


Abstract
The proteins in a cell often assemble into complexes to carry out their functions and play an essential role of biological processes. The PCFamily server identifies template-based homologous protein complexes [called protein complex family (PCF)] and infers functional modules of the query proteins. This server first finds homologous structure complexes of the query using BLASTP to search the structural template database (11 263 complexes). PCFamily then searches the homologous complexes of the templates (query) from a complete genomic database (Integr8 with 6 352 363 protein sequences in 2274 species). According to these homologous complexes across multiple species, this sever infers binding models (e.g. hydrogen-bonds and conserved amino acids in the interfaces), functional modules, and the conserved interacting domains and Gene Ontology annotations of the PCF. Experimental results demonstrate that the PCFamily server can be useful for binding model visualizations and annotating the query proteins. We believe that the server is able to provide valuable insights for determining functional modules of biological networks across multiple species. The PCFamily sever is available at http://pcfamily.life.nctu.edu.tw.
INTRODUCTION
Protein complexes are fundamental units of macromolecular organization and their composition is also known to vary according to cellular requirements (1). To identify and characterize the protein complexes, genome-scale interaction discovery approaches, such as two-hybrid system or affinity purification (2,3), have been proposed. However, these methods are often unable to respond how a protein interacts with others. Based on increasing protein–protein interactions (PPI) (4–7) and structure complexes (8), previous studies have suggested that the total number of protein–protein interaction types are limited (approximately 10 000 types) (9) and the quaternary structures (QS) can be clustered into 3151 QS families (10).
A known 3D-structure complex provides physical protein interaction topology, interacting domains and atomic detailed binding models of interactions. Recently, some studies utilized template-based methods [i.e. comparative modeling (11) and fold recognition (12)], which search a 3D-complex library to model a large set of yeast complexes (13,14). These methods are time-consuming to search all possible homologous PPIs or complexes, which are useful to explore interface evolutions of a specific 3D-structure complex, from a large complete genomic database (e.g. Integr8) with many species (15).
To address these issues, we numerously enhanced and modified both PPI family search [sequence-based PPI search method (16)] and 3D-domain interologs with template-based scoring function [3D-template PPI prediction method (17)]. According to our knowledge, PCFamily is the first public server that identifies homologous complexes (two or more proteins) and module evolution of the query. For a set of query protein sequences, this server provides the template-based homologous complexes [called protein complex family (PCF)] in multiple species, graphic visualization of conserved interacting residues and binding models (interfaces), conserved Gene Ontology (GO) annotations (18) and interacting domains. Our results demonstrate that this server achieves high agreements on interacting domains and GO annotations between query proteins and their respective homologous complexes.
METHOD AND IMPLEMENTATION
Figure 1 shows the details of the PCFamily server to search the template-based homologous complexes (PCF) of a set of query protein sequences by following steps (Figure 1A). First, the server uses BLASTP to search template candidates from structural template database [11 263 structure complexes selected from Protein Data Bank (PDB)]. Then we utilize template-based scoring function (17) to statistically evaluate the complex similarity (joint Z-value ≥3.0) between query proteins and candidates (Figure 1B and C). After a template was selected, the server searches the PPI family of each interface of template with Z-value ≥3.0 from a complete genomic database (Integr8 version 103, containing 6 352 363 protein sequences in 2274 species) (15) (Figures 2A and 1D). These PPI families are combined into homologous complexes with the significant complex similarity (joint Z-value ≥3.0) according to the interfaces of the 3D-complex template (Figure 1E). For this PCF including the query, we measured the conservation ratio (CR) of the domain composition (DC) and CRs of biological processes (BPs), cellular components (CCs) and molecular functions (MF) using GO annotations. Finally, this server provides homologous complexes, graphic visualization of complex topology, detailed residues interactions, interface alignments across multiple species (Figure 2) and conservations with GO annotations and DCs.
Figure 1.

Overview of the PCFamily server for homologous complexes search using proteins Skp1, Skp2 and Cks1 of R. norvegicus as the query. (A) The main procedure. (B) Identify the template candidate (PDB code 2ast) of the query using BLASTP and template-based scoring function to scan the structural template database. (C) The topology of the template. (D) The homologous PPI families of interfaces A–B and B–C of the template searching on Integr8 database. (E) Template-based homologous complexes of the query.
Figure 2.

Binding models and MSAs of PPI family in Skp1–Skp2–Cks1 complex (PDB code 2ast). (A) The atomic binding model with hydrogen-bonds (red dash lines) for each interface of the template. (B) MSAs of PPI family of the interface A (Skp1)–B (Skp2), respectively.
Homologous complex
The concept of homologous complex (two or more proteins) is extended from homologous PPIs (16) and 3D-domain interologs with template-based scoring function (17). Here, we used a 3D-trimer template T (proteins A, B and C) with two interfaces A–B and B–C as a simple case to define the homologous complex of T as follows: (i) A′, B′ and C′ are the homologous proteins of A, B and C, respectively, with the significant sequence similarity (BLASTP E-values ≤10−10) (19,20); (ii) A′–B′ and B′–C′ are the template-based homologous PPIs of A–B and B–C, respectively, with the significant interface similarity (Z-value ≥3.0) (17); and (iii) significant complex similarity (joint Z-value ≥3.0) between complexes A′–B′–C′ and A–B–C. The joint Z-value of the complex similarity is defined as
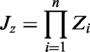 |
(1) |
where n is the number of interfaces of a template (T); Zi is the Z-value (interface similarity) of the template-based homologous PPI i (e.g. A′–B′) based on the template interface (e.g. A–B). Here, JZ ≥ 3.0 is considered as significant similarity according to the statistical analysis of 941 3D-structure complexes with 2 138 123 homologous complexes.
Template-based scoring function
We have recently proposed a template-based scoring function to determine the reliability of the PPI derived from a 3D-dimer structure (17). For a predicted template-based PPI, this scoring function assigned a score, including residue–residue interacting scores, which consist of the steric (Evdw) and hydrogen-bond (ESF) energies, and sequence consensus scores which the couple-conserved residue score (Econs) and contact-residue similarity score (Esim). Finally, we calculated the Z-value of the score for this PPI using the mean and standard deviation (SD) of 10 000 random interfaces by mutating 60% interface residues.
Annotations of homologous complexes
A 3D-complex template and its homologous complexes can be considered as a PCF. The concept of the PCF is analogous to the notions of protein sequence family (21), protein structure family (22) and PPI family (16). We believe that PCFs can be applied widely in biological investigations. We assume that the members of a PCF are conserved on GO annotations, interacting domain(s) and binding model(s). Using these conservations of a PCF, the PCFamily server can annotate the GO terms (BP, CC and MF) and DCs of query proteins. To statistically evaluate the agreement of GO terms and DCs between the template and its PCF (with N homologous complexes), we define the agreement ratio (AR) using the CR (CR = Na/N), where Na is the number of homologous complexes with the same GO term (or DC) in a PCF. The AR is given as
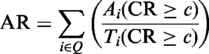 |
(2) |
where Q is a set of query templates; Ti (CR ≥ c) is the total number of the GO terms (or DCs) of template i when CR ≥ c; and Ai (CR ≥ c) is the number of the agreement GO terms (or DCs) of template i when CR ≥ c.
INPUT, OUTPUT AND OPTIONS
PCFamily is an easy-to-use web server (Figure 3). Users input a single or a set of protein sequence(s) in FASTA format or a 3D-complex protein structure (PDB code) (Figure 3A). Typically, the PCFamily server yields structural template candidates within 25 s when querying three sequences and the numbers of amino acids are lesser than 450 (Figure 3B). For the query, this server shows the template candidate and its PCF: detailed atomic interactions of the interfaces and binding models by using Jmol (23); protein interaction topology (Figure 3C); multiple sequence alignments (MSA) with hydrogen-bond residues and conserved residues (Figure 3D); and CRs of DCs and GO terms (BP, CC and MF; Figure 3E).
Figure 3.

The PCFamily server search results using proteins Epor, Epo and Epor of M. musculus as the query. (A) The user interface for inputting the query protein sequences or PDB code. (B) The template candidate of the query. (C) The numbers of conserved domains and GO-term conservations, interfaces, protein interaction topology and homologous complexes of the query (selected template). (D) MSAs and interacting residue conservations of homologous PPIs of the interface A (Epo)–B (Epor), respectively. (E) Conserved domain and GO-term compositions of the PCF.
Example analysis
The complex of Skp1, Skp2 and Cks1
Figure 1 shows search results using S-phase kinase-associated protein 1 (Skp1; UniProt accession number: Q6PEC4), S-phase kinase-associated protein 2 (Skp2; B2GUZ0) and RGD1561797 protein (Cks1, B2RZ99) of Rattus norvegicus as the query. Skp1 and Skp2 are subunits of the SCFSkp2 ubiquitin ligase complex that regulates proteolysis of the p27Kip1 protein in cell cycle progression (24,25). Recognition and ubiquitination of p27Kip1 requires the accessory protein Cks1 by the SCFSkp2 ubiquitin–ligase complex (24). According to KEGG pathway database (26), Skp1–Skp2 and Skp2–Cks1 in R. norvegicus are recorded in the ubiquitin-mediated proteolysis pathway and the small-cell lung cancer pathway, respectively. For this query, the PCFamily server found the template candidate [PDB code 2ast (24)] (Figure 1C) and 43 homologous complexes (called SCF complex family) from nine species [e.g. Homo sapiens, R. norvegicus and Bos taurus (Figure 1E)]. Among these 43 homologous complexes, one complex (H. sapiens) is recorded in the IntAct database (7) and three homologous complexes, including the query in R. norvegicus, Q9WTX5 (Skp1)–Q9Z0Z3 (Skp2)–P61025 (Cks1b) in Mus musculus and Q3ZCF3 (SKP1)–A7MB09 (SKP2)–Q0P5A5 (CKS1B) in B. taurus, are recorded in KEGG pathway. In addition, six members are Skp1–Skp2–Cks1b (or Cks2) complexes, which are highly relative to the query and the template. All members of this PCF have the same DC PF01466 (Skp1)–PF00646 (F–box)–PF01111 (CKS) and a high consensus DC PF03931 (Skp1_POZ)–PF00646–PF01111 (CR = 0.95). The query proteins consist of these two DCs (Figure 1E).
The PCFamily server provides the binding model and MSAs of each interface (Figure 2 and Supplementary Figure S1) based on the template. Interface A–B (Figure 2A) contains three main hydrogen-bonds, including Gln1097–Trp2097, Glu1156–Tyr2128 and Asn1157–Ser2121. These six residues are conserved in mammals (Figure 2B). Additionally, PCFamily identifies six sidechain–sidechain hydrogen-bonds forming the network to stabilize the interface B–C (24) (Supplementary Figure S1). All interacting residues forming the hydrogen-bonds are often highly conserved and useful for observing the interface evolution across multiple species.
Epor–Epo–Epor complex
Erythropoietin (Epo) stimulates the proliferation and differentiation of the cells (e.g. erythroid precursor cells) (27,28). Epo binds and orientates two cell-surface erythropoietin receptors (Epor) to activate cells and trigger an intracellular phosphorylation cascade (29). Using M. musculus Epor (P14753), Epo (P07321) and Epor (P14753) as the query proteins (Figure 3A), the PCFamily server found the template candidate (PDB code 1eer; Figure 3B) and its six homologous Epor–Epo–Epor complexes in three species (Figure 3C). Among these six complexes, three complexes, P19235–P01588–P19235 (H. sapiens), P14753–P07321–P14753 (M. musculus) and Q5FVS4–P29676–Q5FVS4 (R. norvegicus) are recorded in KEGG. Two complexes are formed by Epo (P29676) binding to Epors Q07303 (27) and O35545 (30), respectively. PCFamily indicates the MSAs with hydrogen-bond and conserved residues in the interfaces A–B (Figure 3D) and A–C (Supplementary Figure S2) of Epor–Epo–Epor PCF.
This PCF includes 65 GO-term compositions. Among these GO term compositions, the CR ratios of two MF compositions and three CC compositions exceed 0.6 (Figure 3E). The query has these five GO term compositions, such as GO:0004900 (erythropoietin receptor activity)–GO:0005128 (erythropoietin receptor binding)–GO:0004900. Additionally, the query and these homologous complexes consistently contain two conserved DCs (CR = 1), including PF00041–PF00758–PF00041 and PF09067–PF00758–PF09067. PF00758–PF00041 and PF00758–PF09067 are recorded in iPfam (21). These results reveal that the PCFamily server can identify homologous complexes for the interface evolution and annotations of the query.
RESULTS
To evaluate the accuracy of the PCFamily server for discovery of homologous complexes and the annotations of query proteins, we selected a non-redundant query structural template set. This set comprising 941 protein complexes (2979 sequences and 2042 interfaces, called NR941; Supplementary Table S1) was selected from the PDB released on February 24, 2006. For searching homologous complexes, NR941 was used to assess PCFamily performance and to determine the threshold of joint Z-value Jz [Equation (1)] on the Integr8 database (Figure 4A). In addition, the NR941 set was applied to calculate CRs of DCs (and GO terms) for each PCF and infer the relations between CRs and ARs [Equation (2)] of DCs and GO terms (Figure 4B).
Figure 4.

Evaluations of the PCFamily server on 941 protein complex families. (A) The distributions of recall (solid) and precision (dot) with different joint Z-value thresholds. (B) The relationships between ARs and the CRs of DCs, BPs, MFs and CCs.
We defined the gold standard positive and negative sets to measure the performance of the PCFamily server. Here, we used a trimer structural template T (proteins A, B and C) with two interfaces A–B and B–C as a simple case to describe a positive complex (A′–B′–C′) of T as follows: (i) A′, B′ and C′ are homologs of A, B and C, respectively, with the significant sequence similarity (BLASTP E-values ≤10−10) (19,20); (2) A′–B′ and B′–C′ are PPIs recorded in annotated PPI databases (e.g. IntAct) and have the same interacting domains of A–B and B–C, respectively. Based on the rules, the gold standard positive set includes 770 complexes (Supplementary Table S2) derived from the Integr8 for the set NR941. On the other hand, the gold standard negative set was generated according to the assumption that proteins, located in the same subcellular localization and acting in the similar BPs, are more likely to form a complex than the proteins involved in different processes. This study applied the relative specificity similarity (RSS) (31) to measure the BP and CC similarities of PPIs based on the GO terms. According to 198 882 interactions in IntAct database, we considered a complex candidate as a negative case, if BP and CC RSS scores of any interface of the complex are <0.4 (Supplementary Figure S3). Here, the negative set consists of 1960 complexes (Supplementary Table S3).
Precision, recall and F-measure were utilized to assess the reliability of the PCFamily server for searching homologous complexes. The F-measure is given as (2× precision × recall)/(precision + recall), where the precision and recall are obtained using the gold standard positive and negative sets. Figure 4A shows the relationships between joint Z-value Jz and recall and precision using 941 complexes on the Integr8 database. The recall significantly decreases when joint Z-value ≥3; conversely, the precision increases slightly when joint Z-value is between 3 and 4. The recall and precision are 0.82 and 0.45, respectively, and the PCFamily server yields the highest F-measure value (0.55) if the threshold of joint Z-value is set to 3.
Figure 4B shows the relationships between ARs and the CRs of DCs, BP, CC and MF. If the CR of DCs is >0.6 (black), the AR between the query and their respective homologous complexes exceeds 0.95 [Equation (2)]. If the CR of GO terms (i.e. BP, CC and MF) is >0.6, the ARs are consistently >0.74 for BP (0.77, green), CC (0.74, yellow) and MF (0.75, red). These experimental results demonstrate that this server achieves high agreements on DCs and GO terms between the query (i.e. template complexes) and their respective homologous complexes.
CONCLUSIONS
This study demonstrates the utility and feasibility of the PCFamily server in identifying homologous complexes and inferring conserved domains and GO terms from PCFs. PCFamily is the first server to provide homologous complexes in multiple species: graphic visualization of the complex topology and detailed atomic residue–residue interactions; interface alignments; and conservations of GO terms and DCs. Our experimental results demonstrate that the query and its homologous complexes achieve high agreements on domains and GO terms. We believe that PCFamily is a fast homologous complexes search server and is able to provide valuable insights for determining functional modules of biological networks across multiple species.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Council (to J.-M.Y.) and ATU plan by MOE (to J.-M.Y., partial).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
Authors are grateful to both the hardware and software supports of the Structural Bioinformatics Core Facility at National Chiao Tung University.
REFERENCES
- 1.Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dumpelfeld B, et al. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- 2.Edwards AM, Kus B, Jansen R, Greenbaum D, Greenblatt J, Gerstein M. Bridging structural biology and genomics: assessing protein interaction data with known complexes. Trends Genet. 2002;18:529–536. doi: 10.1016/s0168-9525(02)02763-4. [DOI] [PubMed] [Google Scholar]
- 3.Kemmeren P, van Berkum NL, Vilo J, Bijma T, Donders R, Brazma A, Holstege FC. Protein interaction verification and functional annotation by integrated analysis of genome-scale data. Mol. Cell. 2002;9:1133–1143. doi: 10.1016/s1097-2765(02)00531-2. [DOI] [PubMed] [Google Scholar]
- 4.Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004;32:D449–D451. doi: 10.1093/nar/gkh086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatr-aryamontri A, Ceol A, Palazzi LM, Nardelli G, Schneider MV, Castagnoli L, Cesareni G. MINT: the Molecular INTeraction database. Nucleic Acids Res. 2007;35:D572–D574. doi: 10.1093/nar/gkl950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mewes HW, Dietmann S, Frishman D, Gregory R, Mannhaupt G, Mayer KF, Munsterkotter M, Ruepp A, Spannagl M, Stumpflen V, et al. MIPS: analysis and annotation of genome information in 2007. Nucleic Acids Res. 2008;36:D196–D201. doi: 10.1093/nar/gkm980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kerrien S, Alam-Faruque Y, Aranda B, Bancarz I, Bridge A, Derow C, Dimmer E, Feuermann M, Friedrichsen A, Huntley R, et al. IntAct–open source resource for molecular interaction data. Nucleic Acids Res. 2007;35:D561–D565. doi: 10.1093/nar/gkl958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deshpande N, Addess KJ, Bluhm WF, Merino-Ott JC, Townsend-Merino W, Zhang Q, Knezevich C, Xie L, Chen L, Feng Z, et al. The RCSB Protein Data Bank: a redesigned query system and relational database based on the mmCIF schema. Nucleic Acids Res. 2005;33:D233–D237. doi: 10.1093/nar/gki057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aloy P, Russell RB. Ten thousand interactions for the molecular biologist. Nat. Biotechnol. 2004;22:1317–1321. doi: 10.1038/nbt1018. [DOI] [PubMed] [Google Scholar]
- 10.Levy ED, Pereira-Leal JB, Chothia C, Teichmann SA. 3D complex: a structural classification of protein complexes. PLoS Comput. Biol. 2006;2:e155. doi: 10.1371/journal.pcbi.0020155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aloy P, Russell RB. Interrogating protein interaction networks through structural biology. Proc. Natl Acad. Sci. USA. 2002;99:5896–5901. doi: 10.1073/pnas.092147999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu L, Lu H, Skolnick J. MULTIPROSPECTOR: an algorithm for the prediction of protein-protein interactions by multimeric threading. Proteins. 2002;49:350–364. doi: 10.1002/prot.10222. [DOI] [PubMed] [Google Scholar]
- 13.Aloy P, Bottcher B, Ceulemans H, Leutwein C, Mellwig C, Fischer S, Gavin AC, Bork P, Superti-Furga G, Serrano L, et al. Structure-based assembly of protein complexes in yeast. Science. 2004;303:2026–2029. doi: 10.1126/science.1092645. [DOI] [PubMed] [Google Scholar]
- 14.Davis FP, Braberg H, Shen MY, Pieper U, Sali A, Madhusudhan MS. Protein complex compositions predicted by structural similarity. Nucleic Acids Res. 2006;34:2943–2952. doi: 10.1093/nar/gkl353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kersey P, Bower L, Morris L, Horne A, Petryszak R, Kanz C, Kanapin A, Das U, Michoud K, Phan I, et al. Integr8 and Genome Reviews: integrated views of complete genomes and proteomes. Nucleic Acids Res. 2005;33:D297–D302. doi: 10.1093/nar/gki039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen CC, Lin CY, Lo YS, Yang JM. PPISearch: a web server for searching homologous protein-protein interactions across multiple species. Nucleic Acids Res. 2009;37:W369–W375. doi: 10.1093/nar/gkp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen YC, Lo YS, Hsu WC, Yang JM. 3D-partner: a web server to infer interacting partners and binding models. Nucleic Acids Res. 2007;35:W561–W567. doi: 10.1093/nar/gkm346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matthews LR, Vaglio P, Reboul J, Ge H, Davis BP, Garrels J, Vincent S, Vidal M. Identification of potential interaction networks using sequence-based searches for conserved protein-protein interactions or “interologs”. Genome Res. 2001;11:2120–2126. doi: 10.1101/gr.205301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu H, Luscombe NM, Lu HX, Zhu X, Xia Y, Han JD, Bertin N, Chung S, Vidal M, Gerstein M. Annotation transfer between genomes: protein-protein interologs and protein-DNA regulogs. Genome Res. 2004;14:1107–1118. doi: 10.1101/gr.1774904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, et al. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andreeva A, Howorth D, Brenner SE, Hubbard TJ, Chothia C, Murzin AG. SCOP database in 2004: refinements integrate structure and sequence family data. Nucleic Acids Res. 2004;32:D226–D229. doi: 10.1093/nar/gkh039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herraez A. Biomolecules in the computer - Jmol to the rescue. Biochem. Mol. Biol. Educ. 2006;34:255–261. doi: 10.1002/bmb.2006.494034042644. [DOI] [PubMed] [Google Scholar]
- 24.Hao B, Zheng N, Schulman BA, Wu G, Miller JJ, Pagano M, Pavletich NP. Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol. Cell. 2005;20:9–19. doi: 10.1016/j.molcel.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 25.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 26.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masuda S, Nagao M, Takahata K, Konishi Y, Gallyas F, Jr, Tabira T, Sasaki R. Functional erythropoietin receptor of the cells with neural characteristics. Comparison with receptor properties of erythroid cells. J. Biol. Chem. 1993;268:11208–11216. [PubMed] [Google Scholar]
- 28.Syed RS, Reid SW, Li C, Cheetham JC, Aoki KH, Liu B, Zhan H, Osslund TD, Chirino AJ, Zhang J, et al. Efficiency of signalling through cytokine receptors depends critically on receptor orientation. Nature. 1998;395:511–516. doi: 10.1038/26773. [DOI] [PubMed] [Google Scholar]
- 29.Damen JE, Krystal G. Early events in erythropoietin-induced signaling. Exp. Hematol. 1996;24:1455–1459. [PubMed] [Google Scholar]
- 30.Yamaji R, Murakami C, Takenoshita M, Tsuyama S, Inui H, Miyatake K, Nakano Y. The intron 5-inserted form of rat erythropoietin receptor is expressed as a membrane-bound form. Biochim. Biophys. Acta. 1998;1403:169–178. doi: 10.1016/s0167-4889(98)00037-8. [DOI] [PubMed] [Google Scholar]
- 31.Wu X, Zhu L, Guo J, Zhang DY, Lin K. Prediction of yeast protein-protein interaction network: insights from the Gene Ontology and annotations. Nucleic Acids Res. 2006;34:2137–2150. doi: 10.1093/nar/gkl219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.