


Abstract
Genome-wide association (GWA) study aims to identify the genetic factors associated with the traits of interest. However, the power of GWA analysis has been seriously limited by the enormous number of markers tested. Recently, the gene set analysis (GSA) methods were introduced to GWA studies to address the association of gene sets that share common biological functions. GSA considerably increased the power of association analysis and successfully identified coordinated association patterns of gene sets. There have been several approaches in this direction with some limitations. Here, we present a general approach for GSA in GWA analysis and a stand-alone software GSA-SNP that implements three widely used GSA methods. GSA-SNP provides a fast computation and an easy-to-use interface. The software and test datasets are freely available at http://gsa.muldas.org. We provide an exemplary analysis on adult heights in a Korean population.
INTRODUCTION
Genome-wide association (GWA) study of a large population offers potential genetic causes of complex disease or the traits of interest (1,2). The typical approach assesses the association of each SNP independently with binary phenotypes (case–control) or continuously represented phenotypes (quantitative trait). However, due to the enormous number of SNPs analyzed, such an individual association analysis produces only a handful of significant SNPs from a stringent cutoff. The problem with this approach is that we may not delineate the underlying biological mechanism from the small number of SNPs beyond individual markers or genes. Moreover, many of those prominent SNPs are not reproducible among independent experiments. Another important problem is that many moderate but meaningful associations are lost below the stringent cutoff. In recent years, the gene set analysis (GSA) methods were taken into account in GWA studies which may address these problems.
GSA methods were originally developed for a transcriptome analysis to assess the differential expression of pre-defined gene sets that share common biological functions. They exhibited stronger statistical power than the individual gene analysis, and have revealed many novel gene sets with ‘subtle but coordinated’ expression patterns (3–5). Given that the basic goal of GWA studies is to prioritize the biological networks or processes associated with the trait of interest, it may be reasonable to consider the pre-defined gene sets or pathways as the units of an association analysis. Indeed, by analyzing SNPs on the gene set level, GSA was able to reveal many coordinated association patterns that might be lost by the individual marker analysis.
Several case–control studies employed GSA methods. Wang et al. (6) devised a GSEA framework for SNP arrays. They assigned the most highly associated SNP (best SNP) to each gene to summarize the association of multiple SNPs in each gene. Using the method, they successfully identified the Parkinson’s disease susceptibility pathways. Wang et al. (7) applied the same methods which implicated the molecular mechanism of autism beyond individual genes. Chen et al. (8) employed a gene set score weighted by the network connectivity in KEGG pathways, and analyzed five complex disease data sets. Lesnick et al. (9) showed the significance of the joint effect of weak variants within a candidate pathway for a brain disorder which may predispose to Parkinson’s disease. Askland et al. (10) using their pathway-based analysis tool, prioritized the ion channel gene set as a strong candidate that contributes to the susceptibility of bipolar disorder.
We present a general approach that enables the application of some state-of-the-art GSA methods in GWA studies. Because our approach uses the P-values of each marker, it is applicable to both case–control and quantitative trait studies. We then offer a JAVA-based stand-alone software, called GSA-SNP, that actualizes the methods. We provide an analysis example for adult heights in a Korean population
MATERIALS AND METHODS
The GWA analysis software such as PLINK (11) produces a list of P-values for each SNP either case–control or quantitative trait study. We present three different GSA methods that make use of the P-values for GWA analysis.
Common procedure
We take ‘–log’ on each P-value. Because we will use the ‘k-th best P’ (k = 1, 2, 3, 4 or 5) in each gene, this will give a more symmetric distribution to the gene scores. Each SNP is assigned to a gene whose extents with some padding encompass the SNP. We considered ±20 K more bps in the neighborhood of each gene. We chose the second best SNP in each gene as a default option to summarize the information of multiple SNPs instead of the best SNP (6). Random associations can make a SNP highly significant by chance, and can be more problematic in a quantitative trait study where GWA analysis softwares such as PLINK apply a linear regression to approximate the significance of each SNP. Using the second best P-values, we expect to evade many spurious associations. Such an effect is demonstrated in the Supplementary Data. Larger k may yield more conservative predictions, but may reduce the power. Therefore, in general, we recommend using both the best and the second best P-values and compare the biological relevance of the predictions.
We compile the gene sets to be analyzed. When the P-values of each gene set are computed, we apply the Benjamini–Hochberg multiple testing correction (12).
Z-statistic method
We employ the Z-statistic method (13). Each gene set (GS) is assessed by the Z-statistic:
where 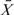 is the average of the gene scores [
is the average of the gene scores [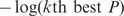 ] in a gene set, m0 and σ are the mean and the standard deviation of all the gene scores, n is the number of genes in the gene set.
] in a gene set, m0 and σ are the mean and the standard deviation of all the gene scores, n is the number of genes in the gene set.
In transcriptome analyses, though simple and sensitive, the Z-statistic method exhibited two drawbacks. First, it assumes that each gene set is a collection of independent samples. This causes some false positives, because many biologically determined gene sets have co-expression patterns. Second, some gene sets may have bi-directional expression changes to maintain homeostasis, but Z-statistic cannot detect such bi-directional patterns because they cancel each other. These problems, however, may be ameliorated in the GWA analysis context. First, the correlations of the gene scores are mostly determined by the linkage disequilibrium (LD); hence the gene scores in a gene set can be at most partially correlated if some of the genes are located within an LD block which is not often the case. Second, only one direction of the scores is meaningful: low P-values, equivalently high – – log(P) scores. If a gene set is associated, we would observe some high gene scores in the set, but the scores of other members are expected to be randomly distributed rather than concentrated at a low score which prevents significant offsets.
Restandardized GSA
We considered two sample-permutation based GSA methods. One is the recently developed restandardization method (14), and the other is GSEA (15) given in the next section. The restandardized GSA has several advantages over existing methods on such as power and reproducibility combined with the maxmean set statistic. Another advantage is that we can compute the P-values accurately from a relatively small number of sample permutations (50–100) if we use the pooled set scores. This saves much computational costs in GWA analysis. See the Supplementary Data for how to compute the P-values of the restandardization GSA (pooled version). We used the maxmean statistic for the gene set score which is defined as follows:
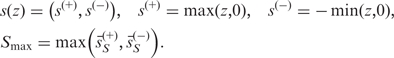 |
However, only the positive parts are meaningful in GWA analysis; hence we use the non-negative mean 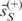 in practice for the gene set score.
in practice for the gene set score.
GSEA
GSEA (15) is of the most widely used class of GSA method and its R code is developed for the case–control studies (16). Our method is based on the P-values of each SNP; hence is also applicable to quantitative trait studies. We employed the maxmean (non-negative mean) set statistic, which has shown favorable properties over the Kolmogorove–Smirnov type statistic in statistical and computational perspectives (14). We also used the Z-statistic for the set score. While the original GSEA used the normalized set scores (denoted NES) that divides each set score by the mean of the randomized ones, we applied the Z-normalization using the mean and standard deviation of the randomized set scores (6).
GSA-SNP SOFTWARE
We developed a JAVA-based stand-alone software, called GSA-SNP, that implements the three GSA methods described above. The detailed user’s manual for the software is available at our web page.
Input and processing methods
Two kinds of input data are required: marker association (typically P-values) and gene sets.
Marker association data
(i) The list of P-values for each SNP which is typically obtained from the computation of a GWA anlaysis software such as PLINK. The SNP ID should be dbSNP RefSNP rs numbers. The P-values represent the association levels of each SNP with the given phenotype. For this single column input values, the Z-statistic method (13) is provided. (ii) The program also reads the lists (50–1000 columns) of randomized P-values each column of which can be obtained by permuting the sample labels of SNP arrays and running a GWA analysis software. These randomized P-values should be attached aside the list of original P-values. For this type of dataset, three widely used GSA methods are available: the Z-statistic method, the restandardized GSA with the maxmean statistic (14), and the GSEA based on maxmean or Z-statistic (14,15). (iii) The program also accepts haplotype association data. The input in this case replaces the SNP ID by chromosomal extents, i.e. the combination of chromosome number, chromosomal start and end points. Each SNP is assigned to the genes whose extents with some padding encompass the SNP. For a given haplotype, the overlapping genes are identified and P-values are assigned to each gene. For this type of dataset, the Z-statistic method is applied. The gene IDs (gene symbol) instead of the SNP IDs can also be used when the user wants to apply another method to summarize the association of SNPs in a gene.
Gene set data
We provide the Gene Ontology gene sets for a default analysis. Therefore, the user need not upload gene sets if (s)he wants to use the Gene Ontology gene sets. Otherwise, the user is required to upload their own gene set data in a tab-separated value format. The gmt format of MSigDB is acceptable (http://www.broadinstitute.org/gsea/msigdb/); hence the user can make use of the rich source of gene sets from MSigDB in our software.
Options
If the user uploads the marker association data, the program detects the data type and automatically shows relevant methods and parameter options. If the SNP data are uploaded, the user can choose the padding size for genes among 0, ±10 000 or ±20 000. The user can also choose the kth best P, k = 1, 2,…, 5 to summarize the SNPs for each gene. The padding of ±20 000, and the second best P are the default options. For an input of a gene list, there is no parameter to choose. For a haplotype input, the user is requested to choose the minimum overlap size between haplotype intervals and genes. The user can also determine the range of gene set sizes and the cutoff for q-values.
Output
The list of significantly associated gene sets with P-values, corrected P-values, and their members that are sorted in the descending order of their association strength. The results are given both on the program window and as a csv file. If the user clicks a gene symbol in the csv file, the web-browser will show its information on GeneCards® (17) (http://www.genecards.org).
Comparison with previous software tools
As the power of GSA in GWA analysis was being conceived, several software tools were developed recently. Holden et al. (16) devised a GSEA method for case–control arrays using R. They used all the SNPs in a gene instead of selecting the best SNP (6). O’Dushlaine et al. (18) developed PERL codes, called SRT, that generate simulated P-values from randomized phenotypes using the PLINK program and perform a sample-randomizing GSA. In this program, they first select a list of significant SNPs from a cutoff threshold and compute the ratio of the significant SNPs in each pathway (gene set). These two methods allow multiple SNPs in a gene to contribute to the set score, which may increase the power of the methods. However, it is possible that significant SNPs in a haplotype block are concentrated in one or two genes of the pathway, which may overly amplify the contribution of a gene. On the other hand, Wang et al.’s method (6) and GSA-SNP choose the kth best P-value to summarize the information of each gene. Therefore, contributions by multiple genes are more emphasized in this approach. Besides, the best P-value is not merely the information of a single SNP because it is determined by comparing the significance of all the SNPs in a gene. One possible concern with using the best P is whether it is from a random association or not. This is why we considered the kth best P-value: the kth P-value can be significant only if all the first k P-values are simultaneously significant which is possible for true positives, but is very difficult to happen by chance.
Because SRT does not pool the set scores of different gene sets, it takes much time to attain a high level of significance. Medina et al. (19) developed a web server, called GeSBAP, that provides a SNP (marker)-randomizing GSA method. It also takes much time for uploading and computing for an SNP input. GSA-SNP provides a fast computation. Several minutes are sufficient on a PC if the input data are properly prepared. It is equipped with an easy GUI that automatically displays relevant options and methods according to the input data types. Because GSA-SNP takes P-values as input data, it is applicable to both case–control and quantitative trait studies. This advantage is also shared with other P-value based methods, SRT and GeSBAP, but GSA-SNP provides three widely used GSA methods if the simulated P-values are prepared. How to choose a GSA method may depend on the preference of the user for each GSA approach, but a general guidance is given in Supplementary Data.
The most time-consuming part for our sample-randomizing methods (restandardized GSA and GSEA) will be to prepare the simulated P-values using a GWA analysis software. Because our methods pool the randomized scores of different gene sets, about a hundred simulations of P-values will be sufficient.
Analysis example
We evaluated the performance of GSA-SNP using a large-scale GWA study dataset, previously reported by the Korea Association Resource (KARE) project (20). The dataset comprised the genotypes of two population-based cohorts recruited in Korea (8842 unrelated individuals), measured using Affymetrix Genome-Wide Human SNP Array 5.0 (352 228 SNPs after QC). Cho et al. (20) reported the results of GWA analyses of eight quantitative traits in the dataset. Among the traits, we focused on height due to the following reasons. Adult human height is a polygenic trait with a highly heritable component (21) and several GWA studies have been reported on this trait (22–27). The comparison of the previous GWA studies shows that the height association signals in a Korean population are weaker than those of European populations: some of the strong association signals in the European populations were also caught in the Korean population, but other signals were lost below the threshold cutoff (see the Supplementary Data). Therefore, it would be interesting to see whether gene-set analysis of the single-marker analyses of Korean dataset would capture biological processes similar to the ones reported by the European studies.
Before applying a GSA, the KARE genotype data were supplemented by imputing SNP genotypes based on the genotypes of the JPT + CHB panel of the International HapMap Phase II (The International HapMap Consortium, 2005). Details of SNP imputation and filtering have been published elsewhere (28) and were summarized in the Supplementary Data. We used this imputed dataset as the input of GSA-SNP. We show the analysis result by the Z-statistic method. A total of 12 Gene Ontology gene sets having corrected P < 0.05 were tabulated. The resulting screen shot is shown in Figure 1. for each of those 12 gene sets, we gathered the P-values of the member genes and compared their distributions with that of all the genes together. As shown in Figure 2, relatively smaller P-values were enriched in the distributions of these 12 gene sets compared to the background distribution. It should be noted that the first quartiles of the gene sets were around 10−2, which implies GSA-SNP successfully identified moderate but consistent association patterns that were not dominated by only a few strong association signals.
Figure 1.

The computation result for height in a Korean population using the Z-statistic method. The genes in each gene set are sorted in the decreasing order of GWA significance.
Figure 2.

Boxplots of P-values of the member genes in each gene set identified by GSA-SNP for height in Korean population. The bottom item ‘All’ means the corresponding distribution of all the genes in the data set.
Many of the gene sets we identified made good biological senses. For example, the GO term ‘skeletal development’ was identified by Gudbjartsson et al. (26) in their GO-based GWA analysis of European adult heights. Similarly, Weedon et al. (25) reported ‘extracellular matrix’ as one of the key biological functions implicated in height regulation. Since collagens are the most abundant proteins in the extracellular matrix, the term ‘collagen’ may be understood in the context of ‘extracellular matrix’. Although ‘glutamate receptor activity’ has not been implicated through GWA studies to our knowledge, one member of that gene set, GRIA1, was implicated near a loci associated with height in Croatian population (27). Endogenous activation of metabotropic glutamate receptors is known to modulate GABAnergic transmission of gonadotropin-releasing hormone (GnRH) neurons (29). Moreover, treatment with a GnRH agonist in short adolescents increased adult height (30). These imply that glutamate receptors might affect adult height in man. The top ranking genes in ‘anion cation symporter activity’ were SLC17A family members, vesicular glutamate transporters (31). Other GO terms such as ‘transmembrane receptor protein phosphatase activity’, ‘golgi stack’ and ‘phosphoric ester hydrolase activity’, were not supported in our literature survey and may deserve further investigation.
CONCLUSION
Here, we proposed a P-value based approach that enabled the application of three widely used GSA methods in both case–control and quantitative trait studies. We also suggested using the kth best P to summarize the association of SNPs in a gene to remove randomly associated signals. However, we also provide the option for the best P-value (k = 1) in our software such that the user can perform the analyses for different k’s, and compare the relevance of the results.
As a stand-alone JAVA program, GSA-SNP provides a fast and secure computation as well as an easy-to-use GUI. Unlike other tools, GSA-SNP provides three powerful GSA methods for GWA analysis. Because it employs methods that pool the randomized set scores, it provides a high level of significance from a relatively small number of simulated analyses.
We expect the demand of GSA in GWA analysis will be increasing rapidly in the near future as has been in the transcriptome analyses, and the GSA-SNP provides a useful tool. GSA methods and tools will be further investigated and optimized in the context of GWA analysis.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute for Mathematical Sciences, Daejeon, Korea (NIMS); Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Korea government (MEST) (grant no. 2010-0016668 to D.N.); MEST, NRF (grant R11-2008-0062293 to S.K.). Funding for open access charge: MEST, NRF (grant R11-2008-0062293).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The KARE genotype and epidemiological data were gratefully made available by National Institute of Health, Korea Center for Disease Control, which supported this work through the KARE Analysis Consortium. We would like to thank the help and support of all the staff at KNIH. Computing cluster used in this study was kindly granted from Korean Bio-information Center (KOBIC).
REFERENCES
- 1.Hirschhorn J.N., Daly M.J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 2.Wang W.Y., Barratt B.J., Clayton D.G., Todd J.A. Genome-wide association studies: theoretical and practical concerns. Nat. Rev. Genet. 2005;6:109–118. doi: 10.1038/nrg1522. [DOI] [PubMed] [Google Scholar]
- 3.Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E., et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 4.Goeman J.J., Bühlmann P. Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics. 2007;23:980–987. doi: 10.1093/bioinformatics/btm051. [DOI] [PubMed] [Google Scholar]
- 5.Nam D., Kim S.Y. Gene-set approach for expression pattern analysis. Brief Bioinform. 2008;9:189–197. doi: 10.1093/bib/bbn001. [DOI] [PubMed] [Google Scholar]
- 6.Wang K., Li M., Bucan M. Pathway-Based Approaches for Analysis of Genomewide Association Studies. Am. J. Hum. Genet. 2007;81:1278–1283. doi: 10.1086/522374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang K., Zhang H., Ma D., Bucan M., Glessner J.T., Abrahams B.S., Salyakina D., Imielinski M., Bradfield J.P., Sleiman P.M., et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459:528–533. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen L., Zhang L., Zhao Y., Xu L., Shang Y., Wang Q., Li W., Wang H., Li X. Prioritizing risk pathways: a novel association approach to searching for disease pathways fusing SNPs and pathways. Bioinformatics. 2009;25:237–242. doi: 10.1093/bioinformatics/btn613. [DOI] [PubMed] [Google Scholar]
- 9.Lesnick T.G., Papapetropoulos S., Mash D.C., Ffrench-Mullen J., Shehadeh L., de Andrade M., Henley J.R., Rocca W.A., Ahlskog J.E., Maraganore D.M. A genomic pathway approach to a complex disease: axon guidance and Parkinson disease. PLoS Genet. 2007;3:e98. doi: 10.1371/journal.pgen.0030098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Askland K., Read C., Moore J. Pathways-based analyses of whole-genome association study data in bipolar disorder reveal genes mediating ion channel activity and synaptic neurotransmission. Hum. Genet. 2009;125:63–79. doi: 10.1007/s00439-008-0600-y. [DOI] [PubMed] [Google Scholar]
- 11.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Royal Statist. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- 13.Kim S.Y., Volsky D.J. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics. 2005;6:144. doi: 10.1186/1471-2105-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Efron B., Tibshirani R. On testing the significance of sets of genes. Ann. Appl. Stat. 2007;1:107–129. [Google Scholar]
- 15.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holden M., Deng S., Wojnowski L., Kulle B. GSEA-SNP: applying gene set enrichment analysis to SNP data from genome-wide association studies. Bioinformatics. 2008;24:2784–2785. doi: 10.1093/bioinformatics/btn516. [DOI] [PubMed] [Google Scholar]
- 17.Rebhan M., Chalifa-Caspi V., Prilusky J., Lancet D. GeneCards: integrating information about genes, proteins and diseases. Trends Genet. 1997;13:163. doi: 10.1016/s0168-9525(97)01103-7. [DOI] [PubMed] [Google Scholar]
- 18.O'Dushlaine C., Kenny E., Heron E.A., Segurado R., Gill M., Morris D.W., Corvin A. The SNP ratio test: pathway analysis of genome-wide association datasets. Bioinformatics. 2009;25:2762–2763. doi: 10.1093/bioinformatics/btp448. [DOI] [PubMed] [Google Scholar]
- 19.Medina I., Montaner D., Bonifaci N., Pujana M.A., Carbonell J., Tarraga J., Al-Shahrour F., Dopazo J. Gene set-based analysis of polymorphisms: finding pathways or biological processes associated to traits in genome-wide association studies. Nucleic Acids Res. 2009;37:W340–W344. doi: 10.1093/nar/gkp481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cho Y.S., Go M.J., Kim Y.J., Heo J.Y., Oh J.H., Ban H.J., Yoon D., Lee M.H., Kim D.J., Park M., et al. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat. Genet. 2009;41:527–534. doi: 10.1038/ng.357. [DOI] [PubMed] [Google Scholar]
- 21.Silventoinen K., Sammalisto S., Perola M., Boomsma D.I., Cornes B.K., Davis C., Dunkel L., De Lange M., Harris J.R., Hjelmborg J.V., et al. Heritability of adult body height: a comparative study of twin cohorts in eight countries. Twin Res. 2003;6:399–408. doi: 10.1375/136905203770326402. [DOI] [PubMed] [Google Scholar]
- 22.Lettre G., Jackson A.U., Gieger C., Schumacher F.R., Berndt S.I., Sanna S., Eyheramendy S., Voight B.F., Butler J.L., Guiducci C., et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat. Genet. 2008;40:584–591. doi: 10.1038/ng.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johansson A., Marroni F., Hayward C., Franklin C.S., Kirichenko A.V., Jonasson I., Hicks A.A., Vitart V., Isaacs A., Axenovich T., et al. Common variants in the JAZF1 gene associated with height identified by linkage and genome-wide association analysis. Hum. Mol. Genet. 2009;18:373–380. doi: 10.1093/hmg/ddn350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang T.L., Xiong D.H., Guo Y., Recker R.R., Deng H.W. Comprehensive association analyses of IGF1, ESR2, and CYP17 genes with adult height in Caucasians. Eur. J. Hum. Genet. 2008;16:1380–1387. doi: 10.1038/ejhg.2008.113. [DOI] [PubMed] [Google Scholar]
- 25.Weedon M.N., Lango H., Lindgren C.M., Wallace C., Evans D.M., Mangino M., Freathy R.M., Perry J.R., Stevens S., Hall A.S., et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat. Genet. 2008;40:575–583. doi: 10.1038/ng.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gudbjartsson D.F., Walters G.B., Thorleifsson G., Stefansson H., Halldorsson B.V., Zusmanovich P., Sulem P., Thorlacius S., Gylfason A., Steinberg S., et al. Many sequence variants affecting diversity of adult human height. Nat. Genet. 2008;40:609–615. doi: 10.1038/ng.122. [DOI] [PubMed] [Google Scholar]
- 27.Polasek O., Marusic A., Rotim K., Hayward C., Vitart V., Huffman J., Campbell S., Jankovic S., Boban M., Biloglav Z., et al. Genome-wide association study of anthropometric traits in Korcula Island, Croatia. Croat Med. J. 2009;50:7–16. doi: 10.3325/cmj.2009.50.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee K., Kim S. A scheme for filtering SNPs imputed in 8,842 Korean individuals based on the International HapMap Project data. Genomics Inform. 2009;7:136–140. [Google Scholar]
- 29.Chu Z., Moenter S.M. Endogenous activation of metabotropic glutamate receptors modulates GABAergic transmission to gonadotropin-releasing hormone neurons and alters their firing rate: a possible local feedback circuit. J. Neurosci. 2005;25:5740–5749. doi: 10.1523/JNEUROSCI.0913-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yanovski J.A., Rose S.R., Municchi G., Pescovitz O.H., Hill S.C., Cassorla F.G., Cutler G.B., Jr Treatment with a luteinizing hormone-releasing hormone agonist in adolescents with short stature. N. Engl. J. Med. 2003;348:908–917. doi: 10.1056/NEJMoa013555. [DOI] [PubMed] [Google Scholar]
- 31.Reimer R.J., Edwards R.H. Organic anion transport is the primary function of the SLC17/type I phosphate transporter family. Pflugers Arch. 2004;447:629–635. doi: 10.1007/s00424-003-1087-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.