


Abstract
SoRT2 is a web server that allows the user to perform genome rearrangement analysis involving reversals, generalized transpositions and translocations (including fusions and fissions), and infer phylogenetic trees of genomes being considered based on their pairwise genome rearrangement distances. It takes as input two or more linear/circular multi-chromosomal gene (or synteny block) orders in FASTA-like format. When the input is two genomes, SoRT2 will quickly calculate their rearrangement distance, as well as a corresponding optimal scenario by highlighting the genes involved in each rearrangement operation. In the case of multiple genomes, SoRT2 will also construct phylogenetic trees of these genomes based on a matrix of their pairwise rearrangement distances using distance-based approaches, such as neighbor-joining (NJ), unweighted pair group method with arithmetic mean (UPGMA) and Fitch–Margoliash (FM) methods. In addition, if the function of computing jackknife support values is selected, SoRT2 will further perform the jackknife analysis to evaluate statistical reliability of the constructed NJ, UPGMA and FM trees. SoRT2 is available online at http://bioalgorithm.life.nctu.edu.tw/SORT2/.
INTRODUCTION
During evolution the gene order in a genome is generally not well conserved because it is subject to be changed by genome rearrangements, such as reversals, transpositions, fusions, fissions and translocations. The studies for analyzing the differences between the gene orders of a set of species genomes have been increasingly recognized as a powerful tool in phylogenetic tree reconstruction, as they have helped biologists to gain a better understanding of the evolution of several groups of genomes, such as animal mitochondria (1), plant chloroplasts (2), bacteria (3) and mammals (4). The combinatorial problems considered in these studies (typically called ‘genome rearrangement problems’) can be formulated as follows. Given the gene (or synteny block) orders of a set of genomes, each represented by a signed permutation, and a set of possible rearrangements, the problem aims to find a shortest series of rearrangements (or a series of minimum weight when rearrangements are weighted according to the probabilities of their occurrences) required to transform (or sort) those genomes into one another (5). The length (or weight) of an optimal series of rearrangements is then called ‘genome rearrangement distance’. The genome rearrangement distance can serve as a measure of an evolutionary distance between species. In contrast to the sequence-based approaches in which local mutations (i.e. substitutions, insertions and deletions of nucleotides/amino acids) accumulate rather quickly, genome rearrangements are global (or large scale) and relatively rare mutations and, therefore, their distances are believed to allow for evolutionary reconstructions of more divergent species.
The genome rearrangements studied in the literature to date can be classified into two categories: (i) ‘intra-chromosomal’ rearrangements, such as reversals, transpositions and block-interchanges (here also called ‘generalized transpositions’), and (ii) ‘inter-chromosomal’ rearrangements, such as fusions, fissions and translocations (5). ‘Reversals’, also called ‘inversions’ in biology, reverse a segment on a chromosome and also exchange its strands (6,7). ‘Transpositions’ move a segment on a chromosome to another location or, equivalently, exchange two adjacent and non-overlapping segments on the chromosome (8,9). ‘Block-interchanges’ are a kind of generalized transpositions that exchange two non-overlapping but not necessarily adjacent segments on a chromosome (10,11). ‘Translocations’ exchange an end segment of a chromosome, which contains a telomere of this chromosome, with an end segment of another chromosome (12,13). ‘Fusions’ join two chromosomes into a bigger one and ‘fissions’ break a chromosome into two smaller ones (14,15). Basically, both fusions and fissions can be considered as special cases of translocations that either act on two chromosomes one of which is empty (i.e. fissions), or result in two chromosomes one of which is empty (i.e. fusions). Currently, exiting web servers involving one or several of the above rearrangement operations include GRIMM (16), MGR (4), ROBIN (17), SPRING (18), DCJ (19) and webMGR (20).
Recently, Yancopoulos et al. (21) introduced and studied the so-called ‘double cut and join’ (DCJ) operation, which cuts the chromosome(s) in two places and rejoins the four cut ends in a new way, as a basis for modeling all the rearrangement operations described above. In this formulation, both reversals and translocations (including fusions and fissions) can be modeled by a DCJ operation, while block-interchanges (including transpositions) by two consecutive DCJ operations, one for generating a small circular chromosome from a chromosome and the other for re-incorporating this circular chromosome at a new site on the same chromosome. In addition, Yancopoulos et al. (21) designed an 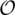 (δn) time algorithm for sorting a genome with linear, multiple chromosomes by reversals, block-interchanges and translocations (including fusions and fissions) with the weight ratio 1:2:1, where n is the number of genes to be considered and δ is the number of needed DCJ operations. Later on, Bergeron et al. (19) reconsidered the DCJ model by allowing the small circular chromosome generated by a DCJ operation not necessarily to be re-incorporated immediately by the following DCJ operation. Since then, this re-formulated DCJ operation has received increased attention, because it can not only provide a unifying model for genome rearrangements, but also result in a relatively simple distance formula that can be calculated by a simpler algorithm (22,23). To the best of our knowledge, however, no software tool has been implemented so far based on the algorithm proposed by Yancopoulos et al. (21).
(δn) time algorithm for sorting a genome with linear, multiple chromosomes by reversals, block-interchanges and translocations (including fusions and fissions) with the weight ratio 1:2:1, where n is the number of genes to be considered and δ is the number of needed DCJ operations. Later on, Bergeron et al. (19) reconsidered the DCJ model by allowing the small circular chromosome generated by a DCJ operation not necessarily to be re-incorporated immediately by the following DCJ operation. Since then, this re-formulated DCJ operation has received increased attention, because it can not only provide a unifying model for genome rearrangements, but also result in a relatively simple distance formula that can be calculated by a simpler algorithm (22,23). To the best of our knowledge, however, no software tool has been implemented so far based on the algorithm proposed by Yancopoulos et al. (21).
More recently, we have proposed two novel algorithms based on permutation groups in algebra (24) to optimally sort a linear and a circular multi-chromosomal genome, respectively, by reversals, generalized transpositions and translocations (including fusions and fissions) in 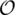 (δn) time, where here δ is the minimum number of rearrangement operations that usually is much less than n. We have implemented these two algorithms into a novel web server called SoRT2 (short for ‘Sorting genomes and reconstructing phylogenetic trees by Reversals, generalized Transpositions and Translocations’) that allows the user to perform the analysis of genome rearrangements by calculating the genome rearrangement distance between any pair of input genomes and displaying a corresponding optimal scenario of rearrangement operations. For more practical applications, we have also implemented and incorporated the following three related algorithms into the SoRT2 web server: (i) sorting by reversals only (6), (ii) sorting by block-interchanges only (17) and (iii) sorting by reversals and block-interchanges (18,24). In addition, we have equipped our SoRT2 with the capability of inferring phylogenetic trees of multiple genomes being considered based on their pairwise genome rearrangement distances and the capability of evaluating the statistical reliability of the tree branches using the jackknife resampling approach (25). For simplicity, when we say ‘gene’ in the rest of the article, it also means ‘synteny block’ or ‘marker’ that represents a conserved sequence region shared by all genomes to be considered.
(δn) time, where here δ is the minimum number of rearrangement operations that usually is much less than n. We have implemented these two algorithms into a novel web server called SoRT2 (short for ‘Sorting genomes and reconstructing phylogenetic trees by Reversals, generalized Transpositions and Translocations’) that allows the user to perform the analysis of genome rearrangements by calculating the genome rearrangement distance between any pair of input genomes and displaying a corresponding optimal scenario of rearrangement operations. For more practical applications, we have also implemented and incorporated the following three related algorithms into the SoRT2 web server: (i) sorting by reversals only (6), (ii) sorting by block-interchanges only (17) and (iii) sorting by reversals and block-interchanges (18,24). In addition, we have equipped our SoRT2 with the capability of inferring phylogenetic trees of multiple genomes being considered based on their pairwise genome rearrangement distances and the capability of evaluating the statistical reliability of the tree branches using the jackknife resampling approach (25). For simplicity, when we say ‘gene’ in the rest of the article, it also means ‘synteny block’ or ‘marker’ that represents a conserved sequence region shared by all genomes to be considered.
METHODS
As mentioned earlier, the program of SoRT2 for sorting a multi-chromosomal genome (that can be linear or circular) into another using reversals, generalized transpositions and translocations (including fusions and fissions) was implemented based on the algorithm that we have recently proposed using permutation groups in algebra (24), where generalized transpositions are weighted 2 and the others are weighted 1. For details, we refer the reader to our paper (24). Notice that the pairwise genome rearrangement distance returned by SoRT2 is the same as the one measured by the DCJ model, both of which actually can be calculated quickly in linear time. Usually, transpositions are observed much less frequently than reversals and translocations in many evolutionary scenarios (21,26). Blanchette et al. (26) have conducted experiments on real biological data to conclude that the most probable weights are 1 for reversals and 2 for transpositions. In addition, Eriksen (27) and his co-workers have used simulations to find that optimal weights for reversals and transpositions are 1 and 2, respectively. On the other hand, if the weight ratio between reversals and transpositions is 1:1, then transpositions are generally favored over reversals, because a reversal (or translocation) removes at most two breakpoints, while a transposition removes at most three breakpoints (and a generalized transposition four breakpoints) (5). According to the above results and discussion, it seems to be biologically meaningful to assign at least twice the weight to generalized transpositions than to the others. However, if generalized transpositions are at least three times the weight of reversals, then there is always an optimal solution for the problem that contains nothing but only reversals and translocations, because a generalized transposition (block-interchange) can be mimicked by three reversals. For example, three consecutive genes (x, y, z) can be transformed into (z, y, x) by a block-interchange or by three reversals with scenario of (x, −z, −y), (z, −x, −y) and (z, y, x). Therefore, it should be reasonable to assign generalized transpositions a weight equal to 2 and the others a weight equal to 1.
In this study, we have implemented and incorporated the following three related algorithms into the SoRT2 web server for its more practical applications: (i) the algorithm proposed by Kaplan et al. (6) for sorting by reversals only, (ii) the algorithm of our ROBIN (17) for sorting by block-interchanges only and (iii) the redesigned algorithm of our SPRING (18) for sorting by reversals and block-interchanges based on permutation groups (24). Furthermore, we have equipped our SoRT2 with the capability of inferring the phylogenetic tree of multiple genomes being considered based on their pairwise genome rearrangement distances using distance-based approaches of building trees, such as neighbor-joining (NJ), unweighted pair group method with arithmetic mean (UPGMA) and Fitch–Margoliash (FM) methods. Finally, we have also adopted the jackknife resampling approach (25), as described as follows, to further calculate statistical reliability of clades (or internal nodes) in the NJ, UPGMA and FM trees. We randomly remove 50% of the input set of genes, while retaining the relative orderings of remaining genes, and calculate the genome rearrangement distance between every pair of genomes. This procedure will be repeated as many times as specified by the user. Suppose that the replicate number specified by the user is 100. We then apply the NEIGHBOR/FITCH program in the PHYLIP package (28) to the 100 matrices of pairwise genome rearrangement distances to obtain 100 jackknife trees. Finally, we apply the CONSENSE program in the PHYLIP package to these 100 jackknife trees to obtain a majority-rule consensus tree with the numbers at each internal node representing the percentage of times that the clade defined by this node appears in the 100 jackknife trees.
TOOL IMPLEMENTATION AND USAGE
The kernel programs of SoRT2 were written in C and its web interface was written in PHP. It is currently installed on IBM PC with 2.8 GHz processor and 3 GB RAM under Linux system and can be freely accessed at http://bioalgorithm.life.nctu.edu.tw/SORT2/. SoRT2 provides a user interface (Figure 1a) that is intuitive and easy to operate. It takes as input two or more linear/circular multi-chromosomal gene orders in a kind of FASTA-like format (see the instance depicted in Figure 1a), which follows the syntax used in GRIMM (16) to represent a genome consisting of n genes that spread over m chromosomes by beginning with a single-line description that starts with a right angle bracket (‘>’), followed by a signed permutation of 1, 2,…, n with m − 1 delimiters ‘$’ inserted between the chromosomes (or with a ‘$’ at the end of each chromosome). When the input is two genomes, SoRT2 will calculate their genome rearrangement distance, as well as a corresponding optimal scenario by highlighting the genes involved in each rearrangement operation (Figure 1b). In the case of multiple genomes, SoRT2 will output a matrix of pairwise genome rearrangement distances (Figure 1c), in which each entry denotes the genome rearrangement distance between its two corresponding genomes and its hyperlink accordingly points to an optimal scenario of used rearrangements. Based on this pairwise rearrangement distance matrix, SoRT2 will further construct a phylogenetic tree of input multiple genomes using the NJ, UPGMA or FM method (Figure 1d). In addition, if the function of computing jackknife support values is selected, SoRT2 will also perform the jackknife analysis according to the replicate number specified by the user to evaluate the statistical reliability of clades in the NJ, UPGMA and FM trees. SoRT2 also provides a hyperlink through which the user can further view a consensus tree and more detailed jackknife support values of clades included or not included in the consensus tree. We refer the user to the help page of SoRT2 for the step-by-step guide of its detailed usage.
Figure 1.

(a) User interface of SoRT2. (b) Display of an optimal rearrangement scenario in which the genes involved in rearrangements are highlighted. (c) A pairwise rearrangement distance matrix obtained when applying SoRT2 to six mammalian genomes with 1360 synteny blocks. (d) A phylogenetic tree of six mammalian genomes produced by SoRT2 with jackknife support values on its clades.
EXPERIMENTAL RESULTS
Below, we tested our SoRT2 on some simulated datasets, as well as three biological datasets of gene orderings from mitochondrial, mammalian and bacterial genomes, respectively, to demonstrate its ability in reconstruction of phylogenetic trees, and also compared it to another similar tool GRIMM (16). Notice that GRIMM utilizes another tool, called MGR (4), to infer its phylogenetic trees, where MGR constructs the phylogenetic trees by using a heuristic of maximum parsimony approach, instead of distance-based approach, based on the genome rearrangement distance involving reversals, fusions, fissions and translocations (4). For a fair comparison, we also used the NJ method to reconstruct the phylogenetic trees based on the pairwise rearrangement distances computed by GRIMM and denoted such a kind of GRIMM by GRIMM-NJ for a distinction from the original GRIMM using MGR for its phylogenetic tree reconstruction. All these testing datasets, as well as their experimental results in details, are available on the help page of SoRT2.
Performance on simulated datasets
First of all, we generated a random rooted binary tree with m multi-chromosomal genomes (or species), where m was varied from 10 to 46 in steps of 4, and assigned a random number x to each edge, where x was an integer between 1 and 5. Then, we evolved the randomly generated tree starting from its root with a uni-chromosomal genome of 200 genes by performing x random rearrangement events to each edge until we obtained the gene orders of all the species genomes at the leaves of the tree. Since transpositions generally occur less frequently than reversals and translocations in real biological data, we used three different ratios in our simulations to randomly generate reversals, transpositions and translocations: (1) 1:0:1, (2) 2:1:2 and (3) 1:1:1. Finally, for each choice of species number and rearrangement ratio, we repeated the experiment 100 times and compared SoRT2 with GRIMM-NJ using their average tree similarity. The tree similarity of a tree reconstruction method was calculated as follows based on the property that each branch (edge) divides the set of species at the leaves of a tree into two groups, with one group connected to one end of the branch and the other group connected to the other end. We first used the TREEDIST program in the PHYLIP package (28) to calculate the symmetric difference, say d, between the randomly generated tree and the tree produced by the method, where the ‘symmetric difference’ is defined as the number of partitions that are not shared between the two trees (i.e. the number of partitions of the first tree that are not present in the second tree plus the number of partitions of the second tree that are not present in the first tree). Next, we converted this symmetric difference to a tree similarity measure using a simple formula that is 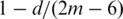 , where 2m − 6 is the maximum symmetric difference between two binary trees (28). The average tree similarities calculated in our experiments for SoRT2 and GRIMM-NJ are shown in Figure 2. In the simulated model without transpositions (whose ratio of randomly selected rearrangements is 1:0:1), the average tree similarities achieved by our SoRT2 are almost the same as those by GRIMM-NJ, as shown in Figure 2a, and their overall average tree similarities are both equal to 99.2%. However, in the models with transpositions, our SoRT2 generally performs better than GRIMM-NJ, as illustrated in Figures 2b and 2c, where the overall average tree similarities of SoRT2 and GRIMM-NJ are 99.4% and 99.2%, respectively, for the simulated dataset with ratio of 2:1:2, and 99.4% and 99.1%, respectively, for that with ratio of 1:1:1.
, where 2m − 6 is the maximum symmetric difference between two binary trees (28). The average tree similarities calculated in our experiments for SoRT2 and GRIMM-NJ are shown in Figure 2. In the simulated model without transpositions (whose ratio of randomly selected rearrangements is 1:0:1), the average tree similarities achieved by our SoRT2 are almost the same as those by GRIMM-NJ, as shown in Figure 2a, and their overall average tree similarities are both equal to 99.2%. However, in the models with transpositions, our SoRT2 generally performs better than GRIMM-NJ, as illustrated in Figures 2b and 2c, where the overall average tree similarities of SoRT2 and GRIMM-NJ are 99.4% and 99.2%, respectively, for the simulated dataset with ratio of 2:1:2, and 99.4% and 99.1%, respectively, for that with ratio of 1:1:1.
Figure 2.

Accuracy comparison of SoRT2 and GRIMM-NJ for their phylogenetic tree reconstruction based on three different ratios of reversals, transpositions and translocations: (a) 1:0:1, (b) 2:1:2 and (c) 1:1:1, where vertical axis indicates average tree similarity (%) and horizontal axis indicates species number.
Table 1 shows the average CPU time of SoRT2 and GRIMM for computing the matrix of pairwise genome rearrangement distances, when applying them to simulated datasets that were randomly generated according to the above simulation method using 10 multi-chromosomal species with 100, 200, 500, 1000, 1500 and 2000 genes, respectively. The experiment was repeated 100 times for each choice of gene number. As indicated in Table 1, both GRIMM and SoRT2 can finish their jobs within a second for multi-chromosomal species with no more than 500 genes. For the species with 1500–2000 genes, GRIMM is clearly faster than our SoRT2, but our SoRT2 still takes only a few seconds to complete its work.
Table 1.
Average CPU time for GRIMM and SoRT2 to compute the matrix of pairwise genome rearrangement distances for 10 multi-chromosomal species with gene number varying from 100 to 2000
| Gene number | GRIMM (S) | SoRT2 (S) |
|---|---|---|
| 100 | 0.19 | 0.31 |
| 200 | 0.19 | 0.46 |
| 500 | 0.21 | 0.90 |
| 1000 | 0.24 | 1.68 |
| 1500 | 0.28 | 2.54 |
| 2000 | 0.31 | 3.46 |
Eleven metazoan mtDNAs
In this experiment, we applied our SoRT2 to a gene order dataset of 11 metazoan mitochondrial DNAs (mtDNAs) with 36 genes that was studied by Blanchette et al. (1), where the 11 metazoan species are human (abbreviated as HU), Asterina pectinifera (sea star, abbreviated as SS), Strongylocentrotus purpuratus (sea urchin, SU), Drosophila yakuba (insect, DR), Artemia franciscana (crustacean, AF), Albinaria coerulea (snail, AC), Cepaea nemoralis (snail, CN), Katharina tunicata (KT, chiton), Lumbricus terrestris (earthworm, LU), Ascaris suum (AS) and Onchocerca volvulus (OV). Although many debating trees for metazoan phylogeny have been proposed, the one shown in Figure 3a is most widely accepted (1) and, therefore, serves as a reference tree for comparing the accuracy of different tools used in this study. According to our experimental results, the NJ tree obtained by SoRT2 (Figure 3b) is the same as the one by GRIMM-NJ (Figure 3c) in topology, in which the species in the same group were placed together as sister taxa, except for three Mollusk species KT, AC and CN. Such an inconsistency also occurred in the phylogenetic tree produced by MGR (Figure 3d), but the two Mollusk AC and CN were placed in the branch of deuterostomes (HU, SS and SU).
Figure 3.

(a) The reference tree of 11 metazoan gene orders adopted from ref. (1), where the 11 metazoan organisms are grouped into six major groupings: Chordate (with HU), Echinoderm (with SS and SU), Arthropod (with DR and AF), Mollusk (with KT, AC and CN), Annelid (with LU) and Nematode (with OV and AS). (b) The NJ tree produced by SoRT2 using a jackknife analysis of 100 replicates, where numbers on internal nodes denote the support values. (c) The NJ tree based on the pairwise rearrangement distances calculated by GRIMM. (d) The phylogenetic tree reconstructed by MGR.
Six mammalian genomes
Zhao and Bourque (29) created a dataset with 1360 synteny blocks of six mammalian genomes (human, chimpanzee, rhesus macaque, mouse, rat and dog) to study how to recover their ancestral rearrangement events on a fixed phylogenetic tree as shown in Figure 4a, where the 1360 synteny blocks in this dataset cover 91.1% of the human genome. In this experiment, we applied our SoRT2, as well as GRIMM-NJ, to this mammalian dataset. As a result, the NJ tree obtained by our SoRT2 (Figure 4b), as well as the GRIMM-NJ tree (Figure 4c), is the same as the one in Figure 4a in topology and has jackknife support values of 100% on almost all its clades. Actually, we had also tested MGR on this mammalian dataset and, unfortunately, MGR was unable to analyze this dataset in a reasonable amount of time so that we did not have its phylogenetic tree in this experiment.
Figure 4.

(a) The reference tree of six mammalian genomes adopted from ref. (29), where its edges were not drawn to scale. (b) The NJ tree created by SoRT2 using a jackknife analysis of 100 replicates, where numbers on internal nodes are the support values. (c) The NJ tree based on the pairwise rearrangement distances returned by GRIMM.
Seven bacterial genomes
In this experiment, we tested our SoRT2, as well as GRIMM-NJ and MGR, on a dataset of seven γ-proteobacterial genomes with 103 genes that came from the study by Belda et al. (3). This dataset consists of Escherichia coli 0157-H7 (abbreviated as ecs, NC_002695), Escherichia coli 0157:H7 EDL933 (ece, NC_002655), Shigella flexneri 2a str. 301 (sfl, NC_004337), Shigella flexneri 2a str. 2457T (sfx, NC_004741), Salmonella typhimurium LT2 (stm, NC_003197), Salmonella enterica subsp. enterica serovar Typhi Ty2 (stt, NC_004631) and Salmonella enterica subsp. enterica serovar Typhi str. CT18 (sty, NC_003198). Basically, these seven γ-proteobacteria are closely related enteric bacteria. Figure 5a shows the NJ tree created by our SoRT2, which clearly and correctly divided the seven γ-proteobacteria into three monophyletic clades. However, both GRIMM-NJ and MGR failed to do that, as shown in Figures 5b and 5c, respectively, because the two E. coli strains and the three Salmonella species did not form mutually exclusive monophyletic clades in their phylogenetic trees.
Figure 5.

(a) The NJ tree constructed by SoRT2 using a jackknife analysis of 100 replicates, where numbers on internal nodes are the support values. (b) The NJ tree based on the pairwise rearrangement distances computed by GRIMM. (c) The phylogenetic tree created by MGR.
SUMMARY
SoRT2 is a web-based tool for the analysis of genome rearrangements involving reversals, generalized transpositions and translocations (including fusions and fissions). It allows the user to quickly calculate pairwise rearrangement distances between input genomes and explore their corresponding optimal scenarios of required rearrangements. In addition, SoRT2 allows the user to quickly infer phylogenetic trees of input multiple genomes based on their pairwise genome rearrangement distances and further evaluate statistical reliability of tree branches. It is worth mentioning that the computation of optimal rearrangement distance involving reversals, generalized transpositions and translocations, and the statistical evaluation of trees are not available in other currently existing web servers. Particularly, as was mentioned in ref. (30), a generalized transposition (block-interchange) acting on a chromosome can be viewed as a process of fragment excision, circularization, linearization and re-incorporation, which exactly happens in the configuration of the immune response in higher animals, although the existence and biological significance of generalized transpositions have not yet been discussed in the current biological literature. Therefore, we believe that SoRT2 can provide interesting insights into the studies of genome rearrangements, particularly involving the generalized transpositions, and phylogenetic reconstruction.
FUNDING
National Science Council of Republic of China (NSC97-2221-E-009-081-MY3 to C.L.L.); NSC97-2221-E-007-080-MY3, NSC97-2221-E-007-081-MY3, NSC98-2627-B-007-011 C.Y.T.). Funding for open access charge: National Science Council of Republic of China.
Conflict of interest statement. None declared.
REFERENCES
- 1.Blanchette M, Kunisawa T, Sankoff D. Gene order breakpoint evidence in animal mitochondrial phylogeny. J. Mol. Evol. 1999;49:193–203. doi: 10.1007/pl00006542. [DOI] [PubMed] [Google Scholar]
- 2.Cosner ME, Jansen RK, Moret BME, Raubeson LA, Wang L, Warnow T, Wyman S. An empirical comparison of phylogenetic methods on chloroplast gene order data in Campanulaceae. In: Sankoff D, Nadeau JH, editors. Comparative Genomics. London: Kluwer Academic Publishers; 2000. pp. 99–121. [Google Scholar]
- 3.Belda E, Moya A, Silva FJ. Genome rearrangement distances and gene order phylogeny in γ-Proteobacteria. Mol. Biol. Evol. 2005;22:1456–1467. doi: 10.1093/molbev/msi134. [DOI] [PubMed] [Google Scholar]
- 4.Bourque G, Pevzner PA. Genome-scale evolution: reconstructing gene orders in the ancestral species. Genome Res. 2002;12:26–36. [PMC free article] [PubMed] [Google Scholar]
- 5.Fertin G, Labarre A, Rusu I, Tannier E, Vialette S. Combinatorics of Genome Rearrangements. Cambridge: The MIT Press; 2009. [Google Scholar]
- 6.Kaplan H, Shamir R, Tarjan RE. Faster and simpler algorithm for sorting signed permutations by reversals. SIAM J. Comp. 1999;29:880–892. [Google Scholar]
- 7.Hannenhalli S, Pevzner PA. Transforming cabbage into turnip: polynomial algorithm for sorting signed permutations by reversals. J. ACM. 1999;46:1–27. [Google Scholar]
- 8.Bafna V, Pevzner PA. Sorting by transpositions. SIAM J. Disc. Math. 1998;11:221–240. [Google Scholar]
- 9.Elias I, Hartman T. A 1.375-approximation algorithm for sorting by transpositions. IEEE/ACM Trans. Comp. Biol. Bioinformatics. 2006;3:369–379. doi: 10.1109/TCBB.2006.44. [DOI] [PubMed] [Google Scholar]
- 10.Christie DA. Sorting permutations by block-interchanges. Inform. Proc. Lett. 1996;60:165–169. [Google Scholar]
- 11.Lin YC, Lu CL, Chang H-Y, Tang CY. An efficient algorithm for sorting by block-interchanges and its application to the evolution of vibrio species. J. Comp. Biol. 2005;12:102–112. doi: 10.1089/cmb.2005.12.102. [DOI] [PubMed] [Google Scholar]
- 12.Hannenhalli S. Polynomial-time algorithm for computing translocation distance between genomes. Disc. Appl. Math. 1996;71:137–151. [Google Scholar]
- 13.Bergeron A, Mixtacki J, Stoye J. On sorting by translocations. J. Comp. Biol. 2006;13:567–578. doi: 10.1089/cmb.2006.13.567. [DOI] [PubMed] [Google Scholar]
- 14.Lu CL, Huang Y-L, Wang TC, Chiu H-T. Analysis of circular genome rearrangement by fusions, fissions and block-interchanges. BMC Bioinformatics. 2006;7:295. doi: 10.1186/1471-2105-7-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feijão P, Meidanis J. SCJ: a variant of breakpoint distance for which sorting, genome median and genome halving problems are easy. Lect. Notes Bioinformatics. 2009;5724:85–96. [Google Scholar]
- 16.Tesler G. GRIMM: genome rearrangements web server. Bioinformatics. 2002;18:492–493. doi: 10.1093/bioinformatics/18.3.492. [DOI] [PubMed] [Google Scholar]
- 17.Lu CL, Wang TC, Lin YC, Tang CY. ROBIN: a tool for genome rearrangement of block-interchanges. Bioinformatics. 2005;21:2780–2782. doi: 10.1093/bioinformatics/bti412. [DOI] [PubMed] [Google Scholar]
- 18.Lin YC, Lu CL, Liu Y-C, Tang CY. SPRING: a tool for the analysis of genome rearrangement using reversals and block-interchanges. Nucleic Acids Res. 2006;34:W696–W699. doi: 10.1093/nar/gkl169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bergeron A, Mixtacki J, Stoye J. A unifying view of genome rearrangements. Lect. Notes Bioinformatics. 2006;4175:163–173. [Google Scholar]
- 20.Lin CH, Zhao H, Lowcay SH, Shahab A, Bourque G. webMGR: an online tool for the multiple genome rearrangement problem. Bioinformatics. 2010;26:408–410. doi: 10.1093/bioinformatics/btp689. [DOI] [PubMed] [Google Scholar]
- 21.Yancopoulos S, Attie O, Friedberg R. Efficient sorting of genomic permutations by translocation, inversion and block interchange. Bioinformatics. 2005;21:3340–3346. doi: 10.1093/bioinformatics/bti535. [DOI] [PubMed] [Google Scholar]
- 22.Lin Y, Moret BME. Estimating true evolutionary distances under the DCJ model. Bioinformatics. 2008;24:i114–i122. doi: 10.1093/bioinformatics/btn148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergeron A, Mixtacki J, Stoye J. A new linear time algorithm to compute the genomic distance via the double cut and join distance. Theor. Comp. Sci. 2009;410:5300–5316. [Google Scholar]
- 24.Huang Y-L, Lu CL. Sorting by reversals, generalized transpositions and translocations using permutation groups. J. Comp. Biol. 2010;17:685–705. doi: 10.1089/cmb.2009.0025. [DOI] [PubMed] [Google Scholar]
- 25.Farris JS, Albert VA, Källersjö M, Lipscomb D, Kluge AG. Parsimony jackknifing outperforms neighbor-joining. Cladistics. 1996;12:99–124. doi: 10.1111/j.1096-0031.1996.tb00196.x. [DOI] [PubMed] [Google Scholar]
- 26.Blanchette M, Kunisawa T, Sankoff D. Parametric genome rearrangement. Gene. 1996;172:GC11–GC17. doi: 10.1016/0378-1119(95)00878-0. [DOI] [PubMed] [Google Scholar]
- 27.Eriksen N. (1+ε)-approximation of sorting by reversals and transpositions. Theor. Comp. Sci. 2002;289:517–529. [Google Scholar]
- 28.Felsenstein J. PHYLIP: phylogeny inference package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- 29.Zhao H, Bourque G. Recovering genome rearrangements in the mammalian phylogeny. Genome Res. 2009;19:934–942. doi: 10.1101/gr.086009.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adam Z, Sankoff D. The ABCs of MGR with DCJ. Evol. Bioinformatics. 2008;4:69–74. [PMC free article] [PubMed] [Google Scholar]