

Abstract
Design of selective ligands for closely related targets is becoming one of the most important tasks in the drug development. New tools, more precise than fast scoring functions and less demanding than sophisticated Free Energy Perturbation methods, are necessary to help accomplish this goal. The methods of intermediate complexity, characterizing individual contributions to the binding energy, have been an area of intense research in the past few years. Our recently developed quantum mechanical/molecular mechanical (QM/MM) modification of the Linear Response (LR) method describes the binding free energies as the sum of empirically weighted contributions of the QM/MM interaction energies and solvent-accessible surface areas for the time-averaged structures of hydrated complexes, obtained by molecular dynamics (MD) simulations. The method was applied to published data on 27 inhibitors of matrix metalloproteinase-3 (MMP-3). The two descriptors explained 90% of variance in the inhibition constants with RMSE of 0.245 log units. The QM/MM treatment is indispensable for characterization of the systems lacking suitable force-field expressions. In this case, it provided characteristics of H-bonds of the inhibitors to Glu202, charges of binding site atoms, and accurate coordination geometries of the ligands to catalytic zinc. The geometries were constrained during the MD simulations, which characterized conformational flexibility of the complexes and helped in the elucidation of the binding differences for related compounds. A comparison of the presented QM/MM LR results with those previously published for inhibition of MMP-9 by the same set of ligands showed that the QM/MM LR approach was able to distinguish subtle differences in binding affinities for MMP-3 and MMP-9, which did not exceed one order of magnitude. This precision level makes the approach a useful tool for design of selective ligands to similar targets, because the results can be safely extrapolated to maximize selectivity.
Keywords: affinity prediction, conformational sampling, free energy of binding, Linear Response methods, metalloproteins, matrix metalloproteinases (MMPs), molecular dynamics, QM/MM methods, structure-based drug design, zinc coordination
INTRODUCTION
Numerous approaches to the prediction of binding affinities have been developed, ranging in complexity from high-speed scoring function to computationally expensive Free Energy Perturbation (FEP) and Thermodynamic Integration (TI) methods. Each group of methods has its own area of application.
The scoring functions1 are classified as force-field based approaches,2,3 empirical scoring methods4–7 utilizing simplified energy functions, and knowledge-based scoring functions,8–10 which are derived from the prevalencies of atom-atom contact pairs in complexes of known structures. The scoring functions were developed for virtual screening and docking applications where the speed is critical. Simplifications introduced to achieve the speed, e.g. the omission of both a thorough conformational sampling and description of some interactions, on the other hand, limit the accuracy of the binding affinity predictions to 2–3 orders of magnitude, which frequently results in ranking violations.
The other end of the spectrum is formed by FEP,11 TI,12 and similar13 methods, representing the most rigorous way of calculating binding free energy through slow, step-wise transformation of the ligand parts.14 FEP is used with molecular dynamics (MD) or Monte Carlo (MC) simulations to produce extensive conformational sampling. In order for the FEP results to converge, a large number of pair-wise interaction evaluations and small perturbation steps are required. Most of the computational time is spent on searching unimportant conformations, making the methods less attractive for applications, which require estimation of binding affinity of a large set of compounds.
The methods of intermediate complexity, bridging the gap between scoring functions and FEP/TI methods, have recently become the subject of intense research. Conformational sampling, as one of the most time-consuming processes, needs to have the extent carefully optimized, to speed up the computations. The Mining Minima approach15 deploys a specialized algorithm16 to find relevant low-energy conformations, which are then used in the computation of the configuration integral. The LR (Linear Response) approximation17–19 and the MM-PBSA approach20,21 methods perform MD conformational sampling only for the initial and final states of the complex formation.
In MM-PBSA method, the binding free energy is calculated as the sum of molecular mechanical (MM) energy, solvation free energy, and entropic contributions averaged over a series of snapshots from MD trajectories. The electrostatic contribution to the solvation term is calculated by solving the Poisson-Boltzmann (PB) equation, and the non-polar contribution to solvation is assumed to be proportional to the solvent-accessible surface area (SA or SASA in Equation (1)).
The LR approach estimates the free energy of binding as a linear combination of the average interaction energies and SASA:22–24
| (1) |
The term Δ for the energies is calculated as the difference of the energy ensemble averages (denoted by angle brackets) of the complex and free components (ligand and free protein). In the QM/MM LR approach, the van der Waals and electrostatic energies, separately scaled by adjustable coefficients α and β, were replaced by the single QM/MM interaction energy for the time-averaged structure of the protein-ligand complex, scaled by α.25 The third Δ-term is the difference in the ensemble averages of SASA for the solvated ligand in the bound and free states. Protein-solvent and solvent-solvent interactions are included in adjustable coefficients α, β, and γ.26–28 The van der Waals coefficient α depends on the studied compounds and/or the used force field (e.g. different values were obtained for thrombin23,26,29). The magnitude of α was analyzed with respect to hydrophobicity of the binding site.26 The coulombic scaling coefficient β, initially assumed to be equal to ½, based on the linear response of the surroundings to electric fields,17–19 varies with the ligand nature and ligand surroundings (protein or water).30–32
Our replacement of the MM-based van der Waals and electrostatic terms by the QM/MM term25 opened a new avenue of applications for the LR approach, especially for the ligand-protein interactions mediated by coordination, covalent, or highly polarized weak bonds. The approach, when applied to zinc-dependent matrix metalloproteinase (MMP) 9, significantly improved description and prediction ability of the LR approach as given by Equation (1). Consequently, it was interesting to examine whether the approach consistently achieves a high performance. In the present study, the QM/MM LR approach was used to describe inhibition of MMP-3 by the same set of ligands as previously used for MMP-9.33 Both MMPs are involved in etiology of cancer invasion, metastasis, inflammation, and necrosis, and MMP-3 is involved in arthritis. Maximization of the differences in binding is required for the development of selective inhibitors. The isoenzymes have quite similar binding sites,34,35 so the additional goal was to examine whether the QM/MM LR approach is capable of distinguishing small differences in binding. The majority of known MMP inhibitors are active against several MMPs and the development of selective inhibitors is a nontrivial task.34 From a broader viewpoint, the development of selective inhibitors for similar isoenzymes is becoming one of the main tasks of computational drug design. This task requires a new class of methods because high-throughput docking and scoring are too coarse to capture the subtle variation in binding to similar targets.
DATA AND METHODS
Structures and affinities of studied ligands
Published inhibitory potencies, characterized by the inhibition constants Ki at 37 °C, of a series of 27 hydroxamate derivatives33 towards MMP-3 were used (Table I below). No experimental errors were published for the inhibitory potencies. Our experience with similar analyses of MMP inhibition shows that better than 90% reproducibility can be achieved in carefully executed experiments with purified enzymes. Coordinates of the MMP-3 catalytic domain were taken from the X-ray crystal structure of the MMP-3 in the complex with N-[[2-methyl-4-hydroxycarbamoyl]but-4-yl-N]-benzyl-P-[phenyl]-P-[methyl] phosphonamide, as deposited in the PDB (file 1B3D).36 Among the 21 available PDB files of MMP-3 inhibitor complexes, the ligand in this file is most similar to the studied compounds.
Table I.
Structures, Inhibitory Potencies, and Simulation Data for Studied Inhibitors of MMP-3.
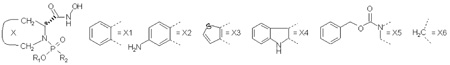 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No | X | R1 | R2 | log(1/Ki) | Linear Response terms in Steps 3 and 4 −Δ<x>, E in kcal/mol and SASA in Å2 | SD a | Bond length (Å) | Angle b | |||||||
| Exp. | Calc. | EvdW | Eel | EQM/MM. | SASA | O1-Zn | O2-Zn | O1-Zn-N2 | O1-Zn-N3 | N2-Zn-N3 | |||||
| 1 | X1 | CH2CH3 | 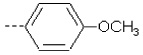 |
8.284 | 8.200 | 11.609 | 50.30 | 1890.75 | 464.46 | 0.155 | 1.844 | 2.017 | 104.6 | 167.5 | 87.9 |
| 2 | X1 | CH3 | 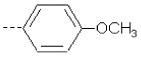 |
8.530 | 8.279 | 388.912 | 90.60 | 1924.28 | 477.20 | 0.765 | 1.956 | 1.947 | 104.9 | 166.3 | 88.8 |
| 3 | X1 |  |
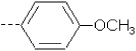 |
8.285 | 7.986 | 178.393 | 34.05 | 1863.53 | 428.70 | 0.456 | 1.946 | 1.948 | 94.8 | 179.0 | 84.3 |
| 4 | X1 | 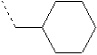 |
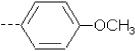 |
8.614 | 8.383 | 20.535 | 32.80 | 2067.29 | 491.82 | 0.213 | 1.965 | 1.953 | 108.0 | 164.1 | 86.9 |
| 5 | X1 | 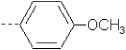 |
7.893 | 7.963 | 45.864 | 89.39 | 2493.80 | 411.07 | 0.318 | 1.947 | 1.947 | 100.6 | 171.4 | 87.8 | |
| 6 | X1 | (CH2)2N(CH2CH3)2 |  |
7.084 | 7.544 | 176.487 | 19.79 | 917.12 | 373.92 | 0.143 | 1.716 | 1.931 | 135.0 | 119.2 | 104.3 |
| 7 | X1 |  |
 |
8.193 | 7.915 | 58.753 | 107.98 | 4776.15 | 353.47 | 0.202 | 1.849 | 2.031 | 102.1 | 164.8 | 91.2 |
| 8 | X1 | (CH2)2OCH2CH3 |  |
8.265 | 8.314 | 80.24 | 60.85 | 4873.99 | 419.29 | 0.322 | 1.947 | 1.943 | 103.9 | 161.6 | 91.1 |
| 9 | X1 | CH2CH3 | 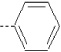 |
7.457 | 7.342 | 20.15 | 11.66 | 1720.62 | 322.14 | 0.26 | 1.946 | 1.946 | 106.6 | 167.2 | 86.6 |
| 10 | X1 | CH2CH3 | 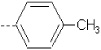 |
7.137 | 7.373 | 354.63 | 38.08 | 741.11 | 348.69 | 0.438 | 1.843 | 2.017 | 105.3 | 161.9 | 89.7 |
| 11 | X1 | CH2CH3 | 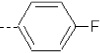 |
7.191 | 7.229 | 40.65 | 78.51 | 717.76 | 324.64 | 0.314 | 1.874 | 2.017 | 110.8 | 161.5 | 86.9 |
| 12 | X1 | CH2CH3 | 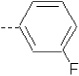 |
7.289 | 7.235 | 199.06 | 82.27 | 1341.75 | 312.27 | 0.743 | 1.867 | 1.987 | 115.2 | 153.8 | 90.3 |
| 13 | X1 | CH2CH3 |  |
7.321 | 7.397 | 193.55 | 20.27 | 1659.06 | 332.94 | 0.395 | 1.875 | 1.99 | 104.5 | 167.8 | 85.7 |
| 14 | X1 | CH2CH3 |  |
6.721 | 7.089 | 11.25 | 82.37 | 366.37 | 308.51 | 0.393 | 1.846 | 2.017 | 99.8 | 165 | 90.7 |
| 15 | X1 | CH2CH3 | 6.790 | 7.035 | 352.53 | 57.27 | 482.85 | 296.74 | 0.112 | 1.866 | 1.988 | 113.3 | 152.9 | 93.5 | |
| 16 | X1 | CH2CH3 |  |
7.174 | 7.131 | 15.46 | 93.94 | 910.25 | 303.89 | 0.246 | 1.797 | 2.071 | 115.0 | 154.3 | 90.4 |
| 17 | X1 | CH2CH3 | 7.529 | 7.382 | 51.40 | 27.01 | 957.79 | 345.61 | 0.541 | 1.81 | 1.593 | 137.4 | 130.7 | 91.9 | |
| 18 | X1 | CH2CH3 |  |
8.361 | 8.555 | 71.37 | 60.94 | 7570.98 | 401.89 | 0.281 | 1.875 | 1.995 | 119.3 | 150.4 | 89.7 |
| 19 | X1 | CH2CH3 | 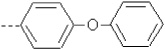 |
9.276 | 9.474 | 408.72 | 24.23 | 8665.43 | 534.54 | 0.101 | 1.849 | 2.026 | 124.0 | 142.8 | 93.2 |
| 20 | X1 | CH2CH3 | 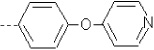 |
8.314 | 8.089 | 30.33 | 46.60 | 1497.50 | 454.18 | 0.131 | 1.758 | 2.039 | 116.0 | 148.8 | 93.5 |
| 21 | X1 | CH2CH3 | 9.301 | 9.432 | 36.51 | 67.38 | 7988.86 | 542.06 | 0.106 | 1.86 | 2.015 | 107.7 | 161.4 | 89.6 | |
| 22 | X1 | CH2CH3 | 7.936 | 7.545 | 46.63 | 135.80 | 3970.28 | 308.13 | 0.165 | 1.83 | 1.961 | 105.4 | 166.6 | 88.0 | |
| 23 | X2 | CH2CH3 | 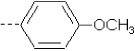 |
7.910 | 7.558 | 50.09 | 111.94 | 3283.05 | 325.14 | 0.17 | 1.864 | 1.988 | 94.7 | 163.6 | 84.1 |
| 24 | X3 | CH2CH3 | 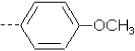 |
8.284 | 8.062 | 41.45 | 13.68 | 4667.35 | 380.83 | 1.78 | 2.052 | 2.028 | 104.8 | 158.2 | 85.5 |
| 25 | X4 | CH2CH3 | 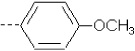 |
8.69 | 9.028 | 213.47 | 134.26 | 4963.11 | 538.76 | 0.399 | 1.821 | 1.875 | 125.2 | 130.7 | 101.1 |
| 26 | X5 | CH2CH3 | 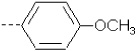 |
7.827 | 7.871 | 22.22 | 33.22 | 1625.96 | 414.26 | 0.213 | 1.861 | 2.057 | 110.4 | 161.0 | 86.0 |
| 27 | X6 | CH2CH3 | 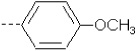 |
6.278 | 6.502 | 27.52 | 82.75 | 326.19 | 209.48 | 0.329 | 1.95 | 1.945 | 105.3 | 158.8 | 87.2 |
Standard deviation for the inhibitor atoms during the MD simulation.
Ideal values are 120° for trigonal bipyramid in all three cases, and 90°, 180°, and 90°, respectively, for the square pyramid. See Figure 1 for atom numbering.
Initial inhibitor/enzyme complexes
Ligand structures were sketched and minimized using the Sybyl 7.1 suite of programs37 running under Irix 6.5, and using the Tripos force field.38 After full geometry optimization using DFT/B3LYP-6-31G** approach,39 the Mulliken atomic charges40 were calculated. The FlexX program4,41 was used to dock the inhibitors into the active site of MMP-3 that was prepared as described below. The bound conformation in the active site was selected from the top 30 poses using, as the primary criterion, the distances between both hydroxamate oxygens and the catalytic zinc atom (2 Å preferred), and the FlexX ranking as the secondary criterion.42
Following hydrogen addition to the catalytic domain of MMP-3 (file 1B3D), the protein structure was relaxed by energy minimization with the coordinates of heavy atoms constrained. The complex was solvated with two layers of TIP3P water molecules,38 the positions of which were optimized by 10,000 cycles of conjugate gradient minimization, with constrained protein and ligand atoms. In the solvent equilibration phase, water molecules were subjected to MD simulation for 20 ps, keeping the protein and ligand atoms fixed. These steps allowed the solvent molecules to create a reasonable hydration shell of the ligand-receptor complex. The flexible regions was set to include protein atoms and water molecules closer than 5 Å to the ligand and minimized using MM with a distance-dependent dielectrics and conjugate gradient algorithm with a convergence criterion of 0.001 kcal/(mol Å).
QM/MM calculations
The QM/MM approach43 was used for the optimization of the best docked structures (Step 2), and for the calculation of the single-point QM/MM energies of the time-averaged structures, to be utilized in QM/MM LR correlations (Step 4). In both cases, the QM region consisted of side chains of His205 and His211, the backbone atoms and side chains of His201 and Glu202, the entire inhibitor, and the catalytic zinc ion. The backbone atoms were included to obtain valid QM/MM cuts. The rest of the protein was treated with OPLS-AA force field,44 including charges, with the protein and water outside 5 Å of the superimposed ligands frozen. For the QM part of the QM/MM calculations, DFT functional B3LYP45 was deployed. The 6-31G* basis set was used for the interface atoms between the QM and MM regions. The LAV3P** basis set was employed for geometry optimization. For Zn, S, and P atoms, this means the Los Alamos effective core potential (ECP)46,47 with all the s functions and the last p and d Gaussian uncontracted; for the remaining atoms, it implies 6-31G** basis set. All calculation converged by reaching the root mean squared change in density matrix elements of 5.0×10−6 or below. The B3LYP functional was selected for structure optimization, because it provides as good or better geometries and energies as those from correlated ab initio methods for the first-row transition metal complexes.48
Molecular dynamics simulations
Sybyl with Tripos force field38 was used to perform the MD simulations under isothermal/isobaric (NPT) conditions The QM/MM-optimized coordination bonds of the catalytic zinc, slightly different for each ligand, were restrained with a harmonic potential with the force constant 200 kcal/(mol×Å2) and power 2. The Mulliken charges40 for the QM/MM optimized structures were used for the QM region, and the Gasteiger-Hükel49charges were used for the rest of the protein and water. The time step of the simulations was 1 fs, with the nonbonded interactions updated every 25 fs, using the cutoff of 12 Ǻ. All residues within 6 Ǻ of any ligand atom were mobile, and the remaining part of the protein and water was kept frozen.
During the heating phase, the temperature of the system was raised from 0 to 300 K in 15 ps. After 100-ps equilibration run, the production phase was carried out at 300 K for 200 ps. To characterize flexibilities of the binding site and those of the ligands, the root mean square deviations (RMSD) of the c-α carbons and the inhibitor atoms, both with respect to the initial conformations, were calculated throughout the MD simulations. The time-averaged structures, obtained from the readings recorded in 100-fs intervals, were collected at appropriate times. A brief minimization, using the Tripos force field with a distance-dependent dielectrics and the Powell conjugate gradient algorithm with a convergence criterion of 0.001 kcal/(mol Å), produced structures with practically standard bond lengths and angles, with the dihedrals representing the ensemble. In each simulation run, at least one reading showed the geometry of the system that was very close to the relaxed time-averaged structure.
The time-averaged structures were previously shown to result in similar LR correlations as those using ensemble averages.50,51 The single-point QM/MM energy calculations on the time-averaged structures provided the substitute of the ensemble averages of the QM/MM energies, which was obtained as the difference between the QM/MM energies of the complex and the free components (ligand and protein). The QM and MM regions were defined in the time-averaged structures in the same way as in the previous part. The SASA terms were calculated using the ProsSat option in the Homology module of the Insight II modeling package.52 The 1.4-Å probe was used, although recent studies recommend smaller probes.53 Molecular images were produced using Chimera54 (Figure 1) and Sybyl38 (Figure 4).
Fig. 1.

The results of QM/MM optimization for the 27 hydroxamate complexes with MMP-3 (Table I). Average Mulliken charges were: 0.351 (H1), 0.438 (H2), O.633 (C1), 0.585 (C2), −0.217 (N1), −0.586 (N2), −0.583 (N3), −0.577 (N4), −0.633 (O1), −0.639 (O2), and 1.160 (Zn). Average bond lengths, in Å, were: 1.323 (C1-N1), 1.260 (C1=O1), 1.305 (C2-O3), 1.237 (C2=O4), 1.023 (N1-H1), 1.376 (N1-O2), 2.180 (N2-Zn), 2.158 (N3-Zn), 2.158 (N4-Zn), 2.072 (O1-Zn), 2.135 (O2-Zn), 1.406 (O2-H2), and 1.055 (03-H2). The maximum standard deviations were 11.0 % for charges and 2.3 % for distances.
Fig. 4.

Binding site superposition of MMP-3 (PDB file 1B3D) and MMP-9 (PDB file 1GKC) using c-α carbon atoms. MMP-9 is shown in atom colors while, for MMP-3, the colors identify individual subsites: S1 (orange), S1’ (magenta), S2 (green), S2’ (blue) and S3 (red). The sequences of the S1 and S2’ sites, with the conserved residues in black, are also shown.
Regression and cross-Validation
The experimental potencies were correlated with the LR terms using the linear least-squares fits55 based on Equation (1). The robustness and predictive abilities of the regression equations were probed by cross-validation. For this purpose, the fits to the potency data were generated leaving out one or more inhibitors from the calibration process. The resulting equation for each fit was used to predict the potencies of the omitted compounds. The predictive ability of Equation (1) was characterized by the root mean square errors (RMSE) of predicted potencies. We used the leave-one-out (LOO) approach and the leave-several-out (LSO) approach, where six inhibitors were randomly omitted, and the process was repeated 200 times.
RESULTS AND DISCUSSION
The aim of this study was to construct the QM/MM LR model for the inhibitory potencies of 27 hydroxamate inhibitors of MMP-3 (Table I) and compare the results with the previously established QM/MM LR correlation25 for MMP-9 inhibition by the same set of ligands. Both enzymes have very similar binding sites, as suggested by a high correlation (r2=0.849) of the experimental activities of the investigated inhibitors (see below).
Our previously developed four-tier approach was applied,25 comprising: (1) docking of ligands with the selection of the bound pose based on the metal binding geometry; (2) QM/MM minimization of the complexes, (3) MD simulation of the complexes, with the zinc coordination bonds harmonically restrained to the optimized geometry from Step 2; and (4) calculation of the single-point QM/MM energy and the SASA term for the time-averaged structures from Step 3. The outcomes of Step 4 were correlated with experimental data using Equation (1).
Catalytic zinc/inhibitors coordination
The inhibitors33 of MMP-3 were docked into the active site of MMP-3 (PDB file 1B3D)56 using FlexX.4,41 The ranking of poses was based upon the distance between the catalytic zinc and both hydroxamate oxygens as the primary criterion, and FlexX score as the secondary criterion. The average distances from catalytic zinc to carbonyl (O1) and hydroxyl (O2) oxygens for the selected complexes were 1.874±0.071 and 1.973±0.088, respectively. To reduce the QM/MM convergence time, for the top complexes of each ligand, the region within 6Å of the ligand was briefly MM-optimized by the conjugate gradient minimization.
The QM/MM geometry optimization resulted in remarkably similar structures of the hydroxamate groups of inhibitors bound to the catalytic zinc. The average bond lengths and average Mulliken charges of zinc and the coordinating atoms are shown in Figure 1. The zinc charge was also fairly similar in all complexes, with the average value of 1.160 |e|. In Toba’s57 and our25 work, the catalytic zinc center adopted a trigonal-bipyramidal geometry, and the charge on zinc was found to be +0.8 |e| and 1.059 |e| respectively. Ryde,58 Hou,59 and Hoop et al.60 found the partial charge on the zinc atom to be 0.488 |e|, 0.549 |e| and |0.688 |e|, respectively. These differences are expected and may be due to different enzyme-inhibitor system, and use of different Hamiltonians or different basis sets in the QM calculations.
The catalytic zinc in the X-ray structure of MMP-3 (PDB file 1B3D) is coordinated to the nitrogens of three histidines, His201Nε2 (N2), His205Nε2 (N3), and His211Nε2 (N4), as seen in Figure 1, with the coordination bond lengths close to 2.1 Å. In the QM/MM optimized structures, the bond lengths of the Zn-O1 and Zn-O2 bonds (Table I) were 2.072±0.033 Å and 2.135±0.067 Å, respectively, indicating similar roles of the two oxygens in zinc coordination. The average coordination bond lengths to histidine nitrogens were even more similar (Figure 1). The hydroxamate O2-bound hydrogen (H2) was in all cases transferred to Glu202 but remained H-bonded to hydroxamate.
To assess coordination geometry, the bond angles (Table I) were compared with ideal values for two possible penta-coordinated geometries: trigonal bipyramid (TB) and square-based pyramid (SP). The average angles O1-Zn-N2, O1-Zn-N3, and O1-Zn-N4 were 110.17±2.06°, 157.46±13.41°, and 84.97±3.27°, respectively (ideal values for TB: 120°, 120°, 90°; and for SP: 90°, 180°, 90°). The average angles O2-Zn-N2, -N3, and -N4 were 101.57±5.47°, 94.28±3.47°, and 156.02±4.79°, respectively (both TB and SP ideally adopt the values 90°, 90°, 180°). The average bond angle of O2-Zn-O1 is 77.90±2.06° , approaching the ideal value of 90° for both TB and SP configurations. The average bond angles for N2-Zn-N3, N3-Zn-N4 and N2-Zn-N4 are 89.85±4.58°, 98.90±4.60° and 98.74±3.81°, respectively (ideal values are 120°, 90°, 90° for TB; and 90°, 90°, 90° for SP). These data indicate that coordination geometry of the catalytic zinc is closer to SP, with N2 forming the out-of-plane vertex, than to TB.
For the free/bound hydroxamate groups, the average bond lengths were (in Å): C1=O1 1.233/1.260, C1-N1 1.357/1.323, N1-O2 1.400/1.376, and O2-H2 0.979/1.406, respectively. In the QM/MM geometry optimization of the complex, the proton from the hydroxamate OH group was transferred to the Glu202 oxygen but remained H-bonded to the parent oxygen. The H-bonds in MMP-3 complexes seem to be stronger than in other MMP complexes: the average distance between the Glu202 oxygen and O2 oxygen of the hydroxamate obtained here (2.461 Å) is shorter than equivalent distances for the hydroxamate complexes with MMP-7,61 MMP-8,62,63 and MMP-9,25 which all exceeded 2.6 Å. The average C1=O1, C1-N1, and N1-O2 bond lengths for the bound state obtained here (1.260, 1.323, and 1.376 Å) are closer to deprotonated hydroxamate (1.294, 1.317, and 1.378 Å) than to neutral hydroxamate with respective distances of 1.279, 1.337, and 1.416 Å.64 This fact is due to the H-bond formation and does not indicate that hydroxamates bind as ionized molecules. The same behavior was observed for the hydroxamate binding to MMP-9.25 The QM/MM approach handled the zinc-ligand charge transfer,65 bond length changes, polarization, and ionization66 upon binding to zinc, which would be difficult to describe by even advanced force fields.66–72
Dynamics of inhibitor complexes
The QM/MM optimized complexes were subjected to MD simulation using the harmonic bond length and angle constraints applied to the zinc coordination sphere. The average root-mean square deviation (RMSD) values of the backbone α-carbons, as compared to the initial conformation, for all the complexes were rather small, 0.150±0.030 Å per α-carbon, throughout the simulation (Figure 2). For all inhibitors, the average RMSD for the center of mass of all inhibitor atoms was 1.494±1.069 Å. Standard deviations (SD) of the individual RMSD values characterized flexibility of individual bound inhibitors. The RMSD and SD values for individual inhibitors are summarized in Table I and plotted in Figure 2. The RMSD values illustrate conformational sampling on the QM/MM optimized structures. The extents of movements, characterized by the SD values, of the α-carbons and of the inhibitor molecules are not correlated (the squared correlation coefficient r2 = 0.009). While inhibitor 24 (Table I) is the most moving ligand, the α-carbons exhibited the greatest mobility when ligand 3 was bound in the active site.
Fig. 2.

Movements of the backbone of MMP-3 and the inhibitor molecules during the MD simulations characterized by the average RMSD±SD Å from initial conformation.
After 200-ps MD simulations for complexes, free ligands, and free protein were completed, the differences, between the complex and free components (ligand and protein), in the van der Waals and electrostatic energies, as well as the differences in the SASA terms of the bound and free ligands, were calculated for the time-averaged structures. In the correlations using Equation (1), the results obtained after 5 ps were used and are listed in Table I. Longer simulation times provided similar samplings that did not improve the correlations using Equation (1) and the results are not shown. The time-averaged structures were in all studied cases similar to at least one structure that was sampled in the MD simulation. For the same time-averaged structures, single-point QM/MM interaction energies were calculated and are summarized in Table I. These quantities explain inhibitor potency trends for groups of closely related analogs.
Compounds 1, 9, and 14 differ in R2 substituents (4-methoxyphenyl, phenyl, and pyridine, respectively), resulting in the activity decreasing in this order. For all three compounds, both the QM/MM and SASA terms increase in this order (Table I).
Compounds 9 – 11 (Ki = 34.9, 72.9, and 64.4 nM, respectively) are structural analogs of compound 1 but exhibit lower activity as compared to 1 (Ki = 5.20 nM). The 4-methoxyphenyl group in R2 position of 1 is replaced with phenyl (9), 4-methylphenyl (10), and 4-fluorophenyl (11) substituents. Compounds 9 – 11 have higher QM/MM energy values than 1; however, they also exhibit diminished burial of SASA (Table I).
The binding affinity decreases stepwise on moving from ortho (13) to meta (12) and para (11) fluorine substituents. A similar trend was also observed for the SASA and QM/MM energy values, which are most favorable for 13 and least favorable for 11 (Table I). However, the fluorine substituent of 13 forms no H-bond interactions, in contrast to fluorines in 12 and 11, which are H-bonded with a water molecule and the backbone NH group of Tyr223, respectively.
QM/MM LR model for MMP-3
The LR correlations with experimental binding affinities using Equation (1) for both QM/MM and MD energies are summarized in Table II, along with the correlations from the preceding steps. Estimation of binding affinities based on simple linear regression of the FlexX scores with experimental activities resulted in poor correlation (r2=0.057). The use of the MM energies obtained from the minimized complexes in Step 1 increased the correlation to r2=0.425, with statistically significant van der Waals and electrostatic coefficients. The QM/MM energy minimization slightly improved the correlation in Step 2 to r2=0.465, thanks to the dominating SASA terms, because the QM/MM energies were not statistically significant. Inclusion of the QM/MM energy for the optimized geometry did not improve the correlation (r2=0.465), and the pertinent regression coefficient α had a negative sign as well as an inflated error. The cross-correlation between the QM/MM energy and the SASA term was weak (r2=0.228).
Table II.
Correlations of Inhibitory Potencies with the Energy and SASA Terms According to Eq. 1.
| Method | Step | α a (×10−3) | β b (×10−3) | γ c (×10−3) | κ | r2 | SD | F | RMSE | |
|---|---|---|---|---|---|---|---|---|---|---|
| LOO d | LSO e | |||||||||
| FlexX f | 1 | 96.46 ± 7.824 | NA g | NA | −5.717 ± 1.736 | 0.057 | 0.750 | 1.519 | 0.776 | 0.720 |
| MM Minimization | 1 | NS h | NS | 9.100 ± 2.120 | −4.533 ± 0.779 | 0.425 | 0.586 | 18.48 | 0.616 | 0.604 |
| QM/MM Minimization | 2 | NS | NS | 11.630 ± 2.490 | −3.400 ± 0.960 | 0.465 | 0.565 | 21.74 | 0.585 | 0.573 |
| MD | 3 | −0.590± 0.545 | 2.08±1.89 | 8.28± 0.819 | −4.592 ± 0.350 | 0.817 | 0.344 | 34.36 | 0.387 | 0.375 |
| MD | 3 | NS | NS | 7.990± 0.806 | −4.765± 0.318 | 0.797 | 0.348 | 98.22 | 0.358 | 0.355 |
| QM/MM MMP-3 | 4 | 0.127 ± 0.025 | NA | 5.880 ± 0.699 | −5.228 ± 0.242 | 0.903 | 0.245 | 112.28 | 0.262 | 0.264 |
| QM/MM MMP-9 | 3.592 ± 0.580 | NA | 7.543 ± 0.727 | −2.623 ±0.394 | 0.900 | 0.318 | 112.8 | 0.331 | 0.319 | |
| QM/MM MMP-3 & -9 | 4 | 0.044 ± 0.031 | NA | 8.360 ± 0.704 | −4.608 ± 0.267 | 0.768 | 0.425 | 86.26 | 0.433 | 0.537 |
| QM/MM MMP-3 & -9 | 4 | NS | NA | 8.680 ± 0.671 | −4.555 ± 0.267 | 0.759 | 0.429 | 167.38 | 0.435 | 0.525 |
Characterizes QM/MM interaction energy in Step 4 or van der Waals energy in Step 3.
Characterizes electrostatic energy.
Characterizes the solvent accessible surface area.
Leave-one-out cross-validation.
Leave-several-out cross-validation: random selection of 6-member test set, repeated 200 times.
The correlation between logKi and the FlexX scores, with the slope α and the intercept κ.
Not applicable.
Not significant.
The use of the energy terms from the MD conformational sampling in Step 3 increased the correlation to r2=0.817 (Table II). The van der Waals coefficient α had a negative value. For both α and β coefficients, the error terms were almost equal to the coefficient estimates. Refitting Equation (1) with only the SASA term after Step 3 resulted in correlation of r2=0.797. Compounds 1, 2, 4, 15, 16, and 25 (Table I), which saw the greatest description improvement in Step 3 (0.4 – 1.2 log units) compared to step 2, exhibit a significant change in the conformation of the bound ligand upon MD sampling. The change in the binding conformation is accompanied by a large increase of the SASA term, except for compounds 15 and 25. The cross-correlation of the van der Waals and SASA terms was very weak (r2=0.045).
The best model, obtained for Step 4, contained just the single point QM/MM energy for the time-averaged structures and the SASA term. This treatment was most beneficial for compounds 6, 7, 22–24 and 26, resulting in improvement of residuals by 0.2 – 0.4 log units. The correlation for all 27 compounds was characterized by r2 = 0.903 and the standard deviation SD = 0.245 log units (Table II), reflecting a good agreement between actual and calculated values (Table I). For each coefficient, the probability grater than the F ratio was less than 0.0001, implying a negligible likelihood of the random occurrence of a significant coefficient magnitude. The cross-correlation between the QM/MM energies and SASA terms was weak as indicated by the r2 value of 0.342. The dominance of the SASA terms, clearly seen in Table I, is probably reflecting the effect of the inhibitor burial in the binding site. This phenomenon was described previously by Luque and Freire in the analysis of binding energies of several ligand-protein complexes.73
The adjustable coefficient κ in Equation (1) yields an attractive term of about −5.228 log units (Table II), providing a base value for the inhibitors, which is then modulated by the QM/MM interaction and SASA terms. The values of the QM/MM terms (Table I) are negative and the associated positive coefficient α (Table II) implies that a strong interaction between the inhibitor and the binding site is important for inhibition. The SASA terms (Table I) are negative, implying burial of the surface area upon binding. The positive value of the associated coefficient γ (Table II) indicates that the removal of mostly hydrophobic surface area from the contact with water upon binding promotes the binding, as could be expected for the hydrophobic effect.74 The obtained values of γ (Table II: 0.0058-0.011) are in the same range as the slopes of the linear dependencies of solvation free energies of various compounds on SASA: 0.006,22 0.007,75 and 0.016.76 The separation of the overall SASA into polar and non-polar components resulted in a slightly better correlation (r2=0.912, Step 4); with the coefficient values (1.106±0.340)×10−3 and (4.570±1.080)×10−3 for polar and nonpolar components, respectively.
The robustness of the regression equations and their predictive abilities were extensively probed by cross-validation. For this purpose, the fits to the potency data are generated leaving out one or more inhibitors from the calibration process. The resulting equation for each fit is used to predict the potencies of the omitted compounds. The leave-one-out (LOO) procedure and especially the leave-several-out (LSO) procedure with 200 random selections of 6-member test sets provided a thorough evaluation. The RMSE values using LOO (0.262) and LSO (0.264) were only slightly higher than the RMSE value of the complete data set (0.245). It appears that the conformational sampling included in the time-averaged structures (Step 3) and a good description of the zinc coordination bonds (Step 4) are jointly required for a good correlation with experimental inhibitory potencies. The improving quality of correlations by addition of individual Steps is documented in Figure 3.
Fig. 3.

Experimental vs. calculated inhibition potencies of hydroxamates against MMP-3 as obtained by FlexX docking with the zinc binding-based selection of modes in Step 1 (green), MM minimization in Step 1 (cyan), QM/MM minimization in Step 2 (blue), MD simulation with constrained zinc bonds in Step 3 (red), and QM/MM energy calculations for the time averaged structures from MD simulation in Step 4 (black). All correlation results are summarized in Table II. The identity line is drawn for visual aid.
QM/MM LR models for MMP-3 and MMP-9
A similar study as described above for MMP-3 was performed earlier for the same set of compounds inhibiting MMP-9.25 The protocols were slightly different: here the free ligands were simulated separately, while in the former study, the ligand ensembles from the MD simulation of the complexes were used, as previously suggested.21,77 When structures of entire binding sites are compared, they are quite similar for both isoenzymes.34,35 The superposition of the binding sites of MMP-3 and MMP-9 using C-α carbon atoms is shown in Figure 4.
As follows from the docking studies, the R1 substituents bind to subsite S1, and the R2 substituents occupy subsite S2’ in all cases. The X group is an extension of the zinc-binding group, and interacts with subsite S1. Most of the inhibitors reported in the literature bind extensively on the primed side. The non-primed side seems to be more open, with less specific binding pockets, where the inhibitor-binding features could be more diverse. This behavior can also be seen in studied hydroxamate complexes. Beyond subsite S1, the protein cleft is wide open, and the inhibitor binding is more flexible and less specific. Binding on the non-primed side may improve overall binding and enhance inhibitor potency.
For MMP-3, subsite S1 consists of residues Gly161, Asn162, Val163, Leu164, Ala165, His166, Ala167, and Tyr168. The residues forming subsite S2’ are Ala217, Leu218, Met219, Tyr220, Pro221, Leu222, and Tyr223. MMP-9 differs from MMP-3 in the following substitutions. In subsite S1, which is the most different of all six subsites, Gly161, Asn162, Val163, and Tyr168 in MMP-3 were replaced with Asp185, Gly186, Leu187, and Phe192, respectively, in MMP-9. In subsite S2’, the only difference is that Leu222 was replaced with Met422.
In spite of the relative similarity of the binding sites and mutual dependence of the inhibitory potencies in the two isoenzymes (Figure 5, statistical indices below), the QM/MM energies and SASA terms are not correlated: r2 = 0.353 and 0.669, respectively. A major difference is seen in the range of the QM/MM energies, which is about an order of magnitude larger for MMP-3 than for MMP-9. In addition to the slightly different protocols, possible reasons include shorter and stronger H-bonds of the inhibitors with Glu202, and different coordination geometries (SP in MMP-3 and TB in MMP-9), resulting in different coordination bond energies. Additional H-bonds of some ligands with the MMP-3 binding site also contribute to the difference in the QM/MM energies. The hydroxamate NH group forms a H-bond with the backbone carbonyl oxygen of Ala165 for all studied compounds except 15 and 17. Compounds 17, 21, and 27 have the NH group of hydroxamate engaged in an intramolecular H-bond with the P=O group. The P-O group of 20 and 21, and the P=O group of all the ligands except 15, 17, 20, and 21 form a H-bond with the backbone amide group of Leu164. The P=O group is also H-bonded with the backbone amide of Ala165 except for 4, 8, 13, 17, 22, 23, and 27. The side chain amino group and alkoxy oxygen of 15 form H-bonds with the backbone carbonyl of Leu222 and the backbone amide group of Leu164, respectively. The pyridine nitrogen of 14 and alkoxy oxygen of 17 form H-bonds with the backbone amide of Tyr223.
Fig. 5.

Inhibitory potencies of MMP-3 vs. those of MMP-9: experimental (full points) and predicted (open points) values for MMP-3, predicted values for MMP-9 (crosses scattered around the dotted identity line). The full line represents the correlation between the experimental MMP-3 and MMP-9 values.
The magnitude of the coefficient α that is scaling the QM/MM energies in Equation (1) is much smaller (1.27×10−4, Table 2) for MMP-3 than the magnitude for MMP-9 (3.59×10−3) that was obtained for the same set of inhibitors using a slightly different protocol. This difference precludes an assessment of the transferability of the energy-scaling coefficient α between the MMP-3 and MMP-9 systems. The desolvation-characterizing coefficients γ remain remarkably similar in the MMP-3 and MMP-9 systems, and fit the range of the experimentally observed values for desolvation of organic molecules.22,75,76
Prediction of the MMP-3 data set using Equation (1) with the coefficients obtained from MMP-9 resulted in RMSE of 0.447 log units, whereas prediction of the MMP-9 data set with MMP-3 coefficients resulted in RMSE of 0.480 log units. The combined model (N=57) using 27 MMP-3 and 28 MMP-9 (the energy and SASA terms values in Table 1 of the original report25) inhibitors exhibits an intermediate agreement with experiment: r2 = 0.759 and RMSE = 0.429.
The ability of the QM/MM LR approach to truly capture small differences of binding affinities for similar metalloproteins is illustrated in Figure 5. The predicted affinities for MMP-9 are scattered around the dotted identity line. The experimental affinities for MMP-3 follow the same trend as the affinities for MMP-9, as indicated by the full line with the slope of 0.744±0.063 and the intercept of −1.845±0.509 (r2 = 0.849). In spite of this similarity, the predictions for MMP-3 (open points) are closely associated with the experimental MMP-3 data. The differences between affinities for MMP-3 and MMP-9 are most significant for the four best MMP-9 inhibitors, grouped in the low left corner (18 – 21, Table I). The predictions for MMP-3 and MMP-9 in this region do not show any tendency to averaging and clearly distinguish between the two targets. As follows from Figure 5, increased affinities to both enzymes can be expected to result in increased selectivities. In addition, selectivity may originate from other factors, as seen for the 3rd and 4th most active MMP-9 inhibitors (18 and 20 in Table I) In case of MMP-3, these inhibitors form H-bonds with Leu164 and Ala-165 whereas, no such H-bonds are observed for MMP-9 (Leu188 and Ala189, equivalent residues).
CONCLUSIONS
A predictive model for the binding affinities of MMP-3 inhibitors was developed using our QM/MM LR method that is based on a combined use of docking, QM/MM calculations, and MD simulations. Binding affinities are described as a linear combination of QM/MM interaction energies and SASA changes upon binding, both calculated for the relaxed time-averaged structures from MD simulations. The application of the approach to the published data on MMP-3 inhibition by 27 hydroxamates resulted in an excellent correlation (r2=0.903) between experimental and calculated values, as well as in stable and accurate predictions. A comparison with the previously developed QM/MM LR correlation for MMP-9 showed that the QM/MM and SASA terms for MMP-3 and MMP-9 are not correlated. The QM/MM energy terms for MMP-3 are about ten times larger than those for MMP-9, due to different coordination geometry of the catalytic zinc, additional or stronger H-bonds, and possibly due to slightly different computational protocols. The QM/MM LR approach captured the subtle differences of about one order of magnitude in the binding affinities for the most active inhibitors binding to both isoenzymes. This level of precision makes the approach a useful tool for the design of specific inhibitors for individual members of enzyme families.
ACKNOWLEDGMENTS
This work was supported in part by the NIH NCRR grants 1P20RR15566 and 1P20RR16741, as well as by the access to resources of the Computational Chemistry and Biology Network and the Center for High Performance Computing, both at the North Dakota State University.
REFERENCES
- 1.Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: Current status and future challenges. Proteins. 2006;65:15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- 2.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 3.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 4.Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J Mol Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 5.Bohm HJ. The development of a simple empirical scoring function to estimate the binding constant for a protein-ligand complex of known three-dimensional structure. J Comput Aided Mol Des. 1994;8:243–256. doi: 10.1007/BF00126743. [DOI] [PubMed] [Google Scholar]
- 6.Jain AN. Scoring noncovalent protein-ligand interactions: a continuous differentiable function tuned to compute binding affinities. J Comput Aided Mol Des. 1996;10:427–440. doi: 10.1007/BF00124474. [DOI] [PubMed] [Google Scholar]
- 7.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 8.Muegge I, Martin YC. A general and fast scoring function for protein-ligand interactions: A simplified potential approach. J Med Chem. 1999;42:791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 9.Gohlke H, Hendlich M, Klebe G. Knowledge-based scoring function to predict protein-ligand interactions. J Mol Biol. 2000;295:337–356. doi: 10.1006/jmbi.1999.3371. [DOI] [PubMed] [Google Scholar]
- 10.Ishchenko AV, Shakhnovich EI. SMall Molecule Growth 2001 (SMoG2001): An improved knowledge-based scoring function for protein-ligand interactions. J Med Chem. 2002;45:2770–2780. doi: 10.1021/jm0105833. [DOI] [PubMed] [Google Scholar]
- 11.Kollman P. Free energy calculations: Applications to chemical and biochemical phenomena. Chem Rev. 1993;93:2395–2417. [Google Scholar]
- 12.van Gunsteren WF. In: Methods for calculation of free energies and binding constants: Success and problems. van Gunsteren WF, Weiner PK, editors. Leiden: Escom: Computer simulation of biomolecular systems; 1989. pp. 27–59. [Google Scholar]
- 13.Radmer RJ, Kollman PA. Free energy calculation methods: A theoretical and empirical comparison of numerical errors and a new method for qualitative estimates of free energy changes. J Comput Chem. 2003;18:902–919. [Google Scholar]
- 14.Helms V, Wade RC. Computational alchemy to calculate absolute protein-ligand binding free energy. J Am Chem Soc. 1998;120:2710–2713. [Google Scholar]
- 15.Chang C, Gilson MK. Free energy, entropy, and induced fit in host-guest recognition: Calculations with the second-generation mining minima algorithm. J Am Chem Soc. 2004;126:13156–13164. doi: 10.1021/ja047115d. [DOI] [PubMed] [Google Scholar]
- 16.Chang C, Gilson MK. TORK: Conformational analysis method for molecules and complexes. J Comput Chem. 2003;24:1987–1998. doi: 10.1002/jcc.10325. [DOI] [PubMed] [Google Scholar]
- 17.Åqvist J, Medina C, Samuelsson JE. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994;7:385–391. doi: 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- 18.Hansson T, Åqvist J. Estimation of binding free energies for HIV proteinase inhibitors by molecular dynamics simulations. Protein Eng. 1995;8:1137–1144. doi: 10.1093/protein/8.11.1137. [DOI] [PubMed] [Google Scholar]
- 19.Åqvist J. Calculation of absolute binding free energies for charged ligands and effects of long-range electrostatic interactions. J Comput Chem. 1996;17:1587–1597. [Google Scholar]
- 20.Srinivasan J, Cheatham TE, III, Cieplak P, Kollman PA, Case DA. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate-DNA helices. J Am Chem Soc. 1998;120:9401–9409. [Google Scholar]
- 21.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc Chem Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 22.Carlson HA, Jorgensen WL. An Extended Linear Response method for determining free energies of hydration. J Phys Chem. 1995;99:10667–10673. [Google Scholar]
- 23.Jones-Hertzog DK, Jorgensen WL. Binding affinities for sulfonamide inhibitors with human thrombin using Monte Carlo simulations with a Linear Response method. J Med Chem. 1997;40:1539–1549. doi: 10.1021/jm960684e. [DOI] [PubMed] [Google Scholar]
- 24.Lamb ML, Tirado-Rives J, Jorgensen WL. Estimation of the binding affinities of FKBP12 inhibitors using a Linear Response method. Bioorg Med Chem. 1999;7:851–860. doi: 10.1016/s0968-0896(99)00015-2. [DOI] [PubMed] [Google Scholar]
- 25.Khandelwal A, Lukacova V, Comez D, Kroll DM, Raha S, Balaz S. A combination of docking, QM/MM Methods, and MD simulation for binding affinity estimation of metalloprotein ligands. J Med Chem. 2005;48:5437–5447. doi: 10.1021/jm049050v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang W, Wang J, Kollman PA. What determines the van der Waals coefficient beta in the LIE (linear interaction energy) method to estimate binding free energies using molecular dynamics simulations? Proteins. 1999;34:395–402. [PubMed] [Google Scholar]
- 27.Sham YY, Chu ZT, Tao H, Warshel A. Examining methods for calculations of binding free energies: LRA, LIE, PDLD-LRA, and PDLD/S-LRA calculations of ligands binding to an HIV protease. Proteins. 2000;39:393–407. [PubMed] [Google Scholar]
- 28.Almlof M, Brandsdal BO, Aqvist J. Binding affinity prediction with different force fields: examination of the linear interaction energy method. J Comput Chem. 2004;25:1242–1254. doi: 10.1002/jcc.20047. [DOI] [PubMed] [Google Scholar]
- 29.Ljungberg KB, Marelius J, Musil D, Svensson P, Norden B, Aqvist J. Computational modelling of inhibitor binding to human thrombin. Euro J Pharm Sci. 2001;12:441–446. doi: 10.1016/s0928-0987(00)00185-8. [DOI] [PubMed] [Google Scholar]
- 30.Hansson T, Marelius J, Åqvist J. Ligand binding affinity prediction by linear interaction energy methods. J Comput Aided Mol Des. 1998;12:27–35. doi: 10.1023/a:1007930623000. [DOI] [PubMed] [Google Scholar]
- 31.Åqvist J, Marelius J. The linear interaction energy method for predicting ligand binding free energies. Comb Chem High Throughput Screen. 2001;4:613–626. doi: 10.2174/1386207013330661. [DOI] [PubMed] [Google Scholar]
- 32.Åqvist J, Luzhkov VB, Brandsdal BO. Ligand binding affinities from MD simulations. Acc Chem Res. 2002;35:358–365. doi: 10.1021/ar010014p. [DOI] [PubMed] [Google Scholar]
- 33.Sawa M, Kiyoi T, Kurokawa K, Kumihara H, Yamamoto M, Miyasaka T, Ito Y, Hirayama R, Inoue T, Kirii Y, Nishiwaki E, Ohmoto H, Maeda Y, Ishibushi E, Inoue Y, Yoshino K, Kondo H. New type of metalloproteinase inhibitor: design and synthesis of new phosphonamide-based hydroxamic acids. J Med Chem. 2002;45:919–929. doi: 10.1021/jm0103211. [DOI] [PubMed] [Google Scholar]
- 34.Lukacova V, Zhang Y, Mackov M, Baricic P, Raha S, Calvo JA, Balaz S. Similarity of binding sites of human matrix metalloproteinases. J Biol Chem. 2004;279:14194–14200. doi: 10.1074/jbc.M313474200. [DOI] [PubMed] [Google Scholar]
- 35.Lukacova V, Zhang Y, Kroll DM, Raha S, Comez D, Balaz S. A comparison of the binding sites of matrix metalloproteinases and tumor necrosis factor-α converting enzyme Implications for selectivity. J Med Chem. 2005;48:2361–2370. doi: 10.1021/jm0491703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen L, Rydel TJ, Gu F, Dunaway CM, Pikul S, Dunham KM, Barnett BL. Crystal structure of the stromelysin catalytic domain at 2.0 Å resolution: Inhibitor-induced conformational changes. J Mol Biol. 1999;293:545–557. doi: 10.1006/jmbi.1999.3147. [DOI] [PubMed] [Google Scholar]
- 37.Sybyl 7.1. St. Louis: Tripos Inc; 2006. [Google Scholar]
- 38.Clark M, Cramer RDI, van Opdenbosch N. Validation of the general purpose Tripos 5.2 force field. J Comput Chem. 1989;10:982–1012. [Google Scholar]
- 39.Jaguar. Portland: Schröinger, LLC; 2003. [Google Scholar]
- 40.Mulliken RS. Electronic population analysis on LCAO-MO [linear combination of atomic orbital-molecular orbital] molecular wave functions. J Chem Phys. 1955;23:1833–1840. [Google Scholar]
- 41.Kramer B, Rarey M, Lengauer T. Evaluation of the FlexX incremental construction algorithm for protein-ligand docking. Proteins. 1999;37:228–241. doi: 10.1002/(sici)1097-0134(19991101)37:2<228::aid-prot8>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 42.Hu X, Balaz S, Shelver WH. A practical approach to docking of zinc metalloproteinase inhibitors. J Mol Graphics Model. 2004;22:293–307. doi: 10.1016/j.jmgm.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 43.QSite. Portland: Schröinger, LLC; 2003. [Google Scholar]
- 44.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS allatom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 45.Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 46.Hay PJ, Wadt WR. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms scandium to mercury. J Chem Phys. 1985;82:270–283. [Google Scholar]
- 47.Wadt WR, Hay PJ. Ab initio effective core potentials for molecular calculations. Potentials for main group elements sodium to bismuth. J Chem Phys. 1985;82:284–298. [Google Scholar]
- 48.Ryde U. Carboxylate binding modes in zinc proteins: a theoretical study. Biophys J. 1999;77:2777–2787. doi: 10.1016/S0006-3495(99)77110-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sybyl Force Field Manual. St Louis: Tripos Inc; 2003. p. 164. [Google Scholar]
- 50.van Vlijmen HWT, Schaefer M, Karplus M. Improving the accuracy of protein pKa calculations: Conformational averaging versus the average structure. Proteins. 1998;33:145–158. doi: 10.1002/(sici)1097-0134(19981101)33:2<145::aid-prot1>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 51.Zoete V, Michielin O, Karplus M. Protein-ligand binding free energy estimation using molecular mechanics and continuum electrostatics. Application to HIV-1 protease inhibitors. J Comput Aided Mol Des. 2003;17:861–880. doi: 10.1023/b:jcam.0000021882.99270.4c. [DOI] [PubMed] [Google Scholar]
- 52.Insight II: Homology Module. San Diego: Accelrys, Inc; 2003. [Google Scholar]
- 53.Doweyko AM, Johnson SR. A novel method to simulate the hydrophobic effect and its application to the ranking of host/guest complexes. J Chem Inf Model. 2006;46:2563–2573. doi: 10.1021/ci600188h. [DOI] [PubMed] [Google Scholar]
- 54.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin CF. UCSF Chimera - A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 55.Origin, Version 6.0. Northampton: Microcal Software, Inc; 1999. [Google Scholar]
- 56.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Toba S, Damodaran KV, Merz KM. J Binding preferences of hydroxamate inhibitors of the matrix metalloproteinase human fibroblast collagenase. J Med Chem. 1999;42:1225–1234. doi: 10.1021/jm980577f. [DOI] [PubMed] [Google Scholar]
- 58.Ryde U. Molecular dynamics simulations of alcohol dehydrogenase with a four- or five-coordinate catalytic zinc ion. Proteins. 1995;21:40–56. doi: 10.1002/prot.340210106. [DOI] [PubMed] [Google Scholar]
- 59.Hou TJ, Zhang W, Xu XJ. Binding affinities for a series of selective inhibitors of gelatinase-A using molecular dynamics with a linear interaction energy approach. J Phys Chem B. 2001;105:5304–5315. [Google Scholar]
- 60.Hoops SC, Anderson KW, Merz KM. Force field design for metalloproteins. J Am Chem Soc. 1991;113:8262–8270. [Google Scholar]
- 61.Browner MF, Smith WW, Castelhano AL. Matrilysin-inhibitor complexes: Common themes among metalloproteases. Biochemistry. 1995;34:6602–6610. doi: 10.1021/bi00020a004. [DOI] [PubMed] [Google Scholar]
- 62.Grams F, Crimmin M, Hinnes L, Huxley P, Pieper M, Tschesche H, Bode W. Structure determination and analysis of human neutrophil collagenase complexed with a hydroxamate inhibitor. Biochemistry. 1995;34:14012–14020. doi: 10.1021/bi00043a007. [DOI] [PubMed] [Google Scholar]
- 63.Grams F, Reinemer P, Powers JC, Kleine T, Pieper M, Tschesche H, Huber R, Bode W. X-ray structures of human neutrophil collagenase complexed with peptide hydroxamate and peptide thiol inhibitors - Implications for substrate binding and rational drug design. Eur J Biochem. 1995;228:830–841. doi: 10.1111/j.1432-1033.1995.tb20329.x. [DOI] [PubMed] [Google Scholar]
- 64.El-Yazal J, Pang YP. Proton dissociation energies of zinc-coordinated hydroxamic acids and their relative affinities for zinc: Insight into design of inhibitors of zinc-containing proteinases. J Phys Chem B. 2000;104:6499–6504. [Google Scholar]
- 65.Raha K, Merz KM. A quantum mechanics-based scoring function: Study of zinc ion-mediated ligand binding. J Am Chem Soc. 2004;125:1020–1021. doi: 10.1021/ja038496i. [DOI] [PubMed] [Google Scholar]
- 66.El-Yazal J, Pang YP. Novel stable configurations and tautomers of the neutral and deprotonated hydroxamic acids predicted from high-level ab initio calculations. J Phys Chem A. 1999;103:8346–8350. [Google Scholar]
- 67.Vedani A. YETI: An interactive molecular mechanics program for small-molecule protein complexes. J Comput Chem. 1988;9:269–280. [Google Scholar]
- 68.Vedani A, Huhta DW. A new force field for modeling metalloproteins. J Am Chem Soc. 1990;112:4759–4767. [Google Scholar]
- 69.Piquemal JP, Williams-Hubbard B, Fey N, Deeth RJ, Gresh N, Giessner-Prettre C. Inclusion of the ligand field contribution in a polarizable molecular mechanics: SIBFA-LF. J Comput Chem. 2003;24:1963–1970. doi: 10.1002/jcc.10354. [DOI] [PubMed] [Google Scholar]
- 70.Sternberg U, Koch FT, Brauer M, Kunert M, Anders E. Molecular mechanics for zinc complexes with fluctuating atomic charges. J Mol Model. 2001;7:54–64. [Google Scholar]
- 71.Burton VJ, Deeth RJ, Kemp CM, Gilbert PJ. Molecular mechanics for coordination complexes: The impact of adding d-electron stabilization energies. J Am Chem Soc. 1995;117:8407–8415. [Google Scholar]
- 72.Deeth RJ. The ligand field molecular mechanics model and the stereoelectronic effects of d and s electrons. Coordin Chem Rev. 2001;212:11–34. [Google Scholar]
- 73.Luque I, Freire E. Structural parameterization of the binding enthalpy of small ligands. Proteins. 2002;49:181–190. doi: 10.1002/prot.10208. [DOI] [PubMed] [Google Scholar]
- 74.Katz RA, Skalka AM. The retroviral enzymes. Ann Rev Biochem. 1994;63:133–173. doi: 10.1146/annurev.bi.63.070194.001025. [DOI] [PubMed] [Google Scholar]
- 75.Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J Am Chem Soc. 1990;112:6127–6129. [Google Scholar]
- 76.Eisenberg D, McLachlan AD. Solvation energy in protein folding and binding. Nature. 1986;319:199–203. doi: 10.1038/319199a0. [DOI] [PubMed] [Google Scholar]
- 77.Kuhn B, Kollman PA. Binding of a diverse set of ligands to avidin and streptavidin: An accurate quantitative prediction of their relative affinities by a combination of molecular mechanics and continuum solvent models. J Med Chem. 2000;43:3786–3791. doi: 10.1021/jm000241h. [DOI] [PubMed] [Google Scholar]