five subsites as identified in A-D. The parts of the ribbon representing individual subsites are marked as S1-red, S1’-yellow, S2-green, S2’-purple, S3’-cyan. The three coordinating His residues are highlighted in colors corresponding to the subsites and the bound ligand is shown in magenta.
The superposition of TACE to MMPs was based on the structural similarity of their binding sites. The catalytic domain of MMP-7 showed the highest sequence identity with TACE, therefore one of the X-ray structures of MMP-7 (PDB file 1MMQ) was selected as a reference structure for superposition with TACE, using the C, N, and O backbone atoms. Remaining MMPs were superimposed on MMP-7 as described previously.31
Superimposed TACE and MMP-7 structures exhibit an almost perfect overlap of the backbones in individual subsites, except S3 subsite (Figure 2). However, the residues forming individual subsites have considerable effect on the overall shapes of the binding sites. As indicated by the different backbone conformations (Figure 2), the major differences will be found in the shapes of S3 subsites. The side chains of some of the amino acids from adjacent S1 subsite also contribute to these differences. In MMP structures, one of the residues outlining S1 subsite is conserved His that coordinates the structural zinc atom and is well embedded in the structure of the protein. This conformation creates only a weak steric barrier between S1 and S3 subsites of MMPs, opening the possibility of a conformational change of bound inhibitor from the bottom of S1 to S3 subsite and vice versa. In TACE, the same position in S1 subsite is occupied by Leu350 residue whose side chain is not incorporated in the protein surface and points toward the solvent. This extended conformation, along with the backbone of S3 subsite, separates S1 and S3 subsites of the TACE binding site making it less likely, even for very flexible inhibitors, to switch the bound conformation from the S1/S3 region (Figure 2) to S3 subsite. The Tyr352 residue at the bottom of S1 subsite in TACE has more open conformation than the corresponding residues in MMP structures (e.g. Phe in MMP-8), creating a larger cavity that is extending from S1 subsite toward S3 subsite (S1/S3 in Figure 2). Significant difference can be found in the shape of S2 subsite, which is broader and deeper in MMPs as compared to TACE. The side chain of Val353 in TACE is pointing toward the S2 subsite, which is making it shallower than in MMP-8, where the side chain of Gln residue is oriented toward the solvent forming a deeper S2 subsite. Another interesting structural difference of TACE is that S1’ and S3’ subsites, separated on the surface by the side chains of residues Ala439 and Leu348, are merged below the surface.25 This feature will be discussed in more detail in section Specificity Determinants and Comparison with Inhibitor Data.
Figure 2.
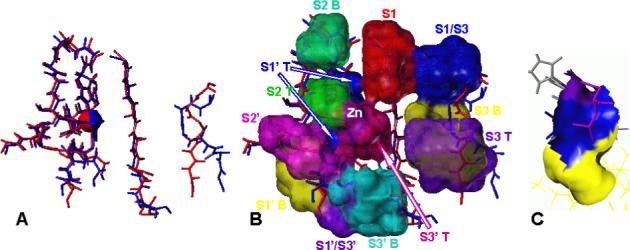
Superimposed structures of TACE (PDB file 1BKC, blue) and MMP-7 (PDB file 1MMQ, red). A - backbone atoms of residues in individual subsites, catalytic zinc (spheres), and side chains of the three coordinating His residues are shown. The backbone structures of five subsites show nearly perfect match, while those of S3 subsites differ and were not used in the superposition. B - the backbones are overlaid with the definitions of the parts of binding site as used in analyzing the differences. S1: red, S1’T: blue, S1’B: yellow, S1’/S3’: purple, S2T: yellow-green, S2B: blue-green, S2’: magenta, S1/S3: blue, S3T: purple, S3B: yellow, S3’T: dark magenta, S3’B: cyan. The parts of individual subsites close to the protein surface and/or to catalytic zinc are marked as top (T), the parts buried in the protein and/or distant from the catalytic zinc are marked as bottom (B). C – the detailed view of one of the subsites (S1’) showing the definition of the top (blue) and bottom (yellow) part of the subsite. The purple color marks the surface of catalytic zinc. The portion of bound ligand interacting with S1’ subsite (magenta) and zinc coordinating His residues are shown.
Comparison of Interaction Energies for Different Enzymes
Binding was examined for five probes representing the atom types most frequently occurring in the peptidic substrates and nonpeptidic inhibitors: negatively charged carbonyl oxygen, neutral sp3 carbon and sp3 oxygen, and positively charged sp3 nitrogen and hydrogen. For the calculation of interaction energies, the superimposed binding sites were enclosed in an irregular lattice of grid points taking actual contours of the binding sites. The lattice originated from a regularly spaced (1.5 Å) grid with the dimensions 18.0, 19.5 and 19.5 Å in x-, y-, and z-directions by dropping the grid points that were not involved in ligand binding inside the binding sites. The probe was placed in each of the grid points, and the Tripos force-field interaction energy35,36 was calculated for individual partially flexible binding sites.31 The interaction energies were compared with two goals: (i) to identify the MMPs and their subsites that are most similar to TACE and (ii) to delineate the properties of the regions responsible for specific interactions of TACE with individual probes.
The relatedness between parts of the binding sites of TACE and MMPs was evaluated by linear regression analysis (LRA) of the interaction energies of probes in pertinent grid points with TACE and MMPs and characterized by the slope, intercept, the correlation coefficient r, and the explained variance r2. For the specific MMP/probe/subsite combinations with strong linear relationships, similarity of the parts of binding sites was examined through the squared correlation coefficient R2 for the identity line between the interaction energies in TACE and MMP: R2 = 1 – SSR/SYY, with SSR = Σ(ETACE – EMMP)2 and SYY = Σ(ETACE-ĒTACE)2; both summations go through pertinent grid points; ETACE and EMMP are interaction energies of the probe with TACE and MMP, respectively; and ĒTACE is the average of interaction energies between the probe in the used grid points and TACE.
For the second goal, to define the regions of the binding sites, which are important for design of specific TACE inhibitors, the differences between interaction energies for TACE and all MMPs for each grid point were calculated and the grid points where the differences exceeded defined levels were considered responsible for specific interactions with ligands.
Overall Subsite Relatedness and Probe Selectivity
For all TACE/MMP pairs, the relationships between the interaction energies were examined for each probe or each subsite using the averages of explained variance r2 from LRA. The negatively charged probe was found to be the least distinctive (on average, 48% of the MMP data could be explained by TACE energies), while the neutral and positive probes were showing more or less similar levels of discrimination between TACE and MMPs (the average r2 was 0.35 for positive probes and 0.32 for neutral probes). For individual subsites, significantly higher relatedness, calculated as average r2 for all TACE/MMP pairs with combined probes, was found in subsites S3’, S1’ and S2 (average r2 = 0.47, 0.46, and 0.42, respectively) than in subsites S2’ and S1 (average r2 = 0.34 and 0.28, respectively), which were followed by the most different subsite S3 (average r2 = 0.17).
Pairwise Relatedness and Similarity of Subsites
To compare the binding sites of TACE and MMPs, pairwise correlations were performed in two phases. First, relatedness characterized by the strength of the linear relationships between the interaction energies of the probe with TACE and MMP was evaluated by LRA. The explained variances (r2) of these relationships (Figure 3) can serve as a guide for finding the most distinct parts of the binding sites, which have weak relationships between the two sets of interactions energies. Such different spots are to be targeted for design of inhibitors specific for TACE and, if necessary, for a selected group of MMPs involved in the same disease state, such as MMPs 1 and 3 in rheumatoid arthritis.12
Figure 3.

Relatedness of subsites in TACE and in individual MMPs expressed as the explained variances of linear relationships between the interaction energies for TACE versus individual MMPs for each probe and subsite. The rows correspond to individual MMPs. The six major columns correspond to individual probes (the 6th column represents combination of all probes), which are further divided into individual subsites. Summary correlations for entire binding site (all) and the binding site excluding S3 subsite (all*) were also calculated. The box colors indicate the level of explained variance (in %): 90−100 (red), 80−89 (amber), 70−79 (tan), 60−69 (gold), 50−59 (yellow), 40−49 (white), 30−39 (aqua), 20−29 (light blue), 10−19 (medium blue), and 0−9 (dark blue).
For the related TACE/MMP pairs with correlated interaction energies (r2 ≥ 0.7), similarity of subsites was evaluated by calculating the correlation coefficient R2 for the identity line between TACE/MMP interaction energies. The results led to identification of a number of MMP/probe/subsite combinations showing significant similarity to TACE (R2 > 0.7): (i) for MMP-1, negative oxygen probe in S1’, neutral sp3 oxygen probe in S2’ and S3’; (ii) for MMP-3, negative oxygen probe in S1’ and neutral sp3 oxygen probe in S3’; (iii) for MMP-10, negative oxygen probe in S3’ and positive hydrogen in S3’; (iv) for MMP-16, positive probes in S3’; (v) for MMPs 2, 11, 13, and 17, negative probe in S1’; and (vi) for MMP-8 neutral sp3 oxygen in S3’. For MMPs with multiple similar probe/subsite selections (MMPs 1, 3, 10, and 16), possible probe/subsite combinations have also been examined and all of them were similar as well.
Many related MMP/probe/subsite combinations with strong linear relationships of interaction energies to those of TACE (explained variance r2 > 0.7, Figure 3) are not significantly similar to their TACE counterparts (R2 < 0.7). This discrepancy is caused by the LRA-fitted line having either (i) the slope close to unity and a significant nonzero intercept or (ii) the slope different from unity. For electrostatic interactions, the first case is observed when the entire subsite has a higher polarity in TACE than in MMP, and in the second case polarity is continually changing along the subsite, i.e., the region in TACE is, compared to MMP, more positive on one side and more negative on the other side or vice versa.
This behavior is most frequently observed in S3’ subsite, where a number of TACE/MMP pairs exhibit strong linear relationships between the interaction energies with positive probes (hydrogen and sp3 nitrogen). However, the LRA-fitted lines often deviate from the identity line and their R2 values are low. A closer look at the data shows that the points could be split into two subsets fitted by different lines, one maintaining the slope close to unity and the other having a significantly lower slope (Figure 4). The spatial positions of the grid points associated with these two lines are different: the points defining the line with unity slope and a significant intercept are located in the part of S3’ subsite that is close, within 4 Å, to catalytic zinc. This result is in agreement with the analyses (see below) showing that this part of S3’ subsite is more polar in TACE than in MMPs. As pointed out previously for several MMPs,26 the second shell residues surrounding the catalytic zinc in TACE comprise three charged residues Glu406, Asp416, and Glu414, while MMPs 1, 3, 7−13, 17−23, and 25−28 contain one conserved Glu residue, and MMPs 2, 14−16, and 24 contain two Glu residues in the same region. As the probe moves further away from the catalytic zinc, polarity of the TACE S3’ subsite changes compared to that of MMPs, which results in the change of the slope. Despite the two outliers from the 4 Å region lying on the line representing the grid points farther from catalytic zinc (Figure 4), the described trends are obvious.
Figure 4.
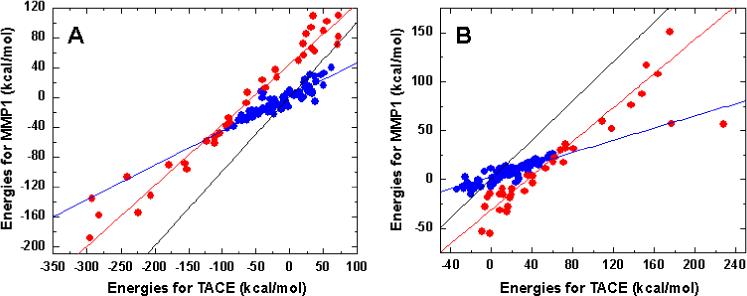
Correlation of interaction energies in S3’ subsite in TACE and MMP-1, illustrating changing properties of the subsite as it extends away from the catalytic zinc. A: negative carbonyl oxygen probe, B: positive sp3 nitrogen probe. Results for positive hydrogen probe were similar as shown in B. Colors of points indicate the distance from the catalytic zinc: close – within 4Å (red points), far – more than 4 Å (blue points). Fitted lines are as follows: identity line (black), region close to catalytic zinc (red), far region (blue).
The relatedness r2 (Figure 3) of individual subsites, in combination with similarity R2 (see above), can serve as a guide for selection of the group of MMPs that should be used to assess specificity of novel TACE inhibitors. If a selective inhibition of TACE versus more similar MMPs is achieved, a good selectivity with respect to the rest of the MMPs can also be expected.
Specificity Determinants and Comparison with Inhibitor Data
TACE and MMP binding sites were analyzed for differences that could aid in design of specific TACE inhibitors and substrates. For individual grid points, the differences between the interaction energies of the probe with MMP and TACE were calculated. The parts of the binding site where the energy differences were higher than a certain level, given as the fraction of the maximum interaction energy for TACE, were considered the points of specific interactions. When is the difference in interaction energies significant and will lead to specific binding? Our previous analysis of binding sites of MMPs showed that in order to obtain a good correlation between the experimental inhibitor or substrate data on two MMPs, the relatedness between the interaction energies of two binding sites had to be at least 70%.31 Here we detected that relatedness r2 ≤ 0.3 corresponds to regions that are showing clear preference for either MMP or TACE. The results for the difference levels 70% and higher are summarized in Figure 5.
Figure 5.
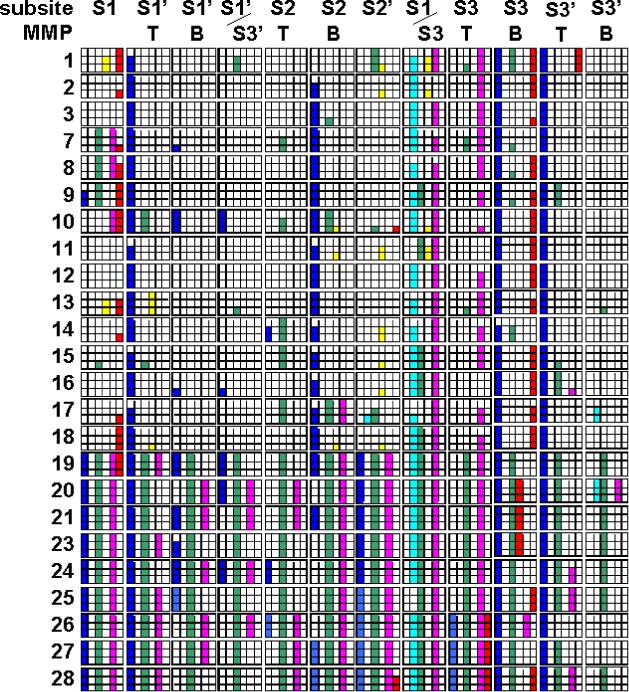
The regions of the binding site responsible for specific interactions of probes with TACE. The 24 major rows correspond to individual MMPs. Each major row is split into three additional rows according to difference level: 90, 80, and 70% (top to bottom). The major columns correspond to specific parts of the binding site (Figure 2). Each major column is split into six columns showing the preference for probe types by TACE or MMP: negative probe preferred by TACE (blue box) or by particular MMP (cyan box), neutral probe preferred by TACE (green box) or by particular MMP (yellow box), positively charged probe preferred by TACE (magenta box) or by particular MMP (red box).
For this analysis, four subsites (S1’, S2, S3, S3’) were split into two halves differing in the properties in TACE and MMPs. The parts marked as T (from top) are the halves of the deep pockets S1’ and S3 lying closer to the protein surface or the halves of the shallow cavities S2 and S3’ that are closer to catalytic zinc. The parts marked as B (from bottom) are the bottom parts of the deep pockets S1’ and S3 protruding into the interior of the protein or the parts of the shallow cavities S2 and S3’ lying farther away from the catalytic zinc. The exact positions of the T and B parts of the subsites are outlined in Figure 2. These subdivisions are in accord with the results of the LRA analyses that showed that the differences in properties of the subsite between TACE and MMP might not apply to the entire subsite but rather to smaller portions of the subsite (Figure 4).
The best target for design of specific TACE inhibitors is S3 subsite, as also indicated by simple superposition of the structures (Figure 2) and by LRA. In S3T part, the positively charged hydrogen and nitrogen probes have much stronger attractive interactions with TACE than with each of the MMPs. The result is corroborated by the much stronger attractive interactions of the negative oxygen probe with MMPs than with TACE in the same parts of the binding site. How are these differences associated with structures of TACE and MMPs? In the part of S3 subsite that continues from S1 subsite (S1/S3, Figure 2), the environment in MMPs is affected by the presence of structural zinc and a coordinating His residue; while in TACE, the Glu319 residue is playing the dominant role. The differences in the S3T part (Figure 2) are caused by the Asp344 residue in TACE and Pro in MMPs. On the other hand, the S3B part exhibits opposite tendencies: preference for the negative probe by TACE caused by the Gln341 residue and for positive probes by a number of MMPs. A common feature in both T and B parts of S3 subsite is the preference for neutral probe by TACE over a number of MMPs. However, as shown in the superposition of the structures (Figure 2), the main differences determining specific interactions with TACE or MMP are the shapes of S3 subsite.
Besides S3 subsite, the analysis of differences in interaction energies uncovered other parts of the binding site that could be targeted in the design of specific TACE inhibitors. Placement of the negatively charged functional group in part S3’T leads to a much stronger binding of ligand to TACE than to the majority of MMPs. As the ligand extends further toward the S3’B part, away from catalytic zinc, the charge should change to neutral or positive to preserve more attractive interactions with TACE over MMPs. The differences in the part S3’B are not as pronounced as in the immediate vicinity of the catalytic zinc. For most MMPs, the difference in attraction of the negative probe to MMP over TACE was only at 60% or lower levels (data not shown). The results obtained on the S3’T part of subsite are in a close agreement with the recently published experimental observation26 that the active site of TACE is more polar than that of MMP-2 as well as with the analysis of the residues surrounding catalytic zinc where TACE contains more charged residues than MMPs (see above).
Significant differences between the interaction of probes with TACE and MMPs are found in S2B part. To obtain preferential binding to TACE, a negatively charged functional group is required for MMPs 2, 3, and 7−18; in contrast, a positively charged substructure is needed for MMPs 20 and 23−25. The S2T part shows only mild preferences for the neutral probe in TACE over most MMPs.
To improve selectivity for TACE compared to specific MMPs, the ligand parts interacting with the S1’B part should be: (i) negatively charged for MMPs 7, 10 and 16; (ii) neutral or negatively charged for MMPs 19, 23, and 25; and (iii) neutral or positively charged for MMPs 20, 26, and 27. The negatively charged functional group in the S1’T part is preferred by TACE over all MMPs. This result is in agreement with several studies. Significant differences in polarity of binding sites, especially in the vicinity of catalytic zinc, have been shown.26 In the series of benzothiadiazepine inhibitors,37 the placement of the NH group in the S1’T part caused significant decrease in inhibition activity against TACE. In the series of peptidic inhibitors,38 the highest overall selectivity toward TACE, as compared to MMPs 1, 3 and 9, has been reported for compounds with thiophene ring in the S1’T part. The carbon atoms in the thiophene ring that are pointing toward the S1’ subsite have more negative partial charges39 than the carbons of the other functional groups, cyclohexane and isobutane, interacting with the S1’T part.
A comparison of interaction energies in S1 subsite indicates that, to obtain better ligand binding to TACE, a positive ligand part is required for MMPs 19−28, while for MMPs 1−10, 13, 14, 17, and 18 the part should be neutral or negative. The study on benzothiadiazepine inhibitors37 supports these results. The inhibitors with the ester or sulfonamide group in S1 subsite showed increased selectivity against TACE and MMPs 1, 2, and 9, while those with the amino group exhibited comparable inhibitory potencies.
The comparison of crystal structures of TACE and MMPs (Figure 6) uncovers another unique structural feature of TACE that is expected to play a significant role in specificity: S3’ subsite is, below the surface, merged with the S1’ pocket.25 The two subsites are on the surface separated by the side chains of residues Ala439 and Leu348.
Figure 6.
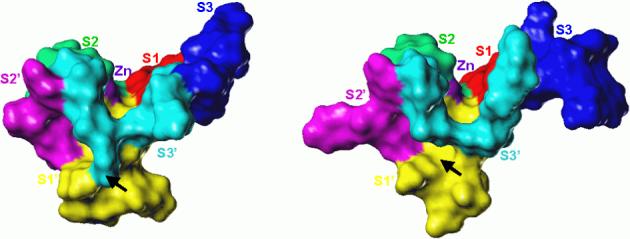
Comparison of S1’/S3’ regions of TACE (PDB file 1BKC, right) and MMP-8 (PDB file 1JAN, left). Colors identify catalytic zinc and individual subsites: zinc (purple), S1 (red), S1’ (yellow), S2 (green), S2’ (magenta), S3 (blue), S3’ (cyan). In TACE, S1’ and S3’ subsites are separated only on the surface by the side chains of the two residues (Ala439 and Leu348), while in the MMP structure the two subsites are completely separated. The arrows mark the regions where the subsites S1’ and S3’ are merged and separated in TACE and MMP, respectively.
Owing to this structural difference and the false31 reputation of S1’ subsite as an MMP specificity pocket, a number of attempts to design of specific TACE inhibitors focused on S1’ subsite. However, some inhibitor data indicate that other parts of the binding site might be involved in the specific interactions. A set of sixteen α-sulfonamide hydroxamates with butynyl group residing in S1’ subsite and variable functional groups interacting with S1 and S2’ subsites show selectivity toward TACE over MMP-1 ranging from none to ∼800 fold. Large scatter in selectivity was also observed for TACE over MMP-9 (none to 100 fold) and MMP-13 (none to ∼ 30) for the same set of compounds.40 A similar result was obtained from a series of lactams with (2-methyl-4-quinolinyl)-methoxy group interacting with S1’ subsite and variable groups interacting with other parts of the binding site.41 Selectivity of inhibitors against TACE versus MMPs 1, 2, and 9 ranges from single digits to thousands for each of the enzymes. The significance of the parts of the binding site other than S1’ subsite is also demonstrated by the data on a set of piperazine analogues.42 In a subset of seven compounds with varying functional groups interacting with S1’ subsite, there was only about a 3-fold difference in specificity of TACE over MMP-1. In contrast, another subset of seven compounds with invariant S1’ functional group and modifications in different parts of the molecule exhibited about a 70-fold change in specificity. Several studies38,43,44 have shown that, for the interaction with S1’ subsite, the size of the functional group alone is insufficient for obtaining selectivity to TACE, even when compared with MMP-1 which has a small S1’ pocket. Among α-substituted hydroxamates,45 selectivity against TACE vs MMP-1 significantly increased by replacing isobutyl in S1’ subsite with larger alkoxy-benzyl substituents. According to our results (Figure 5), a neutral probe, in this case a benzene ring, is highly preferred in TACE over MMP-1 in the region where S1’ subsite connects to S3’ subsite.
Presented results are useful for design of specific inhibitors of TACE or MMPs that is complicated by apparent high shape similarity of the binding sites. When chemical nature and flexibility of the binding sites are brought into analysis, overall similarity of binding sites of TACE and MMPs decreases to the point that can lead to an impression that the majority of synthesized inhibitors will be selective either to TACE or individual MMPs. However, interactions of inhibitors with multiple subsites require that similarities of all participating subsites be taken into account. The contributions from several subsites may mutually weaken or even cancel the overall effect. As shown in Figure 5, the selectivity of an inhibitor for TACE could be increased by placing positive or neutral functional group in S1 subsite, while in S3’ subsite the same functional groups would have no effect on selectivity. The binding modes of inhibitors should be identical in the targets assessed for selectivity. The physical barrier between S1 and S3’ subsites in MMP-7 is so negligible that simple, less anchored inhibitors could switch the orientations between the subsites. The inhibitors would be selective for TACE if binding in S1 subsites of both TACE and MMP-7. However, if they prefer S3’ subsite in MMP-7, selectivity for TACE is eliminated. We believe that only a careful selection of targeted subsites and properties of the functional groups interacting with them will lead to highly specific binding.
The guidelines for design of ligands specific for TACE or for TACE and a small group of MMPs, depending on the targeted disease, cannot be generalized into few simple rules. The results summarized in Figures 3 and 5 can be used to guide synthesis of inhibitors and selection of the group of MMPs that are similar to TACE and, consequently, provide a critical evaluation of the ligand selectivity. All analyses were performed without allowing the catalytic domains to do any large-scale motions. This fact limits the applicability of the results to small inhibitors and substrates. The bound conformations of inhibitors are determined by the most attractive interactions in the binding sites and may differ in individual enzymes. A difference in binding modes in the assessed enzymes precludes the selectivity estimation.
Methods
Structures
For TACE and nine types of MMPs, X-ray and/or NMR structures have been obtained from PDB database32 as follows: TACE – 1BKC, MMP-1 - 1AYK, 1CGL, 1HFC, 2AYK, 2TCL, 3AYK, 4AYK, 966C, 1CGE, 1CGF; MMP-2 - 1HOV, 1QIB; MMP-3 - 1B3D, 1B8Y, 1BIW, 1BM6, 1BQO, 1C3I, 1CAQ, 1CIZ, 1CQR, 1D5J, 1D7X, 1D8F, 1D8M, 1G49, 1G4K, 1GO5, 1HFS, 1HY7, 1SLM, 1SLN, 1UEA, 1UMS, 1UMT, 1USN, 2SRT, 2USN, 1QIA, 1QIC; MMP-7 - 1MMP, 1MMQ, 1MMR; MMP-8 – 1A85, 1A86, 1BZS, 1I73, 1I76, 1JAN, 1JAO, 1JAP, 1JAQ, 1JHI, 1JJ9, 1KBC, 1MMB, 1MNC; MMP-9 – 1GKC; MMP-12 – 1JIZ, 1JK3; MMP-13 – 1EUB, 1FLS, 1FM1, 456C, 830C; MMP-14 – 1BQQ, 1BUV. In the NMR files (shown in italics) containing multiple structures, only the first structure was selected. Comparative models of MMPs 2 and 9−18 were obtained from Mobashery et al.46 For MMPs 19−21 and 23−28, comparative models were constructed in our group as described in detail previously.31 For protonation states of ionizable residues, the Gasteiger charges for default atom definitions as implemented in the Sybyl/Biopolymer dictionary35 were used for all structures. Carboxyl groups of Glu and Asp are ionized, with delocalized electrons leading to two equivalent oxygen atoms with charges −0.550. The Lys residue contains a terminal sp3 nitrogen forming NH3+ group with overall charge +0.816. Arg has two equivalent terminal nitrogens with slightly positive partial charges (+0.205). All other residues are treated as nonionized.
The comparative modeling procedure has been validated by construction of the comparative model for MMP-2, for which X-ray structure as well as the comparative model prepared in different group46 was available. The final root-mean-squared deviation (RMSD) values between the X-ray structure and our comparative model, RMSD=1.614, and between X-ray structure and the published comparative model, RMSD=1.564, as well as the close correlation between their interaction energies with all five probes warrants their use for MMPs where no experimental structures are available. For a number of MMPs, multiple experimental structures, X-ray and/or NMR, were available from PDB database. For these types of MMPs, a representative set of interaction energies was created by averaging the interaction energies for all similar experimental structures. The experimental structures were considered similar if the correlation of their interaction energies was R2 > 0.70. For MMPs with multiple groups of similar structures, the groups including structures originating from more different laboratories were used to create the representative set of energies. This rule was implemented in order to capture the real similarities or differences between individual types of MMPs and TACE and not the subtle differences in individual experimental structures caused by different technique used for their determination.
Superposition
The alignment of catalytic domains of TACE and MMPs using sequence homology in Sybyl 6.9135 shows very low sequence identity. Superposition of TACE (PDB file 1BKC) and MMP-8 (PDB file 1A85, used previously31 as a template for superposition of MMP structures) guided by the alignment results was not satisfactory: the RMSD of the entire catalytic domain was about 12 Å, while RMSDs of the aligned catalytic domains of MMP structures were between 1 and 2 Å. For a direct comparison of the interaction energies, the TACE structure (PDB file 1BKC) had to be superimposed manually on one of the MMP structures. The catalytic domain of MMP-7 showed highest sequence identity with TACE, hence it was selected as a reference structure for superposition with TACE. The residues of five subsites (S1, S1’, S2, S2’, and S3’) in TACE were easily identified based on the positions of basic structural elements such as catalytic zinc and coordinating His residues (His405, His409, His415), the ligand bound in the catalytic domain, and the peptide backbone structure in this part of the binding site (Figure 1). Due to significant differences in residues forming the five subsites in MMP-7 and TACE, only the heavy backbone atoms were used for superposition. The RMSD for the atoms used for superposition was 1.024 Å, compared to RMSD=3.587 Å of the same set of atoms when the entire catalytic domain was superimposed onto MMP-8 as was done for all MMP structures.31 The superposition of the side chains was also satisfactory as was verified through RMSD for the side chains that are identical in catalytic sites of TACE and MMP-7. When only heavy backbone atoms of the five subsites were superimposed, RMSD = 0.906 compared to RMSD = 3.420 for the whole domain superposition. Superposition with other MMPs was satisfactory, as indicated by RMSD=1.065 for side chains identical to MMP-8 (PDB file 1A85), which was used previously31 as a template for superposition of MMP structures. It is worth noting that some of the side chains, which are identical in TACE and MMP-7 were different from the side chains identical in TACE and MMP-8.
Interaction Energy Calculations
Five probes imitating the most frequently occurring atoms in metzincin substrates and inhibitors were selected: carbonyl oxygen with a charge of −1.5, neutral sp3 oxygen and sp3 carbon, and sp3 nitrogen and hydrogen with charges of +1. The interaction energies between the TACE catalytic domain and the probes were calculated as described previously.31 Briefly, the probes were placed into each grid point of regularly spaced (1.5 Å) grid enclosing the binding site and the geometry of the complex was optimized. Overall size of the grid was 18.0, 19.5 and 19.5 Å in x-, y-, and z-directions. Due to the irregular shape of the binding site, a large portion of the grid points was omitted during calculation and the interaction energies of probes with the binding site were calculated only in the grid points pertinent to ligand binding. The coordinates of actual grid points used in calculation are listed in Supporting Information. During optimization, all residues including the backbone atoms with at least one atom within 2 Å distance from the probe were treated as flexible, and the rest of the protein and the position of the probe were fixed. For geometry optimization, the Tripos force field35,36 with Gasteiger charges47 on protein atoms and a charge of +2 on the zinc atom were used. After geometry optimization and energy calculation, the probe was removed and energy of the protein without the probe was calculated. The interaction energy of the probe with the catalytic domain was obtained as the difference of the force-field energies of the optimized protein with and without the probe.
Supplementary Material
Acknowledgment
This work was supported in part by the NIH NCRR grants 1 PP20 RR 15566 and 1 P20 RR 16471, as well as by the access to resources of the Computational Chemistry and Biology Network and the Center for High Performance Computing, both at the North Dakota State University.
Footnotes
Supporting Information Available: Coordinates and charges of the used structures of MMP and TACE catalytic domains, superimposed for the calculations, in the mol2 format, and coordinates of the used grid points. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Sternlicht MD, Coussens LM, Vu TH, Werb Z. Biology and regulation of the matrix metalloproteinases. In: Clendeninn NJ, Appelt K, editors. Matrix Metalloproteinase Inhibitors in Cancer Therapy. Humana Press; Totowa, NJ: 2000. pp. 1–37. [Google Scholar]
- 2.Chakraborti S, Mandal M, Das S, Mandal A, Chakraborti T. Regulation of matrix metalloproteinases: An overview. Mol. Cell. Biochem. 2003;253:269–285. doi: 10.1023/a:1026028303196. [DOI] [PubMed] [Google Scholar]
- 3.Gee JMW, Knowlden JM. ADAM metalloproteases and EGFR signaling. Breast Cancer Res. 2003;5:223–224. doi: 10.1186/bcr637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stahl M, Taroni C, Schneider G, Hoffmann F. Mapping of protein surface cavities and prediction of enzyme class by a self-organizing neural network. Protein Eng. 2000;13:83–88. doi: 10.1093/protein/13.2.83. [DOI] [PubMed] [Google Scholar]
- 5.Blobel CP. Functional and biochemical characterization of ADAMs and their predicted role in protein ectodomain shedding. Inflamm. Res. 2002;51:83–84. doi: 10.1007/BF02684007. [DOI] [PubMed] [Google Scholar]
- 6.Crisman JM, Zhang B, Norman LP, Bond JS. Deletion of the mouse meprin β metalloprotease gene diminishes the ability of leukocytes to disseminate through extracellular matrix. J. Immunol. 2004;172:4510–4519. doi: 10.4049/jimmunol.172.7.4510. [DOI] [PubMed] [Google Scholar]
- 7.Villa JP, Bertenshaw GP, Bylander JE, Bond JS. Meprin proteolytic complexes at the cell surface and in extracellular spaces. Biochem. Soc. Symp. 2003;70:53–63. doi: 10.1042/bss0700053. [DOI] [PubMed] [Google Scholar]
- 8.Morimoto Y, Nishikawa K, Ohashi M. KB-R7785, A novel matrix metalloproteinase inhibitor, exerts its antidiabetic effect by inhibiting tumor necrosis factor-α production. Life Sci. 1997;61:795–803. doi: 10.1016/s0024-3205(97)00561-4. [DOI] [PubMed] [Google Scholar]
- 9.Belec L, Meillet D, Hernvann A, Gresenguet G, Gherardi R. Differential elevation of circulating interleukin-1β, tumor necrosis factor α, and interleukin-6 in AIDS-associated cachectic states. Clin. Diagn. Lab. Immunol. 1994;1:117–120. doi: 10.1128/cdli.1.1.117-120.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsuji F, Oki K, Okahara A, Suhara H, Yamanouchi T, Sasano M, Mita S, Horiuchi M. Differential effects between marimastat, a TNF-α converting enzyme inhibitor, and anti-TNF-α antibody on murine models for sepsis and arthritis. Cytokine. 2002;17:294–300. doi: 10.1006/cyto.2002.1015. [DOI] [PubMed] [Google Scholar]
- 11.Holmbeck K, Bianco P, Caterina J, Yamada S, Kromer M, Kuznetsov SA, Mankani M, Robey PG, Poole AR, Pidoux I, Ward JM, Birkedal-Hansen H. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 1999;99:81–92. doi: 10.1016/s0092-8674(00)80064-1. [DOI] [PubMed] [Google Scholar]
- 12.MacNaul KL, Chartrain N, Lark M, Tocci MJ, Hutchinson NI. Discoordinate expression of stromelysin, collagenase, and tissue inhibitor of metalloproteinases-1 in rhemautoid human synovial fibroblasts. J. Biol. Chem. 1990;265:17238–17245. [PubMed] [Google Scholar]
- 13.Leib SL, Clements JM, Lindberg RLP, Heimgartner C, Loeffler JM, Pfister LA, Tauber MG, Leppert D. Inhibition of matrix metalloproteinases and tumour necrosis factor α converting enzyme as adjuvant therapy in pneumococcal meningitis. Brain. 2001;124(Part 9):1734–1742. doi: 10.1093/brain/124.9.1734. [DOI] [PubMed] [Google Scholar]
- 14.Meli DN, Loeffler JM, Baumann P, Neumann U, Buhl T, Leppert D, Leib SL. In pneumococcal meningitis a novel water-soluble inhibitor of matrix metalloproteinases and TNFα converting enzyme attenuates seizures and injury of the cerebral cortex. J. Neuroimmunol. 2004;151:6–11. doi: 10.1016/j.jneuroim.2004.01.026. [DOI] [PubMed] [Google Scholar]
- 15.Skovronsky DM, Moore DB, Milla ME, Doms RW, Lee VMY. Protein kinase C-dependent α-secretase competes with β-secretase for cleavage of amyloid-β precursor protein in transgolgi network. J. Biol. Chem. 2000;275:2568–2575. doi: 10.1074/jbc.275.4.2568. [DOI] [PubMed] [Google Scholar]
- 16.Deb S, Zhang JW, Gottschall PE. Activated isoforms of MMP-2 are induced in U87 human glioma cells in response to β-amyloid peptide. J. Neurosci. Res. 1999;55:44–53. doi: 10.1002/(SICI)1097-4547(19990101)55:1<44::AID-JNR6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 17.Nelson AR, Fingleton B, Rothenberg ML, Matrisian LM. Matrix metalloproteinases: Biologic activity and clinical implications. J. Clin. Oncol. 2000;18:1135–1149. doi: 10.1200/JCO.2000.18.5.1135. [DOI] [PubMed] [Google Scholar]
- 18.McCawley LJ, Matrisian LM. Matrix metalloproteinases: Multifunctional contributors to tumor progression. Mol. Med. Today. 2000;6:149–156. doi: 10.1016/s1357-4310(00)01686-5. [DOI] [PubMed] [Google Scholar]
- 19.Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pozzi A, Moberg PE, Miles LA, Wagner S, Soloway P, Gardner HA. Elevated matrix metalloprotease and Angiostatin levels in integrin α 1 knockout mice cause reduced tumor vascularization. Proc. Natl. Acad. Sci. U.S.A. 2000;97:2202–2207. doi: 10.1073/pnas.040378497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sunnarborg SW, Hinkle CL, Stevenson M, Russell WE, Raska CS, Peschon JJ, Castner BJ, Gerhart MJ, Paxton RJ, Black RA, Lee DC. Tumor necrosis factor-α converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J. Biol. Chem. 2002;277:12838–12845. doi: 10.1074/jbc.M112050200. [DOI] [PubMed] [Google Scholar]
- 22.Coussens LM, Fingleton B, Matrisian LM. Cancer therapy: Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 23.Close DR. Matrix metalloproteinase inhibitors in rheumatic diseases. Ann. Rheum. Dis. 2001;60:iii62–iii67. doi: 10.1136/ard.60.90003.iii62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moss MJ, Becherer D, Millia M, Pahel G, Lambert M, Andrews R, Frye S, Haffner C, Cowan D, Maloney P, Dixon EP, Jansen M, Vitek MP, Mitchell J, Leesnitzer T, Warner J, Conway J, Bickett DM, Bird M, Priest R, Reinhard J, Lin P. TNF-α converting enzyme. In: Bottomley KMK, Bradshaw D, Nixon JS, editors. Metalloproteinases as Targets for Antiinflammatory Drugs. Birkhauser; Berlin: 1999. pp. 187–204. [Google Scholar]
- 25.Maskos K, Fernandez-Catalan C, Huber R, Bourenkov GP, Bartunik H, Ellestad GA, Reddy P, Wolfson MF, Rauch CT, Castner BJ, Davis R, Clarke HRG, Petersen M, Fitzner JN, Cerretti DP, March CJ, Paxton RJ, Black RA, Bode W. Crystal structure of the catalytic domain of human tumor necrosis factor-α-converting enzyme. Proc. Natl. Acad. Sci. U.S.A. 1998;95:3408–3412. doi: 10.1073/pnas.95.7.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Solomon A, Rosenblum G, Gonzales PE, Leonard JD, Mobashery S, Milla ME, Sagi I. Pronounced diversity in electronic and chemical properties between the catalytic zinc sites of TACE and MMPs despite their high structural similarity. J. Biol. Chem. 2004;279:31646–31654. doi: 10.1074/jbc.M401310200. [DOI] [PubMed] [Google Scholar]
- 27.Wasserman ZR, Duan JJW, Voss ME, Xue C-B, Cherney RJ, Nelson DJ, Hardman KD, Decicco CP. Indentification of selectivity determinant for inhibition of tumor necrosis factor-α converting enzyme by comparative modeling. Chem. Biol. 2003;10:215–223. doi: 10.1016/s1074-5521(03)00044-9. [DOI] [PubMed] [Google Scholar]
- 28.Matter H, Schwab W. Affinity and selectivity of matrix metalloproteinase inhibitors: A chemometrical study from the perspective of ligands and proteins. J. Med. Chem. 1999;42:4506–4523. doi: 10.1021/jm990250u. [DOI] [PubMed] [Google Scholar]
- 29.Terp GE, Cruciani G, Christensen IT, Jorgensen FS. Structural differences of matrix metalloproteinases with potential implications for inhibitor selectivity examined by the GRID/CPCA Approach. J. Med. Chem. 2002;45:2675–2684. doi: 10.1021/jm0109053. [DOI] [PubMed] [Google Scholar]
- 30.Kastenholz MA, Pastor M, Cruciani G, Haaksma EE, Fox T. GRID/CPCA: A new computational tool to design selective ligands. J. Med. Chem. 2000;43:3033–3044. doi: 10.1021/jm000934y. [DOI] [PubMed] [Google Scholar]
- 31.Lukacova V, Zhang Y, Mackov M, Baricic P, Raha S, Calvo JA, Balaz S. Similarity of binding sites of human matrix metalloproteinases. J. Biol. Chem. 2004;279:14194–14200. doi: 10.1074/jbc.M313474200. [DOI] [PubMed] [Google Scholar]
- 32.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagase H. Substrate specificity of MMPs. Matrix Metalloproteinase Inhibitors in Cancer Therapy. 2000:39–66. [Google Scholar]
- 34.Yamamoto M, Tsujishita H, Hori N, Ohishi Y, Inoue S, Ikeda S, Okada Y. Inhibition of membrane-type 1 matrix metalloproteinase by hydroxamate inhibitors: An examination of the subsite pocket. J. Med. Chem. 1998;41:1209–1217. doi: 10.1021/jm970404a. [DOI] [PubMed] [Google Scholar]
- 35.Tripos Inc. Sybyl 6.9. 1699 South Hanley Road; St. Louis, MO: 2003. p. 63144. [Google Scholar]
- 36.Clark M, Cramer RD, III, van Op den Bosch N. Validation of the general purpose Tripos 5.2 force field. J. Comput. Chem. 1989;10:982–1012. [Google Scholar]
- 37.Cherney RJ, Duan JJW, Voss ME, Chen L, Wang L, Meyer DT, Wasserman ZR, Hardman KD, Liu RQ, Covington MB, Qian M, Mandlekar S, Christ DD, Trzaskos JM, Newton RC, Magolda RL, Wexler RR, Decicco CP. Design, synthesis, and evaluation of benzothiadiazepine hydroxamates as selective tumor necrosis factor-α converting enzyme inhibitors. J. Med. Chem. 2003;46:1811–1823. doi: 10.1021/jm020475w. [DOI] [PubMed] [Google Scholar]
- 38.Rabinowitz MH, Andrews RC, Becherer JD, Bickett DM, Bubacz DG, Conway JG, Cowan DJ, Gaul M, Glennon K, Lambert MH, Leesnitzer MA, McDougald DL, Moss ML, Musso DL, Rizzolio MC. Design of selective and soluble inhibitors of tumor necrosis factor-α converting enzyme (TACE). J. Med. Chem. 2001;44:4252–4267. doi: 10.1021/jm0102654. [DOI] [PubMed] [Google Scholar]
- 39.Belenkii LI, Suslov IA, Chuvylkin ND. Substrate and positional selectivity in electrophilic substitution reactions of pyrrole, furan, thiophene, and selenophene derivatives. Chem. Heterocycl. Comput. 2003;39:36–48. [Google Scholar]
- 40.Levin JI, Chen JM, Cheung K, Cole D, Crago C, Delos Santos E, Du X, Khafizova G, MacEwan G, Niu C, Salaski EJ, Zask A, Cummons T, Sung A, Xu J, Zhang Y, Xu W, Ayral-Kaloustian S, Jin G, Cowling R, Barone D, Mohler KM, Black RA, Skotnicki JS. Acetylenic TACE Inhibitors. Part 1. SAR of the acyclic sulfonamide hydroxamates. Bioorg. Med. Chem. 2003;13:2799–2803. doi: 10.1016/s0960-894x(03)00514-6. [DOI] [PubMed] [Google Scholar]
- 41.Duan JJ, Lu Z, Xue CB, He X, Seng JL, Roderick JJ, Wasserman ZR, Liu RQ, Covington MB, Magolda RL. Discovery of N-Hydroxy-2-(2-oxo-3-pyrrolidinyl)acetamides as potent and selective inhibitors of tumor necrosis factor-α converting enzyme (TACE). Bioorg. Med. Chem. Lett. 2003;13:2035–2040. doi: 10.1016/s0960-894x(03)00313-5. [DOI] [PubMed] [Google Scholar]
- 42.Letavic MA, Axt MZ, Barberia JT, Carty TJ, Danley DE, Geoghegan KF, Halim NS, Hoth LR, Kamath AV, Laird ER, Lopresti-Morrow LL, McClure KF, Mitchell PG, Natarajan V, Noe MC, Pandit J, Reeves L, Schulte GK, Snow SL, Sweeney FJ, Tan DH, Yu CH. Synthesis and biological activity of selective pipecolic acid-based TNF-α converting enzyme (TACE) inhibitors. Bioorg. Med. Chem. Lett. 2002;12:1387–1390. doi: 10.1016/s0960-894x(02)00183-x. [DOI] [PubMed] [Google Scholar]
- 43.Sawa M, Kiyoi T, Kurokawa K, Kumihara H, Yamamoto M, Miyasaka T, Ito Y, Hirayama R, Inoue T, Kirii Y, Nishiwaki E, Ohmoto H, Maeda Y, Ishibushi E, Inoue Y, Yoshino K, Kondo H. New type of metalloproteinase inhibitor: Design and synthesis of new phosphonamide-based hydroxamic acids. J. Med. Chem. 2002;45:919–929. doi: 10.1021/jm0103211. [DOI] [PubMed] [Google Scholar]
- 44.Kottirsch G, Koch G, Feifel R, Neumann U. β-aryl-succinic acid hydroxamates as dual inhibitors of matrix metalloproteinases and tumor necrosis factor-α converting enzyme. J. Med. Chem. 2002;45:2289–2293. doi: 10.1021/jm0110993. [DOI] [PubMed] [Google Scholar]
- 45.Barlaam B, Bird TG, van der Brempt L, Campbell D, Foster SJ, Maciewicz R. New α-substituted succinate-based hydroxamic acids as TNF-α convertase inhibitors. J. Med. Chem. 1999;42:4890–4908. doi: 10.1021/jm990377j. [DOI] [PubMed] [Google Scholar]
- 46.Massova I, Kotra LP, Mobashery S. Structural insight into the binding motifs for the calcium ion and the noncatalytic zinc in matrix metalloproteases. Bioorg. Med. Chem. Lett. 1998;8:853–858. doi: 10.1016/s0960-894x(98)00128-0. [DOI] [PubMed] [Google Scholar]
- 47.Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity: A rapid access to atomic charges. Tetrahedron. 1980;36:3219–3228. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.