


Abstract
The protein–ligand interacting mechanism is essential to biological processes and drug discovery. The SiMMap server statistically derives site-moiety map with several anchors, which describe the relationship between the moiety preferences and physico-chemical properties of the binding site, from the interaction profiles between query target protein and its docked (or co-crystallized) compounds. Each anchor includes three basic elements: a binding pocket with conserved interacting residues, the moiety composition of query compounds and pocket–moiety interaction type (electrostatic, hydrogen bonding or van der Waals). We provide initial validation of the site-moiety map on three targets, thymidine kinase, and estrogen receptors of antagonists and agonists. Experimental results show that an anchor is often a hot spot and the site-moiety map can help to assemble potential leads by optimal steric, hydrogen bonding and electronic moieties. When a compound highly agrees with anchors of site-moiety map, this compound often activates or inhibits the target protein. We believe that the site-moiety map is useful for drug discovery and understanding biological mechanisms. The SiMMap web server is available at http://simfam.life.nctu.edu.tw/.
INTRODUCTION
As the number of protein structures increases rapidly, structure-based drug design and virtual screening approaches are becoming important and helpful in lead discovery (1–4). A number of docking and virtual screening methods (5–8) have been utilized to indentify lead compounds, and some success stories have been reported (9–13). However, identifying lead compounds by exploiting thousands of docked protein–compound complexes is still a challenging task. The major weakness of virtual screenings is likely due to incomplete understandings of ligand-binding mechanisms and the subsequently imprecise scoring algorithms (2–4).
Most of docking programs (5–7) use energy-based scoring methods which are often biased toward both the selection of high-molecular weight compounds and charged polar compounds (14,15). These approaches generally cannot identify the key features (e.g. pharmacophore spots) that are essential to trigger or block the biological responses of the target protein. Although pharmacophore techniques (16) have been applied to derive the key features, these methods require a set of known active ligands that were acquired experimentally. Therefore, the more powerful techniques for post-screening analysis to identify the key features through docked compounds and to understand the binding mechanisms provide a great potential value for drug design.
To address these issues, we presented the SiMMap server to infer the key features by a site-moiety map describing the relationship between the moiety preferences and the physico-chemical properties of the binding site. According to our knowledge, SiMMap is the first public server that identifies the site-moiety map from a query protein structure and its docked (or co-crystallized) compounds. The server provides pocket–moiety interaction preferences (anchors) including binding pockets with conserved interacting residues, moiety preferences and interaction type. We verified the site-moiety map on three targets, thymidine kinase, and estrogen receptors of antagonists and agonists. Experimental results show that an anchor is often a hot spot and the site-moiety map is useful to identify active compounds for these targets. We believe that the site-moiety map is able to provide biological insights and is useful for drug discovery and lead optimization.
METHOD AND IMPLEMENTATION
Figure 1 presents an overview of the SiMMap server for identifying the site-moiety map with anchors, describing moiety preferences and physico-chemical properties of the binding site, from a query protein structure and docked compounds. The server first uses checkmol (http://merian.pch.univie.ac.at/∼nhaider/cheminf/cmmm) to recognize the compound moieties and utilizes GEMDOCK (8) to generate a merged protein–compound interaction profile (Figure 1B), including electrostatic (E), hydrogen bonding (H) and van der Waals (V) interactions. According to this profile, we infer anchor candidates by identifying the pockets with significant interacting residues and moieties with Z-score ≥1.645. The neighbor anchor candidates, which are the same interaction type and the distances between their centers are <3.5Å, are grouped into one anchor. These anchors form the site-moiety map describing interaction preferences between compound moieties and the binding site of the query (Figures 1C and D). Finally, this server provides graphic visualization for the site-moiety map; anchors with moiety structures and compositions, pocket–moiety interactions and the relationship between anchors and moieties of query compounds.
Figure 1.

Overview of the SiMMap server for the site-moiety map using herpes simplex virus type-1 thymidine kinase (TK) and 1000 docked compounds as the query. (A) Main procedure; (B) the merged protein–compound interaction profile; (C) the pocket–moiety interaction preferences of three anchors: E1 (electrostatic), H2 (hydrogen-bonding) and V1 (van der Waals). Each anchor consists of a binding pocket with conserved interacting residues, the moiety composition and anchor type; (D) the site-moiety map with four anchors.
Site-moiety map, anchor and pocket
The anchor (pocket–moiety interaction preference) is the core of a site-moiety map. An anchor possesses three essential elements: (i) a binding pocket with conserved interacting residues and specific physico-chemical properties; (ii) moiety preferences of the pocket; (iii) pocket–moiety interaction type (E, H or V). An anchor can be considered as ‘key features’ for representing the conserved binding environment element or a ‘hot spot’ that involves biological functions. In addition, we regard a binding pocket, which consists of several residues significantly interacting to compound moieties, as a part of the binding site. The binding pocket often possesses specific physico-chemical properties and geometric shape to bind preferred moieties. The site-moiety map, which can help to assemble potential leads by optimal steric, hydrogen bonding, and electronic moieties, is useful for drug discovery and understanding biological mechanisms.
Data sets
To describe and evaluate the utility of the SiMMap server, we tested the server on three target proteins for virtual screening. These proteins are herpes simplex virus type-1 thymidine kinase [TK, PDB code 1kim(17)], estrogen receptor α for antagonists [ER, PDB code 3ert(18)] and estrogen receptor α for agonists [ERA, PDB code 1gwr(19)]. Each compound set consists of 10 known active ligands and 990 compounds selected randomly from available chemical directory (ACD) proposed by Bissantz et al. (20). Currently, the docked conformations of these 1000 compounds were generated by the in-house GEMDOCK program (8) which is comparable to some docking methods (e.g. DOCK, FlexX and GOLD) on the 100 protein–ligand complexes and some screening targets (8,14). In addition, GEMDOCK has been successfully applied to identify inhibitors and binding sites for some targets (10,13,21,22).
Main procedure
The SiMMap server performs six main steps for a query (Figure 1A). Here, we used TK as an example for describing these steps. First, users input a protein structure and its docked compounds. The server used checkmol to identify moieties of docked compounds and GEMDOCK to generate E, H and V interaction profiles. For each profile, the matrix size is N×K where N and K are the numbers of compounds and interacting residues of query protein, respectively. An interaction profile matrix P(I) with type I (E, H or V) is represented as
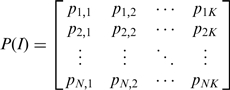 |
where pi,j is a binary value for the compound i interacting to the residue j (Figure 1B). For H and E profiles, pi,j is set to 1 (green) if an atom pair between the compound i and the residue j forms hydrogen bonding or electrostatic interactions, respectively; conversely, the interaction is set to 0 (black). For van der Waals (vdW) interaction, an interaction is set to 1 when the energy is less than −4 (kcal/mol).
SiMMap identified consensus interactions between residues and compound moieties with similar physical-chemical properties through the profiles. For each interacting residue [a column of the matrix P(I); (Figure 1B)], we used Z-score value to measure the interacting conservation between this residue and moieties. The standard deviation (σ) and mean (μ) were derived by random shuffling 1000 times in a profile. The Z-score of the residue j is defined as 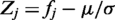 , where fj is the interaction frequency and given as
, where fj is the interaction frequency and given as 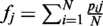 .
.
Spatially, neighbor interacting residues and moieties with statistically significant Z-score ≥1.645 were referred as an anchor candidate. Neighbor anchor candidates, which are spatially overlapped and the same anchor type, were clustered as an anchor and the anchor center is the weighted geometric center of their interacting compound moieties. Here, two anchors were merged if the distance of two anchor centers is <3.5 Å. In each anchor, top three residues with the highest Z-score values were regarded as key residues forming a binding pocket. For each anchor, we identified its moieties of docked compounds according to the moiety library derived from checkmol, and calculated the moiety composition (Figure 1C). These anchors form the site-moiety map (Figure 1D) of the query.
SiMMap can be applied to identify active compounds for structure-based virtual screening. One of weaknesses of virtual screening is likely incomplete understanding of the chemistry involved in ligand binding and the subsequently imprecise scoring algorithms. When a compound highly agrees with the anchors of the site-moiety map, this compound often activates or inhibits the target. The SiMMap server scores a compound by combining predicted binding energy of GEMDOCK and the anchor score between the map and the compound. The SiMMap score, S(i), for a compound i is defined as
| (1) |
where ASa(i) is the anchor score of compound i in the anchor a, n is the number of anchors, E(i) is the docked energy of compound i and M is the atom number of compound i. The anchor score is set to 1 when the compound i agrees the moiety preference of the anchor a. Here, the anchor score and the term M0.5 are useful to reduce the deleterious effects of selecting high-molecular weight compounds (23). Based on SiMMap scores, we can obtain new ranks of query compounds.
INPUT AND OUTPUT
SiMMap is an easy-to-use web server (Figure 2). Users input a protein structure without ligands in PDB format and its docked or co-crystallized compounds in MDL mol, SYBYL mol2 or PDB format (Figure 2A). These docked compounds should be generated by any external docking methods (e.g. DOCK, FlexX, GOLD and GEMDOCK) before users uploaded these compounds. Typically, the SiMMap server yields a site-moiety map within 5 min if the number of query compounds is less than 100. This server provides the graphic visualization of the site-moiety map and anchors elements, including a binding pocket with interacting residues, moiety compositions and structures, numbers of involved compounds, and anchor types (Figure 2B). For each anchor, this server shows docked conformations of compounds and the detailed atomic interactions between pocket residues and moieties (Figure 2C). In addition, SiMMap shows the new rank and compound moiety structures fitting the anchors for each query compound (Figure 2D). SiMMap uses two open source tools for graphic visualization: Jmol (http://www.jmol.org/) for displaying 3D protein and compound structures with anchors and OASA (http://bkchem.zirael.org/oasa_en.html) for visualizing compound structures. The server allows users to download the anchor coordinates in the PDB format; interaction profiles; new ranks and anchor scores of query compounds.
Figure 2.

The SiMMap server analysis results using estrogen receptor (ER) and 1000 docked compounds as the query. (A) The user interface for uploading target protein structure and docked compounds. (B) The site-moiety map has one hydrogen-bonding and three van der Waals anchors for ER. Each anchor contains the moiety structures and composition, anchor type, and key residues in the binding pocket. (C) The details of moiety structures and residue–moiety interactions in the H1 anchor. (D) The SiMMap scores, ranks and the relationships between anchors and moieties of query compounds.
EXAMPLE ANALYSIS
Thymidine kinase
The SiMMap server inferred the site-moiety map of TK. This map consisted of four anchors [i.e. E1, H1, H2 and V1 (Figure 1D)] and the moiety composition and conserved interacting residues of each anchor (Figure 1C). The E1 anchor possesses a binding pocket with residue R222, and three moiety types [i.e. sulfuric acid monoester (40%), carboxylic group (35%) and phosphoric acid monoester (25%)] derived from 57 compounds. The E1 includes the phosphate moiety of ATP and its residue R222 playing a major role to interact with the substrate (24,25). Furthermore, the H1 anchor is a polar pocket with three residues (H58, R222 and E225) that often form hydrogen bonds with polar moiety types among 308 compounds, for example, hydroxyl group (22%), carboxylic acid (8%), ketone (8%), ether (7%) and carboxylic amide (7%). The H2 anchor consists of the residue Q125 and 157 moieties divided into five major moiety types, including hydroxyl group (38%), carboxylic amide (14%), ketone (9%), amine (8%) and sulfuric acid monoester (6%). Finally, the V1 anchor has a binding pocket with residues W88, R163, Y172 and bulky moieties, such as aromatic ring (42%), heterocyclic group (23%), phenol (9%) and oxohetarene (5%).
The preferred moiety types of an anchor are suitable groups interacting to conserved residues of the binding pocket. The moiety preference is able to guide the suggestion of functional group substitutions for lead structures. For example, the moiety preferences of these four anchors (Figure 1D) cover the moiety types derived from 15 TK co-crystal ligands (Supplementary Table S1). In addition, these compounds contain carboxylic amide or amine groups in the H1 anchor. This result shows that the pocket–moiety interactions of these 15 complexes are highly consistent with the pocket–moiety interaction preferences obtained from 1000 docked complexes.
Estrogen receptor
We used estrogen receptor (ER), a therapeutic target for osteoporosis and breast cancer (26), as the second example. Based on 1000 docked compounds and ER, the SiMMap server identifies four anchors (H1, V1, V2 and V3) and provides moiety preferences and compositions in these anchors (Figure 2B). The H1 anchor comprises three residues (E353, L387 and R394) and five main moiety types: hydroxyl group (36%), carboxylic acid (16%), amine (7%), ketone (7%) and sulfuric acid monoester (6%) summarized from 319 compounds. Furthermore, three residues (L346, T347 and L525) and 839 compounds are involved in the V1 anchor, preferring five moiety types [i.e. aromatic ring (49%), heterocyclic group (22%), alkenes (11%), phenol (8%) and oxohetarene (4%)]. The anchor V2 is a hydrophobic pocket containing L346, F404 and L387, and the former two residues are highly conserved (27). These hydrophobic residues interact with aromatic ring (52%), heterocyclic group (23%), phenol (12%), alkenes (5%) and oxohetarene (3%). Finally, aromatic rings (55%), heterocyclic groups (17%), alkenes (11%) and phenols (9%) summarized from 560 compounds often form vdW contacts with the long side chains of M343, M421 and L525 in the anchor V3. The ring groups of antagonists are often stabilized by the side chains of M343, L346, T347, L387, M421 and L525. In this case, most selective estrogen receptor modulators of ER [e.g. EST_01 (raloxifene), EST_06 (LY-326315,) and EST_05 (EM-343)] agree with these four anchors (Figure 2D).
RESULTS
Anchors identified by the SiMMap server often contain key pockets and moieties. To initially validate the anchors for biological mechanisms (e.g. ligand binding and catalysis mechanisms), we selected 15 TK and 22 ER co-crystallized ligands (Figure 3, Supplementary Table S1 and Figure S1). The corresponding moieties of these co-crystallized ligands were highly matched the anchors derived from 1000 docked compounds (10 known active ligands and 990 randomly selected compounds described in ‘Data sets’ section). The site-directed mutagenesis shows that the conserved interacting residues of the anchors are often essential for ligand binding and catalysis mechanisms. For example, the positive-charged residue R222 in E1 interacts with the phosphate group of TK substrates for phosphorylation (28; Figure 3A and B). The site-directed mutagenesis indicates that Q125 in H2 is essential for the substrate specificity (24) and the triple mutant, H58L/M128F/Y172F (H1 and V1), shows the drug resistance to the compound acyclovir (29). In addition, the hydrogen-bonding interaction between E225 and the hydroxyl group of the substrates is able to help stabilize the LID region for the catalytic reaction (29). For ER target, 22 ER co-crystallized ligands contain three consistent moieties that are hydroxyl group and aromatic rings (Supplementary Figure S1). The hydroxyl group forms hydrogen bonds with R394 and E353 in H1, and the aromatic ring yields vdW contacts with L346, L387 and F404 in V2. The other consistent aromatic ring forms vdW contacts with L346, T347 and L525 in V1. These results show that an anchor is often a hot spot and involved in biological functions.
Figure 3.

The relationships between the site-moiety map and 15 co-crystallized ligands of TK. (A) The mapping between four inferred anchors (binding pocket with conserved interacting residues) and these 15 ligands in the active site. (B) The moieties of these 15 ligands in each anchor. (C) The moiety compositions of 1000 docked compounds (SiMMap) and these 15 ligands.
To provide initial validation of the SiMMap server for virtual screening, we selected TK, ER and ERA with 1000 compounds as test sets. First, we compared the accuracies of SiMMap with those of GEMDOCK on these three targets based on true positive rates (Supplementary Figure S2). SiMMap, combining anchor scores and docking energies (Equation 1), outperforms GEMDOCK on these cases. We then compared SiMMap with other three programs (DOCK, FlexX and GOLD) on TK and ER sets. All approaches were tested using the same proteins and compound sets (Supplementary Table S2). When the positive rate was 90%, the false positive rates were 6.8% (SiMMap), 25.5% (DOCK), 13.3% (FlexX) and 9.1% (GOLD) for TK and were 1.1% (SiMMap), 17.4% (DOCK), 70.9% (FlexX) and 8.3% (GOLD) for ER.
The compound, which agrees with anchors of the site-moiety map, is often able to activate or inhibit the target protein (Supplementary Tables S1, S3 and S4). In addition, the anchor score [i.e. AS(i) defined in Equation 1] of SiMMap can be used to reduce the ill-effect of the energy-based scoring methods which are often biased toward both the selection of high molecular weight compounds and charged polar compounds (14,15). For example, according to the SiMMap scores (Equation 1), the ranks of MFCD0005750 (adenylic acid), MFCD0005753 (deoxyadenylic acid) and MFCD0005763 (3′-guanylic acid) are 1, 3 and 9, respectively. These three compounds are thymidine analogs and agree with the four anchors of TK (Figure 1 and Supplementary Table S3). For the top ranks of ER, MFCD0002206 (masoprocol) and MFCD00012748 were also the analogs of the active compounds (Supplementary Table S4). The anchor score of SiMMap was helpful to reduce the highly polar compounds (e.g. MFCD00011393 and MFCD00003569 in TK; MFCD00004690 and MFCD00013089 in ER) whose anchor scores are low. The anchor score of SiMMap can easily combine with other energy-based scoring functions.
CONCLUSION
This work demonstrates the utility and feasibility of the SiMMap server for statistically inferring the site-moiety map describing the relationship between the moiety preferences and physico-chemical properties of the binding site. Our experimental results show that the site-moiety map is useful to reflect biological functions and identify active compounds from thousands of compounds. In addition, the site-moiety map can guide to assemble potential leads by optimal steric, hydrogen-bonding, and electronic moieties. We believe that the SiMMap serve is able to provide the biological insights of protein–ligand binding models, enrich the screening accuracy, and guide the processes of lead optimization.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Council (to J.-M.Y.); ATU plan by MOE (in part).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
Authors are grateful to both the hardware and software supports of the Structural Bioinformatics Core Facility at National Chiao Tung University.
REFERENCES
- 1.Hajduk PJ, Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat. Rev. Drug. Discov. 2007;6:211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 2.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 3.Lyne PD. Structure-based virtual screening: an overview. Drug Discov. Today. 2002;7:1047–1055. doi: 10.1016/s1359-6446(02)02483-2. [DOI] [PubMed] [Google Scholar]
- 4.Tanrikulu Y, Schneider G. Pseudoreceptor models in drug design: bridging ligand- and receptor-based virtual screening. Nat. Rev. Drug Discov. 2008;7:667–677. doi: 10.1038/nrd2615. [DOI] [PubMed] [Google Scholar]
- 5.Ewing TJ, Makino S, Skillman AG, Kuntz ID. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001;15:411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- 6.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 7.Kramer B, Rarey M, Lengauer T. Evaluation of the FLEXX incremental construction algorithm for protein-ligand docking. Proteins. 1999;37:228–241. doi: 10.1002/(sici)1097-0134(19991101)37:2<228::aid-prot8>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 8.Yang JM, Chen CC. GEMDOCK: a generic evolutionary method for molecular docking. Proteins. 2004;55:288–304. doi: 10.1002/prot.20035. [DOI] [PubMed] [Google Scholar]
- 9.An J, Lee DC, Law AH, Yang CL, Poon LL, Lau AS, Jones SJ. A novel small-molecule inhibitor of the avian influenza H5N1 virus determined through computational screening against the neuraminidase. J. Med. Chem. 2009;52:2667–2672. doi: 10.1021/jm800455g. [DOI] [PubMed] [Google Scholar]
- 10.Hung HC, Tseng CP, Yang JM, Ju YW, Tseng SN, Chen YF, Chao YS, Hsieh HP, Shih SR, Hsu JT. Aurintricarboxylic acid inhibits influenza virus neuraminidase. Antiviral Res. 2009;81:123–131. doi: 10.1016/j.antiviral.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Powers RA, Morandi F, Shoichet BK. Structure-based discovery of a novel, noncovalent inhibitor of AmpC beta-lactamase. Structure. 2002;10:1013–1023. doi: 10.1016/s0969-2126(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 12.Schapira M, Raaka BM, Das S, Fan L, Totrov M, Zhou Z, Wilson SR, Abagyan R, Samuels HH. Discovery of diverse thyroid hormone receptor antagonists by high-throughput docking. Proc. Natl Acad. Sci. USA. 2003;100:7354–7359. doi: 10.1073/pnas.1131854100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang JM, Chen YF, Tu YY, Yen KR, Yang YL. Combinatorial computational approaches to identify tetracycline derivatives as flavivirus inhibitors. PLoS ONE. 2007;2:e428. doi: 10.1371/journal.pone.0000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang JM, Shen TW. A pharmacophore-based evolutionary approach for screening selective estrogen receptor modulators. Proteins. 2005;59:205–220. doi: 10.1002/prot.20387. [DOI] [PubMed] [Google Scholar]
- 15.Pan Y, Huang N, Cho S, MacKerell AD., Jr Consideration of molecular weight during compound selection in virtual target-based database screening. J. Chem. Inform. Comp. Sci. 2003;43:267–272. doi: 10.1021/ci020055f. [DOI] [PubMed] [Google Scholar]
- 16.Tafi A, Bernardini C, Botta M, Corelli F, Andreini M, Martinelli A, Ortore G, Baraldi PG, Fruttarolo F, Borea PA, et al. Pharmacophore based receptor modeling: the case of adenosine A3 receptor antagonists. An approach to the optimization of protein models. J. Med. Chem. 2006;49:4085–4097. doi: 10.1021/jm051112+. [DOI] [PubMed] [Google Scholar]
- 17.Champness JN, Bennett MS, Wien F, Visse R, Summers WC, Herdewijn P, de Clerq E, Ostrowski T, Jarvest RL, Sanderson MR. Exploring the active site of herpes simplex virus type-1 thymidine kinase by X-ray crystallography of complexes with aciclovir and other ligands. Proteins. 1998;32:350–361. doi: 10.1002/(sici)1097-0134(19980815)32:3<350::aid-prot10>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 18.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 19.Warnmark A, Treuter E, Gustafsson JA, Hubbard RE, Brzozowski AM, Pike AC. Interaction of transcriptional intermediary factor 2 nuclear receptor box peptides with the coactivator binding site of estrogen receptor alpha. J. Biol. Chem. 2002;277:21862–21868. doi: 10.1074/jbc.M200764200. [DOI] [PubMed] [Google Scholar]
- 20.Bissantz C, Folkers G, Rognan D. Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. J. Med. Chem. 2000;43:4759–4767. doi: 10.1021/jm001044l. [DOI] [PubMed] [Google Scholar]
- 21.Yang M-C, Guan H-H, Yang J-M, Ko C-N, Liu M-Y, Lin Y-H, Chen C-J, Mao SJT. Rational design for crystallization of beta-lactoglobulin and vitamin D-3 complex: revealing a secondary binding site. Crystal Growth Design. 2008;8:4268–4276. [Google Scholar]
- 22.Chin KH, Lee YC, Tu ZL, Chen CH, Tseng YH, Yang JM, Ryan RP, McCarthy Y, Dow JM, Wang AH, et al. The cAMP receptor-like protein CLP is a novel c-di-GMP receptor linking cell-cell signaling to virulence gene expression in Xanthomonas campestris. J. Mol. Biol. 2010;396:646–662. doi: 10.1016/j.jmb.2009.11.076. [DOI] [PubMed] [Google Scholar]
- 23.Yang JM, Chen YF, Shen TW, Kristal BS, Hsu DF. Consensus scoring criteria for improving enrichment in virtual screening. J. Chem. Inf. Model. 2005;45:1134–1146. doi: 10.1021/ci050034w. [DOI] [PubMed] [Google Scholar]
- 24.Kussmann-Gerber S, Kuonen O, Folkers G, Pilger BD, Scapozza L. Drug resistance of herpes simplex virus type 1–structural considerations at the molecular level of the thymidine kinase. Eur. J. Biochem. 1998;255:472–481. doi: 10.1046/j.1432-1327.1998.2550472.x. [DOI] [PubMed] [Google Scholar]
- 25.Wild K, Bohner T, Folkers G, Schulz GE. The structures of thymidine kinase from herpes simplex virus type 1 in complex with substrates and a substrate analogue. Protein Sci. 1997;6:2097–2106. doi: 10.1002/pro.5560061005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou HB, Sheng S, Compton DR, Kim Y, Joachimiak A, Sharma S, Carlson KE, Katzenellenbogen BS, Nettles KW, Greene GL, et al. Structure-guided optimization of estrogen receptor binding affinity and antagonist potency of pyrazolopyrimidines with basic side chains. J. Med. Chem. 2007;50:399–403. doi: 10.1021/jm061035y. [DOI] [PubMed] [Google Scholar]
- 27.Maeda M. The conserved residues of the ligand-binding domains of steroid receptors are located in the core of the molecules. J. Mol. Graph Model. 2001;19:543–551, 601-546. doi: 10.1016/s1093-3263(01)00087-0. [DOI] [PubMed] [Google Scholar]
- 28.Evans JS, Lock KP, Levine BA, Champness JN, Sanderson MR, Summers WC, McLeish PJ, Buchan A. Herpesviral thymidine kinases: laxity and resistance by design. J. Gen. Virol. 1998;79(Pt 9):2083–2092. doi: 10.1099/0022-1317-79-9-2083. [DOI] [PubMed] [Google Scholar]
- 29.Pilger BD, Perozzo R, Alber F, Wurth C, Folkers G, Scapozza L. Substrate diversity of herpes simplex virus thymidine kinase. Impact Of the kinematics of the enzyme. J. Biol. Chem. 1999;274:31967–31973. doi: 10.1074/jbc.274.45.31967. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.