


Abstract
The discrimination between functionally neutral amino acid substitutions and non-neutral mutations, affecting protein function, is very important for our understanding of diseases. The rapidly growing amounts of experimental data enable the development of computational tools to facilitate the annotation of these substitutions. Here, we describe a Random Forests-based classifier, named Mutation Detector (MuD) that utilizes structural and sequence-derived features to assess the impact of a given substitution on the protein function. In its automatic mode, MuD is comparable to alternative tools in performance. However, the uniqueness of MuD is that user-reported protein-specific structural and functional information can be added at run-time, thereby enhancing the prediction accuracy further. The MuD server, available at http://mud.tau.ac.il, assigns a reliability score to every prediction, thus offering a useful tool for the prioritization of substitutions in proteins with an available 3D structure.
INTRODUCTION
The human population contains approximately 10 million single nucleotide polymorphism (SNP) sites (1). The non-synonymous SNPs (nsSNPs) account for a large portion of the known genetic variations associated with human diseases (2). Many experimental mutagenesis studies have been dedicated to the identification of disease-causing amino acid (AA) substitutions among SNP sites. However, experimental mutagenesis is time-, labor- and cost-demanding. Thus, numerous computational tools have been developed to predict effects of AA substitutions on protein function.
The reported methods have significantly different input requirements. (i) One set of tools focuses only on sequence-based features (3–7). For example, Ng and Henikoff (5) developed a homology-based algorithm (SIFT; sorting intolerant from tolerant) to estimate the viability of substitutions according to the profiles of AA residues in alignment columns. (ii) A number of approaches extend beyond the use of alignments to sequence-based prediction of structural features (3,5,7). For instance, Bromberg and Rost (3) trained neural networks using, among other features, predicted secondary structure and residue solvent accessibility. (iii) To reflect the differences between the wild type AA and the mutant, several methods utilize physicochemical features (3,6,7), illustrating the differences in the hydropathy, secondary structure propensities, etc. (iv) With the growing number of solved structures, several tools choose to utilize observed structural data such as solvent accessibility (8–11), distance to the ligand (9–11), statistical knowledge-based potentials (9) and micro-environment description (8). (v) A number of studies show that prediction can be improved by combining information from various sources (3,7,11). For example, PolyPhen (11) employs a rule-based system that incorporates information from the UniProtKB/Swiss-Prot annotations (12) together with data extracted from solved 3D-structure and sequence alignment.
AA substitution prediction algorithms are usually contingent on a data set of substitution variants that have been experimentally annotated as neutral or non-neutral. The available data sets can be broadly divided into four categories. (i) Relatively clean substitution data collected from extensive mutagenesis studies (13–15). These studies probe nearly all substitutions over whole proteins to reflect the whole spectrum of effects. (ii) Comprehensive collections of naturally occurring substitutions annotated through association studies and targeted laboratory mutagenesis experiments (UniProtKB/Swiss-Prot (12), HGMD (2), etc). Unfortunately, this data might be biased by investigator interest and some of the annotated neutrals are likely non-neutral mutations whose disease associations were overlooked. Moreover, the number of false non-neutral annotations obtained from association studies is also relatively high (16). (iii) The Protein Mutant Database (PMD) (17), representative of the third type of data, avoids the problem of false non-neutrals by reporting substitutions that have been experimentally validated. (iv) The fourth category includes evolutionary model (EM)-based substitution data sets that are a relatively reliable set of neutral mutations created by analyzing single residue substitutions between orthologous proteins (3,7).
Herein, we present a web-based tool, named MuD, aimed at distinguishing functionally neutral and non-neutral AA substitutions. MuD utilizes a machine learning algorithm and a set of structural- and sequence-based features. A benchmark experiment using a cross-validation on a subset of the Bromberg and Rost (3) data set (Sub-BR data set) showed similar performance as SNAP (screening for non-acceptable polymorphisms). However, the performance on a test set of three proteins (3-PRO), which have previously been used for benchmarking, confirmed the importance of using reliable experimentally verified structural data (e.g. naturally occurring ligands pertinent to the function of the protein and the biological oligomerization state of the protein). Given this improvement in performance, we enabled the MuD server to allow users to guide the prediction scheme using select structural features. The web server is free and open to all users and there is no login requirement.
METHODS
We employed a Random Forests machine-learning algorithm to differentiate between non-neutral and neutral substitutions. The model was trained and tested on the Sub-BR data set (described below) using cross-validation. In addition, we evaluated the performance on the 3-PRO data set after training on the complete Sub-BR data set.
Data sets
MuD’s model was trained on substitutions extracted from the Bromberg and Rost (3) data set. Since MuD requires both sequence and structural data, we excluded proteins for which the structure was not available. We also excluded proteins with fewer than five homologs, and substitutions in positions with low information content, as measured by the ConSurf (18) confidence interval. This filtration procedure resulted in a set of 19 615 substitutions (70% non-neutral). A balanced data set contributes to the prediction accuracy (19). Thus, we randomly removed approximately a third of the non-neutral substitutions, yielding a balanced set (Sub-BR) comprising 12 133 substitutions from 1178 proteins. Of this subset, 10 253 substitutions originated from PMD and 2065 were EM substitutions. The classification of the Sub-BR proteins according to the Structural Classification of Proteins (SCOP) database (20) can be found on the web server.
We extended our examination to the 3-PRO data set, comprised of the Escherichia coli lac I repressor (15), HIV protease (13) and T4 lysozyme (14,21) mutagenesis data. After applying the filtration procedure (as explained previously), we retained subsets with 1773/2230, 148/157 and 315/1475 non-neutral to neutral mutants ratios, respectively; constituting 99%, 90% and 89% of the original data sets, respectively. The original mutagenesis experiments classified each substitution into four categories: no phenotypic effect and three levels of severity of phenotypic change. We followed the reduced binary classification of Ref. (9), where the substitutions with no effect were deemed neutral and the rest were non-neutral.
Machine learning
Random Forests is a classifier consisting of an ensemble of tree-structured classifiers (22). We used the R software implementation of Random Forests (23). The number of trees to grow was set to 650 and the number of random features to be searched at each tree node was the square-root of the number of features.
Data gathering
Both the sequences and the PDB file names required for all data sets were extracted from the corresponding UniProtKB/Swiss-Prot entries (12). The multiple sequence alignments (MSAs) and the PDB (24) files were downloaded from the ConSurf-DB database (25) and from the protein quaternary structure (PQS) server (26), respectively. We excluded proteins sharing <30% sequence identity with the query protein to ensure that the sequences in the MSA would all belong to the same protein fold (27).
Feature set
We used a total of 14 features (with 41 dimensions). The novel features are presented here, whereas traditional descriptors such as secondary structure assignment, UniProtKB/Swiss-Prot annotations, physicochemical and AA preference (using SIFT), number of sequences in the alignment, stability prediction change and evolutionary conservation are included in the Supplementary Data.
Structure-based features
Solvent accessibility and oligomerization interface
The solvent accessibility was calculated according to the structure (among all the protein’s structures) in which the query position had the least area of side chain solvent accessibility. This structure was used to calculate two solvent accessibility features: Cβ density (10) and side chain accessible surface area (using NACCESS (28)). To measure the involvement of the query position in the oligomerization interfaces, we performed a search over all available structures for the maximal difference between the heavy atom density of the query position in its single chain and complex forms.
Ligand distance and binding site conservation
To identify residues involved in ligand binding or catalytic activity, we measured the shortest distance between the heavy atoms of the query position and of the ligands in all available structures. A ligand was defined as a hetero atom compound, dinucleotide chain or a protein chain shorter than 15 AAs. Since crystal structures often include ingredients of the crystallization solution, which are not naturally present, we strove to rank the ligands according to their biological relevance. We assumed that the evolutionary conservation of the binding site indicates whether the ligand bound to the site is indeed naturally present. Therefore, we ranked the ligands according to the ConSurf conservation score assigned to the most conserved AA with a distance of 4 Å from the ligand. This value was selected empirically (Supplementary Data).
Fold three-dimensional residue environment
Fold three-dimensional residue environment (F3DRE) was calculated following Ref. (8), but taking into account the residue environment of homologs (inferred from the MSA). Given a query position, we retrieved from the protein structure all the AA positions Ψ whose Cα atoms were within a distance of 9 Å from the Cα of the query position. Next, according to the alignment of the AA sequence of the query protein in the MSA we defined MSA columns T that correspond to Ψ. For every column t|t ∈ T, we calculated the AA composition vector Caa,t|1 ≤ aa ≤ 20, t ∈ T (Supplementary Equation S1), while disregarding gaps in the column. Next, we calculated the F3DRE for every AA as the average AA composition over all T (Supplementary Equation S2).
Sequence-based features
Sequence identity to the closest homolog bearing the substitution (SIDCH)
Counterintuitively, in some of the non-neutral mutations, the mutant AA appeared naturally in the aligned positions in homologous proteins, and we hypothesized that they are likely to occur mostly in distant homologs. Therefore, we added a feature that specifies the sequence identity of the query protein to the closest homolog bearing the mutant AA. For example, the mutation I104S in the human protein transthyretin (UniProtKB/Swiss-Prot ID: TTHY_HUMAN) is known to cause amyloidosis Type 1 (29). Two homologous proteins (A7UIU9_PERFV and Q9PTT3_SPAAU) with sequence identities of 58% and 53% to TTHY_HUMAN feature the AA serine in the corresponding position, and the value of I104S SIDCH was set to 58%.
SNAP
The prediction scheme presented here is based on a solved crystal structure of the query protein, reducing the size of the learning data set considerably. Thus, we also used SNAP, a prediction scheme based on sequence alone. The predictions utilized during the performance assessment were obtained from the testing procedures described in Ref. (3).
Performance measurements
The performance of MuD on the Sub-BR data set was examined using a leave-one-out cross-validation test. To empirically estimate how well the method can be generalized to unseen substitutions it is important that the training and testing sets are as dissimilar as possible. Therefore, during each iteration of the leave-one-out cross-validation, all substitutions from a single protein were designated for testing, whereas the substitutions belonging to proteins with HSSP distances ≤0 (27) to the test protein were designated for training. For alignments of >250 residues this HSSP-distance threshold infers that no pair of proteins had over 21% pairwise sequence identity.
To calculate the average and SD for the performance measures, we used a bootstrap procedure with 1000 iterations. At each iteration we randomly selected 60% of the data set while maintaining a balanced ratio of non-neutral to neutral substitutions. The performance measures on each subset were calculated according to predictions obtained during the cross-validation.
To compare PolyPhen’s performance, we defined the ‘benign’ and ‘possibly damaging’ predictions as neutral and the prediction ‘probably damaging’ as non-neutral. This binary classification gave the highest Matthew’s correlation coefficient (MCC) value for the PolyPhen predictions on the Sub-BR data set (MCC of 0.39 ± 0.01). The alternative definition, setting ‘benign’ predictions as neutral and ‘possibly damaging’ predictions and ‘probably damaging’ as non-neutral gave a lower MCC of 0.35 ± 0.01.
To further measure MuD’s performance, we trained on the Sub-BR data set and tested on the 3-PRO data set in fully- and semi-automatic schemes. As in the cross validation procedure, only proteins with an HSSP-value ≤0 to any of the 3-PRO proteins were retained in the training set. In the semi-automatic scheme, all ligands not present naturally were disregarded and the oligomerization state of the protein was determined according to the literature. Specifically, (i) the T4 lysozyme was predicted as a dimer by the PQS. However, according to the literature its biological unit is a monomer (30). As the T4 lysozyme has been used as a scaffold for the creation of an artificial binding site, most of its solved structures include biologically non-relevant ligands (30). Hence, we disregarded all ligands found in the T4 lysozyme structures. (ii) Most of the HIV protease structures have an inhibitor located at the active site. All other ligands and additional chains were removed. (iii) At the semi-automatic prediction of the lac I repressor substitutions, we considered only the DNA segments and ligands that were located at the known repressor binding site.
Performance measures
We used several standard measures [Equations (1–5), and Supplementary Table S1 and Equation S5]
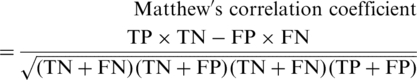 |
(1) |
| (2) |
| (3) |
| (4) |
| (5) |
to evaluate the performance of MuD and to compare it with other prediction tools. To assess the overall performance, we utilized both the receiver operating characteristic (ROC) area under curve (AUC) and the MCC. The ROC curve analysis does not require the determination of a decision threshold and as such can better describe the performance of the prediction. However, although the SIFT predictions are numeric they have been optimized for a default cutoff. Furthermore, PolyPhen predictions are not numeric. Therefore, the comparison of performance of SIFT and PolyPhen to MuD was only possible using MCC.
RESULTS
Cross-validation performance
According to the MCC and ROC AUC, MuD and SNAP exhibit similar performance and are both better than SIFT and PolyPhen (Table 1, and Supplementary Table S2 and Figure S1) on the Sub-BR data set.
Table 1.
The performance of MuD, SIFT, PolyPhen and SNAP on the Sub-BR, and the Sub-PMD data sets
| Sub-BR data set |
Sub-PMD data set |
|||||||
|---|---|---|---|---|---|---|---|---|
| MuD | SNAP | SIFT | PolyPhen | MuD | SNAP | SIFT | PolyPhen | |
| Precision | 72.8 ± 0.5 | 68.7 ± 0.4 | 69.6 ± 0.5 | 66.6 ± 0.5 | 73.3 ± 0.4 | 69.5 ± 0.4 | 70.6 ± 0.5 | 67.8 ± 0.5 |
| True positive rate | 76.6 ± 0.4 | 79.9 ± 0.4 | 73.5 ± 0.5 | 73.5 ± 0.5 | 76.6 ± 0.4 | 79.9 ± 0.4 | 73.6 ± 0.5 | 73.5 ± 0.5 |
| Specificity | 71.8 ± 0.5 | 64.1 ± 0.5 | 68.4 ± 0.5 | 63.9 ± 0.5 | 58.8 ± 0.6 | 48.3 ± 0.6 | 55.9 ± 0.6 | 48.9 ± 0.7 |
| MCC | 49.5 ± 0.4 | 48.3 ± 0.8 | 42.9 ± 0.1 | 39.2 ± 0.8 | 45.2 ± 0.1 | 41.0 ± 1.1 | 36.0 ± 1.0 | 29.7 ± 1.1 |
| ROC AUC | 81.9 ± 0.3 | 78.8 ± 0.3 | NR | NR | 74.8 ± 0.4 | 70.9 ± 0.4 | NR | NR |
The average and SD of the performance measures were obtained by a bootstrap procedure run for 1000 iterations performed on the cross-validation predictions. The results on the Sub-PMD data set are a subset of the results obtained during the cross-validation on the entire data set. According to the MCC, MuD and SNAP perform better than SIFT and PolyPhen both on the entire data set and on the PMD subset. According to the ROC AUC, MuD performed better than SNAP. However, according to the MCC both methods exhibited similar performance. Although all methods exhibited a decline in the performance on the Sub-PMD data set relative to the performance on the Sub-BR data set, MuD surpassed all methods on the Sub-PMD data set. Values in the table have been multiplied by 100.
The Sub-BR data set consists of two subsets: substitutions extracted from the PMD database and neutral EM substitutions. We reviewed the performance of MuD on each of these subsets separately. MuD did better than SIFT, PolyPhen and SNAP on the Sub-PMD data set. However, the performance of all four methods on this set was lower than on the entire data set. MuD had a high FP rate of 41.1 ± 0.6% on the substitutions extracted from the PMD compared to only 2.0 ± 0.2% on the EM subset. The implications of this are discussed in the Supplementary Data.
Prediction performance may also depend on the protein’s structural class. To examine this possibility, we analyzed the cross-validation results of SNAP and MuD and the predictions made by SIFT and PolyPhen on the Sub-BR data set according to the SCOP class assignment of the query proteins (Supplementary Figure S2). Ninty seven percent of the Sub-BR data set mutations occur at proteins assigned by SCOP to one of the seven ‘true’ SCOP classes. Across all SCOP classes MuD performed as good as any of the other methods or better. The performance of MuD, SNAP and SIFT on the ‘membrane and cell surface proteins and peptides’ and ‘small proteins’ classes showed a decline in the performance relative to the four main SCOP classes (all α, all β, α / β, α + β). These classes comprise a small number of substitutions (437 and 391, respectively), but the decline may be indicative of a true difficulty.
Performance on the 3-PRO data set
We also evaluated performance on additional data sets that have previously been used for benchmarking. Our results of the fully automated and of the semi-automatic schemes were compared with SIFT, PolyPhen and SNAP (Table 2). A comparison between all the fully automated methods indicated that neither was favorable over the others. SNAP performed best on the T4 lysozyme, SIFT performed best on the HIV protease and MuD performed best on the lac I repressor data set. However, semi-automatic MuD surpassed all other methods in all performance measures.
Table 2.
Introduction of reliable structural data improves the prediction performance
| HIV proteasea |
T4 lysozyme |
Lac repressor |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SA MuD | MuD | SNAP | SIFT | SA MuD | MuD | SNAP | SIFT | PP | SA MuD | MuD | SNAP | SIFT | PP | |
| Precision | 67.6 | 61.6 | 54.3 | 66.0 | 37.0 | 24.4 | 34.9 | 29.8 | 31.1 | 79.2 | 81.83 | 64.1 | 65.2 | 83.9 |
| TP rate | 93.2 | 95.3 | 98.0 | 93.2 | 92.1 | 97.1 | 81.0 | 91.4 | 89.9 | 77.9 | 77.83 | 74.6 | 70.1 | 67.9 |
| Specificity | 58.0 | 44.3 | 22.3 | 54.8 | 66.5 | 35.9 | 67.8 | 54.1 | 58.0 | 83.8 | 83.00 | 66.9 | 70.3 | 85.7 |
| MCC | 54.4 | 45.6 | 30.7 | 51.7 | 45.0 | 27.4 | 37.7 | 34.8 | 36.4 | 61.8 | 60.93 | 41.2 | 40.2 | 54.2 |
| ROC AUC | 89.9 | 81.2 | 78.2 | NR | 87.4 | 83.7 | 83.8 | NR | NR | 89.5 | 89.0 | 79.3 | NR | NR |
The performance of PolyPhen (PP), SNAP, SIFT, the semi-automatic (SA MuD) and the fully-automatic MuD (MuD) on the 3-PRO data set. The performance of the automatic predictors appears to depend on the evaluation criterion and data set. Nevertheless as indicated by all measures, the application of the semi-automatic prediction scheme improved the performance, thus surpassing all the fully-automatic methods. In the semi-automatic prediction, we removed from the solved crystal structures of the query proteins, the structures of the non-naturally present ligands and selected the appropriate dimerization state for each of the proteins. Values in the table have been multiplied by 100.
aPolyPhen did not produce predictions for the HIV-1 protease.
The web server
The MuD web server implementation encourages the user to introduce into the prediction scheme specific biological data about the target protein. The graphical user interface and an example of the results page are depicted in Supplementary Figure S6.
DISCUSSION
We tested MuD using cross-validation analysis on the Sub-BR data set and found automatic MuD to be as good as SNAP, and better than PolyPhen and SIFT. However, the assessment also indicates that MuD might be less suitable for the prediction of substitutions in non-globular and small proteins.
The incorporation of structural features such as solvent accessibility, ligand proximity and oligomerization interfaces into the automatic MuD might not always be advantageous. This is due to (i) the presence of non-naturally present ligands, such as ingredients of the crystallization solution and (ii) incorrect oligomerization state assignments that may hinder the prediction accuracy. To alleviate these problems, the MuD web server implementation offers a semi-automatic scheme. It enables the user to incorporate additional data about the target protein in order to improve the prediction accuracy. This procedure aims reducing the erroneous features that might be extracted from the crystal structure. Specifically, the graphical interface of the server allows the user to filter out irrelevant ligands and to select the structure of the biological unit. Additionally, the user can change the indication of functionally important residues.
Detailed analysis of three well-characterized proteins showed that the incorporation of target-specific data on each protein via the semi-automatic scheme improved the prediction performance, surpassing current methods. The largest difference in the performance between the semi-automatic and automatic MuD is manifested in the T4 lysozyme data set. This is not surprising since many of the PDB T4 lysozyme structures contain non-relevant ligands and do not present the protein in its correct biological shape.
An important aspect of MuD is the ascription of a reliability score to every prediction (Supplementary Data). The reliability score offers the researcher a qualitative assessment of the prediction, indicating its expected accuracy, sensitivity and precision. The reliability score can be used to prioritize substitutions in a given set according to their likelihood of affecting the protein function.
We are hopeful that this tool will assist researchers in the annotation of disease causing substitutions in proteins with a solved crystal structure.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Commission FP6 Integrated Project EuroHear LSHG-CT- 20054-512063 (to N.B.-T., K.B.A.); NATO traveling grant No. CBP.MD.CLG 983009 (to T.H. and N.B-T); National Library of Medicine (grant 2-R01-LM007329 to Y.B.). Funding for open access charge: European Commission FP6 Integrated Project EuroHear LSHG-CT- 20054-512063.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Guy Nimrod for helpful discussions.
REFERENCES
- 1.Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, Sherry S, Mullikin JC, Mortimore BJ, Willey DL, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- 2.Stenson PD, Ball E, Howells K, Phillips A, Mort M, Cooper DN. Human Gene Mutation Database: towards a comprehensive central mutation database. J. Med. Genet. 2008;45:124–126. doi: 10.1136/jmg.2007.055210. [DOI] [PubMed] [Google Scholar]
- 3.Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35:3823–3835. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Capriotti E, Arbiza L, Casadio R, Dopazo J, Dopazo H, Marti-Renom MA. Use of estimated evolutionary strength at the codon level improves the prediction of disease-related protein mutations in humans. Hum. Mutat. 2008;29:198–204. doi: 10.1002/humu.20628. [DOI] [PubMed] [Google Scholar]
- 5.Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu. Rev. Genomics Hum. Genet. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- 6.Stone EA, Sidow A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. 2005;15:978–986. doi: 10.1101/gr.3804205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrer-Costa C, Orozco M, de la Cruz X. Sequence-based prediction of pathological mutations. Proteins. 2004;57:811–819. doi: 10.1002/prot.20252. [DOI] [PubMed] [Google Scholar]
- 8.Capriotti E, Fariselli P, Calabrese R, Casadio R. Predicting protein stability changes from sequences using support vector machines. Bioinformatics. 2005;21(Suppl. 2):ii54–58. doi: 10.1093/bioinformatics/bti1109. [DOI] [PubMed] [Google Scholar]
- 9.Chasman D, Adams RM. Predicting the functional consequences of non-synonymous single nucleotide polymorphisms: structure-based assessment of amino acid variation. J. Mol. Biol. 2001;307:683–706. doi: 10.1006/jmbi.2001.4510. [DOI] [PubMed] [Google Scholar]
- 10.Saunders CT, Baker D. Evaluation of structural and evolutionary contributions to deleterious mutation prediction. J. Mol. Biol. 2002;322:891–901. doi: 10.1016/s0022-2836(02)00813-6. [DOI] [PubMed] [Google Scholar]
- 11.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.The Universal Protein Resource (UniProt) in 2010. Nucleic acids research. 38:D142–148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loeb DD, Swanstrom R, Everitt L, Manchester M, Stamper SE, Hutchison CA., 3rd Complete mutagenesis of the HIV-1 protease. Nature. 1989;340:397–400. doi: 10.1038/340397a0. [DOI] [PubMed] [Google Scholar]
- 14.Alber T, Sun DP, Nye JA, Muchmore DC, Matthews BW. Temperature-sensitive mutations of bacteriophage T4 lysozyme occur at sites with low mobility and low solvent accessibility in the folded protein. Biochemistry. 1987;26:3754–3758. doi: 10.1021/bi00387a002. [DOI] [PubMed] [Google Scholar]
- 15.Markiewicz P, Kleina LG, Cruz C, Ehret S, Miller JH. Genetic studies of the lac repressor. XIV. Analysis of 4000 altered Escherichia coli lac repressors reveals essential and non-essential residues, as well as “spacers” which do not require a specific sequence. J. Mol. Biol. 1994;240:421–433. doi: 10.1006/jmbi.1994.1458. [DOI] [PubMed] [Google Scholar]
- 16.Emahazion T, Feuk L, Jobs M, Sawyer SL, Fredman D, St Clair D, Prince JA, Brookes AJ. SNP association studies in Alzheimer’s disease highlight problems for complex disease analysis. Trends Genet. 2001;17:407–413. doi: 10.1016/s0168-9525(01)02342-3. [DOI] [PubMed] [Google Scholar]
- 17.Kawabata T, Ota M, Nishikawa K. The Protein Mutant Database. Nucleic Acids Res. 1999;27:355–357. doi: 10.1093/nar/27.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben-Tal N. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33:W299–302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dobson RJ, Munroe PB, Caulfield MJ, Saqi MA. Predicting deleterious nsSNPs: an analysis of sequence and structural attributes. BMC Bioinformatics. 2006;7:217. doi: 10.1186/1471-2105-7-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andreeva A, Howorth D, Chandonia JM, Brenner SE, Hubbard TJ, Chothia C, Murzin AG. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res. 2008;36:D419–425. doi: 10.1093/nar/gkm993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rennell D, Bouvier SE, Hardy LW, Poteete AR. Systematic mutation of bacteriophage T4 lysozyme. J. Mol. Biol. 1991;222:67–88. doi: 10.1016/0022-2836(91)90738-r. [DOI] [PubMed] [Google Scholar]
- 22.Breiman L. Random forests. Machine Learning. 2001;45:5–32. [Google Scholar]
- 23.Liaw A, Wiener M. Classification and regression by randomForest. R News. 2002;2:18–22. [Google Scholar]
- 24.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldenberg O, Erez E, Nimrod G, Ben-Tal N. The ConSurf-DB: pre-calculated evolutionary conservation profiles of protein structures. Nucleic Acids Res. 2009;37:D323–327. doi: 10.1093/nar/gkn822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henrick K, Thornton JM. PQS: a protein quaternary structure file server. Trends Biochem. Sci. 1998;23:358–361. doi: 10.1016/s0968-0004(98)01253-5. [DOI] [PubMed] [Google Scholar]
- 27.Rost B. Enzyme function less conserved than anticipated. J. Mol. Biol. 2002;318:595–608. doi: 10.1016/S0022-2836(02)00016-5. [DOI] [PubMed] [Google Scholar]
- 28.Hubbard SJ, Campbell SF, Thornton JM. Molecular recognition. Conformational analysis of limited proteolytic sites and serine proteinase protein inhibitors. J. Mol. Biol. 1991;220:507–530. doi: 10.1016/0022-2836(91)90027-4. [DOI] [PubMed] [Google Scholar]
- 29.Berni R, Malpeli G, Folli C, Murrell JR, Liepnieks JJ, Benson MD. The Ile-84–>Ser amino acid substitution in transthyretin interferes with the interaction with plasma retinol-binding protein. J. Biol. Chem. 1994;269:23395–23398. [PubMed] [Google Scholar]
- 30.Heinz DW, Matthews BW. Rapid crystallization of T4 lysozyme by intermolecular disulfide cross-linking. Protein Eng. 1994;7:301–307. doi: 10.1093/protein/7.3.301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.