


Abstract
Recombination signal sequences (RSSs) flanking V, D and J gene segments are recognized and cut by the VDJ recombinase during development of B and T lymphocytes. All RSSs are composed of seven conserved nucleotides, followed by a spacer (containing either 12 ± 1 or 23 ± 1 poorly conserved nucleotides) and a conserved nonamer. Errors in V(D)J recombination, including cleavage of cryptic RSS outside the immunoglobulin and T cell receptor loci, are associated with oncogenic translocations observed in some lymphoid malignancies. We present in this paper the RSSsite web server, which is available from the address http://www.itb.cnr.it/rss. RSSsite consists of a web-accessible database, RSSdb, for the identification of pre-computed potential RSSs, and of the related search tool, DnaGrab, which allows the scoring of potential RSSs in user-supplied sequences. This latter algorithm makes use of probability models, which can be recasted to Bayesian network, taking into account correlations between groups of positions of a sequence, developed starting from specific reference sets of RSSs. In validation laboratory experiments, we selected 33 predicted cryptic RSSs (cRSSs) from 11 chromosomal regions outside the immunoglobulin and TCR loci for functional testing.
INTRODUCTION
V(D)J recombination is a mechanism of vertebrate genetic recombination that assembles gene segments into functional immunoglobulin (Ig) and T-cell receptor (TCR) genes. This site-specific recombination reaction generates the enormous repertoire of TCR and Ig molecules that are necessary for the recognition of diverse antigens from bacterial, viral and parasitic invaders. This reaction is directed by recombination signal sequences (RSSs), which flank each of the hundreds of potential donor gene segments. The V(D)J recombinase, comprised of the RAG1 and RAG2 proteins, introduces double-strand DNA breaks at the junction between a RSS and the flanking gene segment. DNA repair activity then re-joins breaks at two distant cuts to generate a functional gene through chromosomal rearrangement. Each RSS is composed of seven conserved nucleotides (a heptamer), residing next to the gene encoding sequence, followed by a spacer (containing either 12 ± 1 or 23 ± 1 poorly conserved nucleotides) and a conserved nonamer (9 bp). The RSSs are present on the 3′-side of a V region, on both sides of D segments, and on the 5′-side of the J region. Assembly of the correct composition of gene segments is directed by spacer length; recombination only joins gene segments flanked by RSSs with different spacer lengths.
Aberrant V(D)J recombination activity has been associated with oncogenic chromosomal translocations in lymphoid leukemia and lymphomas. The mechanisms of translocation remain unclear, but appear to include aberrant cutting of RSS-like sequences (cryptic RSSs) by the V(D)J recombinase at sites outside the Ig and TCR loci. Joining of these breaks to recombinase cuts within the Ig or TCR loci potentially leads to high expression of an oncogene in lymphocytes and leukemic transformation (1,2).
Considering the impact of this process in the context of translocation related to leukemia, a tool for predicting cRSSs on a genome-wide scale would be very useful to identify potential recombination sites. This task is complicated by the modular nature of RSSs and the variability of nucleotide sequences within the heptamer, spacer and nonamer elements. Using correlations between nucleotides at several positions within the 12 and 23 spacers and correlations between the identities of nucleotides at key positions, it is possible to predict the overall recombination efficiency of a RSS (3–5). It is now recognized that while RSSs are defined by a strict requirement for highly conserved nucleotides in the heptamer and nonamer, the quality of RSS function is determined in an analog manner by numerous complex interactions between the RAG proteins and the less-well conserved nucleotides in the heptamer, the nonamer, and, importantly, the spacer. For the latter, the importance of consensus nucleotides in defined points is emerging as a determinant for the efficient recognition of RSSs by RAG proteins (5).
THE DNAGrab ALGORITHM: GENOME-WIDE SEARCH AND CLASSIFICATION OF RSSs
To accomplish the prediction of cryptic RSS, we developed a software tool, DNAGrab, which scans the whole genome to identify candidate cRSSs. This algorithm makes use of probability models, which can be recasted to Bayesian networks, taking into account the correlations between groups of positions of a sequence, developed starting from specific reference sets of physiological RSSs (3). These Bayesian models are created by searching statistical correlations for each position in RSS patterns with all the other positions, placing no restrictions on the number of correlations or in the spatial positions relationship in the sequence. This approach determines, from all possible combinations of disjoint probability distributions, the set of distributions that most effectively distinguishes functional sites from non-functional sequences. Although the family of models to consider is very large, the use of mutual information allows a fast model selection by maximizing the mean recombination information content (RIC) for physiological RSSs (3). The RIC score, defined as the natural logarithm of the joint probability function of mutually correlated positions, is finally used to predict the possible functional cryptic RSSs.
The prediction capability of this score with respects to the effective recombination efficiency of RSSs has been long investigated and validated with in vitro testing (3–5). However, some recent experiments for determining how well RIC scores correlated with the levels of RAG-mediated cleavage and V(D)J recombination activity demonstrate that the prediction capability of the RIC score with respect to in vivo testing is quite weak (6). This aspect is explained, at least partially, by experiments showing the effect of the chromatin structure on the RAG cleavage efficiency, which confirms the role of chromatin in discriminating the RSS functionality (7). This information is actually not included in the RIC score, which functional prediction capability must be considered, in the light of this limitation, only as a preliminary screening of genome-wide predictions.
The implementation of DNAGrab, written in C++ and based on modern computational optimization techniques, allows a time-efficient screening of genome-wide data. To perform a genome scale analysis, we started from the previously developed models for RSSs with 12 and 23 spacer nucleotides (4), which define the groups of spatial positions to be considered for correlation screening. While other models can be developed which take into account different nucleotide spacers, in a genome-wide perspective our choice was to focus on 12 and 23 spaced RSS, since only these models are experimentally validated.
Concerning the mouse genome, we used a non-redundant version of the reference datasets employed in the original work (4), for a total of 143 RSS12 and 145 RSS23 sequences. Regarding the human genome, the reference sets of 168 RSS12 and 178 RSS23 were compiled in the context of this study, selecting sequences available from the IMGT database (8). The human and mouse RSS reference datasets can be downloaded from the RSSsite homepage.
These models were used by DNAGrab to scan all human and mouse chromosomes, searching for candidate cRSS sequences. To provide a reasonable dataset of cRSSs predicted to be possible functional substrates for the V(D)J recombinase, we scored the DNAGrab-provided RSSs using the RIC score. In detail, the algorithm computes a score for each sequence starting with CA and compares it with a score that has been experimentally correlated with the RSS function. In the current version of the system, pass/fail RIC thresholds are set accordingly to the work of Cowell and colleagues (3,4): RSS12 are scored as functional (pass) with RIC ≥ −38.81, while RSS23 pass with RIC ≥ −58.45.
Using DNAGrab, the RSS reference models and the RIC pass thresholds, a total of 3 089 308 and 1 833 319 potential RSS12 sequences were identified in human and mouse genomes, respectively, while for RSS23 3 218 664 and 2 091 561 RSSs passing RIC were identified and mapped. Considering that ∼5% of cRSS locations are scored as both 12 and 23 RSS in both genomes, these values indicate a global density of about 1 cRSS per 500 bp for human and 1 cRSS per 720 bp for mouse. These cRSS densities compare with the estimates from Lewis and colleagues (9) of 1 cRSS per 600 bp, which were based only on inference from functional testing of plasmid DNA. Statistics of the genome-wide distribution of cRSSs, which are predicted to be functional according to this scoring scheme, are reported in Tables 1 and 2. There is no significant difference in the number of 12 and 23 cRSSs within the human and the mouse genome. On the other hand, the difference in the frequency of 12 and 23 cRSSs between these two eukaryotic genomes can be attributed to the different reference datasets. There seems to be no apparent selection between intragenic and extragenic regions.
Table 1.
Number and density of HUMAN putative cRSSs for intragenic and extragenic regions
| Chr. | RSS12 |
RSS23 |
||||
|---|---|---|---|---|---|---|
| RSS count | Intrag. dens. | Extrag. dens. | RSS count | Intrag. dens. | Extrag. dens. | |
| chr1 | 248 570 | 890.41 | 1002.74 | 261 241 | 872.17 | 954.10 |
| chr2 | 251 136 | 858.59 | 968.40 | 258 916 | 924.03 | 939.30 |
| chr3 | 214 721 | 872.26 | 922.23 | 205 105 | 939.34 | 965.47 |
| chr4 | 205 738 | 875.10 | 929.12 | 183 816 | 1001.06 | 1039.92 |
| chr5 | 197 938 | 864.89 | 914.01 | 186 426 | 940.04 | 970.44 |
| chr6 | 183 662 | 860.50 | 931.68 | 175 554 | 951.26 | 974.71 |
| chr7 | 169 932 | 858.72 | 936.48 | 174 965 | 876.46 | 909.55 |
| chr8 | 155 781 | 863.59 | 939.55 | 153 573 | 930.73 | 953.06 |
| chr9 | 126 912 | 874.46 | 1112.69 | 137 272 | 873.30 | 1028.71 |
| chr10 | 144 072 | 856.05 | 940.74 | 154 524 | 868.69 | 877.11 |
| chr11 | 141 666 | 874.92 | 952.99 | 148 507 | 876.23 | 909.09 |
| chr12 | 139 185 | 881.05 | 961.68 | 146 332 | 876.15 | 914.71 |
| chr13 | 99 259 | 865.34 | 1160.30 | 96 400 | 972.04 | 1194.71 |
| chr14 | 89 376 | 859.31 | 1201.10 | 99 218 | 873.30 | 1081.96 |
| chr15 | 81 216 | 882.69 | 1262.45 | 96 756 | 864.90 | 1059.69 |
| chr16 | 78 857 | 895.37 | 1145.81 | 106 180 | 727.97 | 850.96 |
| chr17 | 73 289 | 922.98 | 1107.88 | 108 155 | 726.65 | 750.73 |
| chr18 | 75 549 | 844.88 | 1033.47 | 80 168 | 890.12 | 973.92 |
| chr19 | 71 216 | 915.51 | 830.28 | 88 168 | 645.67 | 670.64 |
| chr20 | 61 670 | 871.31 | 1021.98 | 77 213 | 781.58 | 816.26 |
| chr21 | 41 415 | 837.22 | 1162.14 | 39 510 | 784.12 | 1218.17 |
| chr22 | 40 042 | 900.60 | 1281.27 | 53 014 | 664.33 | 967.76 |
| chrX | 164 182 | 833.67 | 945.72 | 159 189 | 949.14 | 975.38 |
| chrY | 33 924 | 774.60 | 750.19 | 28 450 | 908.62 | 986.94 |
| TOT | 3 089 308 | 3 218 664 | ||||
Table 2.
Number and density of MOUSE putative cRSSs for intragenic and extragenic regions
| Chr. | RSS12 |
RSS23 |
||||
|---|---|---|---|---|---|---|
| RSS count | Intrag. dens. | Extrag. dens. | RSS count | Intrag. dens. | Extrag. dens. | |
| chr1 | 136 509 | 1426.42 | 1444.56 | 154 144 | 1175.16 | 1279.29 |
| chr2 | 127 417 | 1407.53 | 1426.40 | 148 619 | 1148.64 | 1222.91 |
| chr3 | 112 623 | 1408.35 | 1417.12 | 121 963 | 1213.85 | 1308.59 |
| chr4 | 108 463 | 1456.20 | 1434.87 | 124 708 | 1168.66 | 1247.96 |
| chr5 | 108 471 | 1495.78 | 1406.25 | 125 747 | 1126.75 | 1213.05 |
| chr6 | 105 388 | 1433.10 | 1418.73 | 120 445 | 1155.72 | 1241.37 |
| chr7 | 102 874 | 1449.90 | 1482.63 | 118 938 | 1123.63 | 1282.39 |
| chr8 | 92 561 | 1392.15 | 1423.27 | 105 324 | 1109.16 | 1250.80 |
| chr9 | 90 497 | 1375.52 | 1371.05 | 104 061 | 1112.28 | 1192.34 |
| chr10 | 95 577 | 1380.14 | 1360.09 | 102 615 | 1187.03 | 1266.81 |
| chr11 | 89 231 | 1310.35 | 1365.49 | 104 088 | 1103.98 | 1170.59 |
| chr12 | 87 244 | 1323.81 | 1389.87 | 96 912 | 1180.01 | 1251.21 |
| chr13 | 87 463 | 1396.70 | 1375.26 | 95 933 | 1175.18 | 1253.84 |
| chr14 | 79 458 | 1403.24 | 1575.61 | 98 595 | 1141.77 | 1269.79 |
| chr15 | 65 347 | 1440.15 | 1583.78 | 82 324 | 1150.06 | 1257.17 |
| chr16 | 62 698 | 1417.32 | 1568.14 | 77 027 | 1174.76 | 1276.42 |
| chr17 | 61 659 | 1408.09 | 1545.15 | 78 012 | 1093.06 | 1221.26 |
| chr18 | 58 031 | 1405.72 | 1564.20 | 71 989 | 1166.83 | 1260.92 |
| chr19 | 48 079 | 1429.58 | 1275.87 | 48 447 | 1158.45 | 1266.18 |
| chrX | 111 850 | 1472.87 | 1489.94 | 109 886 | 1414.72 | 1516.57 |
| chrY | 1906 | 1448.93 | 1343.42 | 1784 | 1596.39 | 1513.99 |
| TOT | 1 833 319 | 2 091 561 | ||||
Using the RSSsite web interface to identify and score cRSSs
Data about cRSS predictions performed on the human chromosome sequences (Hg18) and mouse chromosome sequences (Mm9), as downloaded from the UCSC Genome Browser (10), have been collected in a MySQL database. A web interface has been developed using HTML and the perl scripting language. Using this interface, it is possible to query the database about predicted human and mouse cRSSs, both with 12 and 23 spacers, and also to analyze user-provided human or murine sequences using the DNAGrab algorithm directly.
Three different interfaces have been provided to query the database and retrieve predicted cRSS subsets: a genomic region ‘Cytoband search’, a position-related ‘Chromosomal search’ and a generic ‘Gene search’ on an all chromosomes. Using the first option, the cRSS search is performed in a specific cytoband of the selected chromosome, according to annotations reported in our local database. Using the second option, the cRSS analysis is restricted to a specific region of a single chromosome, according to the UCSC genomic coordinates. Gene search in all chromosomes is based on the string entered by the user in a text area, which is compared with all the gene symbols, RefSeq and protein accession identifiers stored in the local annotation database. All the cRSSs predicted within the user-specified gene will be displayed with the relative genomic coordinates and RIC score. The query region extends from the transcription start site to the transcription end site of the gene and can be expanded upstream and downstream using the menu in the text area.
The output results can be obtained in two different formats: tab-delimited text (Figure 1) or UCSC-uploadable format. The results can also be downloaded as a compressed file. If the UCSC-uploadable format option is selected, from the generated report it will be possible to display the RSS predictions as user-generated tracks into the UCSC Genome Browser. The system creates a temporary text file in bed format and uploads it directly to the UCSC web site (10), hence this option may be slow depending on the number of sequences selected. An example of a cRSS track displayed on the UCSC annotation background of the Gnb211 mouse gene is shown in Figure 2 .
Figure 1.

Sample tabular output from a cRSS ‘Chromosomal search’ database query on a region of human chr8 (nt. 128 817 497–28 822 855). Shown are the chromosome range, organism, type of cRSS requested and the putative signal sequences (7) with the sequence, the location, the RIC score and a clickable link to UCSC.
Figure 2.

Sample graphical output from a cRSS ‘Gene search’ query for the mouse gene Gnb2l1. Shown are the location of 12 (red) and 23 (black) cRSSs passing the RIC score and mapping in a window encompassing the specified gene±1000 bp.
The ‘Analyse your own sequence’ section of the web interface enables users to identify the presence of putative cRSSs within any sequence. The user can choose to use the human or the murine model for searching both for 12 and 23 spaced RSSs. For multiple analyses only sequences in FASTA format are accepted while, for single sequence analysis, sequences with or without the FASTA definition line are accepted. The DnaGrab algorithm is used to predict cRSSs within user-provided sequences. While predicted cRSSs stored in the database are all considered functional according to the defined thresholds, this section provides a RIC score, with respect to the selected model, for all the substrings starting with CA, providing information also for sequences that did not pass the RIC filter. The output is also available in tabular form from the web page (Figure 3).
Figure 3.

Sample tabular output of the ‘Analyse your own sequence’ section of the RSSsite. The sequence has been analyzed using both the human and murine models, searching for 12 and 23 spaced cRSS. In the output, the start and end position are reported, with the strand and the RIC score. The last column provides, according to the RIC thresholds, an indication about the possibility of the sequence to be a DNA breaking point.
EXPERIMENTAL VALIDATION AND SUMMARY
The RSSsite web server provides a valuable tool to build preliminary in silico hypothesis on the sequence-based mechanisms regulating both physiological and aberrant V(D)J recombination. For example, an earlier version of RSSsite was used in a study by Dik and colleagues (11) focusing on the (11;14)(p13;q11) translocation, which is presumed to arise from an erroneous T-cell receptor delta TCRD V(D)J recombination and to result in LMO2 activation. Using our algorithm, this group was able to determine RIC scores for LMO2 cRSSs, which were used to drive functional experimentation. Furthermore, they analyzed the LMO2 locus (−10 kb to +30 kb) for the occurrence of 12- and 23-bp cRSSs predicted to be functional according to the RIC score.
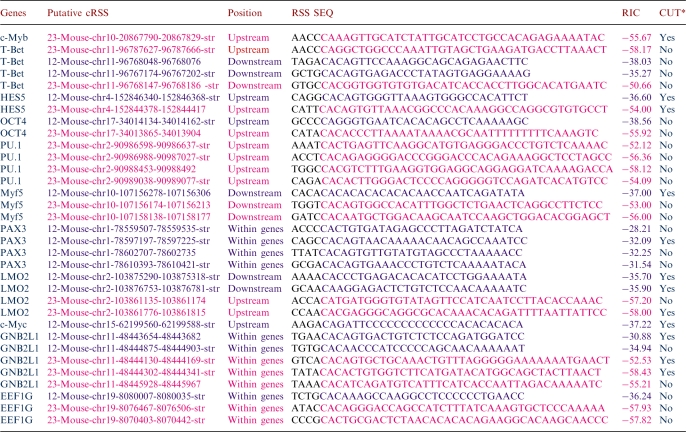
In our laboratory experiments linked with this work, we selected 33 RSSs from 11 chromosomal regions outside the immunoglobulin and TCR loci for functional testing. The dataset is fully described in Table 3. Using ligation-mediated PCR (LM-PCR), V(D)J recombinase-mediated DNA breaks at RSSs were analyzed in genomic DNA prepared from mouse primary thymocytes, where RAG1 and RAG2 were expressed. We observed breaks of putative cRSSs in 12 out of the 33 sites tested (7 out of 15 for RSS12 and 5 out of 18 for RSS23). Concerning the distribution of these 12 RSSs with respects to genes, four of them are intragenic, while eight are outside genes (five upstream and three downstream). Breaks were detected in ∼1% of genomic DNA. DNA sequencing of LM-PCR products confirmed that breaks had occurred precisely at the 5′ boundary of RSSs, which is consistent with bona fide V(D)J recombinase-mediated cuts. V(D)J recombinase-mediated breaks were detected at 8 out of the 11 chromosomal sites tested, including previously uncharacterized RSSs. No translocation products involving these break sites were detected in the assays performed so far. No selection was made for live cells, therefore many unrepaired breaks may have led to cell death.
Table 3.
Functionally tested sequences using putative cRSSs, according to the RIC score, predicted by the DNAGrab algorithm available at the RSSsite
 |
|---|
RSS12 sequences are in blue, RSS23 sequences are in pink.
CUT* sequences are defined according to the visualization by LMPCR of a cleavage product at the 5′-end of the RSS.
As discussed earlier in the ‘Algorithmic’ section of this work, while in vitro experiments have moderate correspondence with functional predictions achieved with the RIC score, in vivo tests demonstrate a lower accordance with the proposed results, which can be partially attributed to the influence of the chromatin structure in recombination signal sequences recognition by RAG proteins. Therefore, the RIC score provided by our algorithm should be considered only as a screening method for a preliminary identification of functional cRSSs in the context of genome-wide analyses, which takes into account the sequence patterns considered outside the chromosomal context. Nonetheless, we believe that our results, as confirmed by the presented experimental validation, can be valuable for the identification of potential recombination sites, although these predictions must be considered taking into account the actual limitations of the developed algorithm that lacks, for example, of information about the chromatin structure.
CONCLUSIONS
The identification and mapping of putative RSSs in the human and mouse genomes has significant applicative potential, given the well-established observation of translocations at cRSS sites in lymphoid malignancies. We described here RSSsite, a web-based database and search tool for the retrieval of predicted cRSSs from the human and mouse genomes, starting from a given chromosome region, a given gene identifier or user-supplied sequences. The software is freely available at the web address http://www.itb.cnr.it/rss.
The surprisingly high frequency of observed V(D)J breaks suggests that many cryptic RSSs may be cut by RAG proteins during lymphocyte development. However, V(D)J-mediated chromosomal translocations remain rare events. The control mechanisms that prevent more frequent involvement of aberrant V(D)J breaks in potentially oncogenic chromosomal translocations are currently unknown. We hope that the RSSsite web server will provide a valuable tool to systematically test genomic and eventually epigenetic mechanisms regulating RSS accessibility and usage at different chromosomal sites.
FUNDING
Italian Fund for Basic Research (FIRB-MIUR) project grants ‘ITALBIONET’ and ‘LITBIO’. Funding for open access charge: National Research Council.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We gratefully acknowledge the help of Lindsay Cowell and Joe Volpe who kindly provided an updated version of their RIC filter. We also thank the former student and collaborators who contributed to this project: Riccardo Fallini (MSc student at IFOM), Lizeta Gjanci (INGENIO Program fellow at ITB-CNR) and Chiara Bishop (Web Designer at ITB-CNR)
REFERENCES
- 1.Lieber MR, Yu K, Raghavan SC. Roles of nonhomologous DNA end joining, V(D)J recombination, and class switch recombination in chromosomal translocations. DNA Repair. 2006;5:1234–1245. doi: 10.1016/j.dnarep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 2.Marculescu R, Vanura K, Montpellier B, Roulland S, Le T, Navarro JM, Jager U, McBlane F, Nadel B. Recombinase, chromosomal translocations and lymphoid neoplasia: targeting mistakes and repair failures. DNA Repair. 2006;5:1246–1258. doi: 10.1016/j.dnarep.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 3.Cowell LG, Davila M, Kepler TB, Kelsoe G. Identification and utilization of arbitrary correlations in models of recombination signal sequences. Genome Biol. 2002;3:RESEARCH0072. doi: 10.1186/gb-2002-3-12-research0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cowell LG, Davila M, Ramsden D, Kelsoe G. Computational tools for understanding sequence variability in recombination signals. Immunol. Rev. 2004;200:57–69. doi: 10.1111/j.0105-2896.2004.00171.x. [DOI] [PubMed] [Google Scholar]
- 5.Lee AI, Fugmann SD, Cowell LG, Ptaszek LM, Kelsoe G, Schatz DG. A functional analysis of the spacer of V(D)J recombination signal sequences. PLoS Biol. 2003;1:E1. doi: 10.1371/journal.pbio.0000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang M, Swanson PC. V(D)J recombinase binding and cleavage of cryptic recombination signal sequences identified from lymphoid malignancies. J. Biol. Chem. 2008;283:6717–6727. doi: 10.1074/jbc.M710301200. [DOI] [PubMed] [Google Scholar]
- 7.Shimazaki N, Tsai AG, Lieber MR. H3K4me3 Stimulates the V(D)J RAG complex for both nicking and hairpinning in trans in addition to tethering in cis: implications for translocations. Mol. Cell. 2009;34:535–544. doi: 10.1016/j.molcel.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lefranc MP, Giudicelli V, Kaas Q, Duprat E, Jabado-Michaloud J, Scaviner D, Ginestoux C, Clément O, Chaume D, Lefranc G. IMGT, the international ImMunoGeneTics information system. Nucleic Acid Res. 2005;33:D593–D597. doi: 10.1093/nar/gki065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewis SM, Agard E, Suh S, Czyzyk L. Cryptic signals and the fidelity of V(D)J joining. Mol. Cell Biol. 1997;17:3125–3136. doi: 10.1128/mcb.17.6.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karolchik D, Kuhn RM, Baertsch R, Barber GP, Clawson H, Diekhans M, Giardine B, Harte RA, Hinrichs AS, Hsu F, et al. The UCSC Genome Browser Database: 2008 update. Nucleic Acids Res. 2008;36:D773–D779. doi: 10.1093/nar/gkm966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dik WA, Nadel B, Przybylski GK, Asnafi V, Grabarczyk P, Navarro JM, Verhaaf B, Schmidt CA, Macintyre EA, van Dongen J.JM, et al. Different chromosomal breakpoints impact the level of LMO2 expression in T-ALL. Blood. 2007;110:388–392. doi: 10.1182/blood-2006-12-064816. [DOI] [PubMed] [Google Scholar]