

Abstract
The capabilities and limitations of the Becke-3–Lee–Yang–Parr (B3LYP) density functional theory (DFT) for modeling proton coupled electron transfer (PCET) in the mixed-valence oxomanganese complex 1 [(bpy)2MnIII(μ-O)2MnIV(bpy)2]3+ (bpy = 2,2'-bipyridyl) are analyzed. Complex 1 serves as a prototypical synthetic model for studies of redox processes analogous to those responsible for water oxidation in the oxygen-evolving complex (OEC) of photosystem II (PSII). DFT B3LYP free energy calculations of redox potentials and pKa's are obtained according to the thermodynamic cycle formalism applied in conjunction with a continuum solvation model. We find that the pKa's of the oxo-ligands depend strongly on the oxidation states of the complex, changing by approximately 10 pH units (i.e., from pH~2 to pH~12) upon III,IV→III,III reduction of complex 1. These computational results are consistent with the experimental pKa's determined by solution magnetic susceptibility and near-IR spectroscopy as well as with the pH dependence of the redox potential reported by cyclic voltammogram measurements, suggesting that the III,IV→III,III reduction of complex 1 is coupled to protonation of the di-μ-oxo bridge as follows: [(bpy)2MnIII(μ-O)2 MnIV(bpy)2]3++H++e−→[(bpy)2MnIII(μ-O)(μ-OH)MnIII(bpy)2]3+. It is thus natural to expect that analogous redox processes might strongly modulate the pKa's of oxo and hydroxo/water ligands in the OEC of PSII, leading to deprotonation of the OEC upon oxidation state transitions.
I. Introduction
Understanding the thermodynamics of proton coupled electron transfer (PCET) in oxomanganese complexes is essential for elucidating the mechanism of oxygen evolution by water oxidation, as catalyzed by the oxygen-evolving complex (OEC) of photosystem II (PSII).1–7 The resulting insight on PCET is also necessary for the rational design of artificial photosynthetic systems.8–11 This paper addresses the PCET mechanism in the mixed-valence oxomanganese dimer (1) [(bpy)2MnIII(μ-O)2MnIV(bpy)2]3+ (bpy = 2,2'-bipyridyl), shown in Figure 1, as computationally characterized at the density functional theory (DFT) level with the Becke-3–Lee–Yang–Parr (B3LYP) hybrid density functional.12,13
Figure 1.
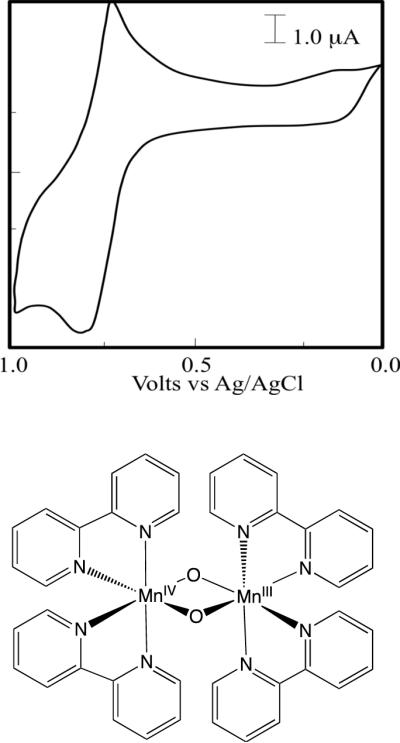
Cyclic voltammogram (top) for a 1 mM solution of complex 1 (bottom) in phosphate buffer at pH 3.78 as reported in Ref. [18].
Several oxomanganese complexes have been suggested as biomimetic models of the OEC of PSII,8,14–19 including the mixed-valence oxomanganese dimer 1 originally synthesized by Nyholm and Turco20 and characterized by X-ray diffraction spectroscopy by Plaksin et at.21 In addition, Mayer et al22 have studied an analogous complex with the 2,2'-bipyridyl ligand substituted by 1,10-phenanthroline. The cyclic voltammogram of 1 includes a reversible one-electron anodic couple at E1/2=1.26 V (vs Ag/AgCl), assigned to the oxidation of the III,IV complex to the IV,IV state.17,23 In addition, an irreversible one-electron cathodic wave with E1/2=0.77 V at pH=3.78 (Fig. 1, top panel)18 is thought to result from reduction of the mixed-valence III,IV complex to the III,III state. Furthermore, the Pourbaix diagram shows a linear dependence of E1/2 with pH in the range pH=3–9, with a ~59 mV/pH slope consistent with the one-electron one-proton couple:18
| (1) |
The availability of electrochemical and spectroscopic data makes complex 1 ideally suited to investigate the capabilities and limitations of the DFT B3LYP level of theory as applied to studies of PCET in oxomanganese complexes. These studies complement our earlier work where we assessed the DFT B3LYP method as applied to the characterization of structural, electronic and magnetic properties of synthetic oxomanganese complexes.8 Our previous studies included Mn dimers, trimers and tetramers where the metal centers are bridged by oxo ligands as well as models of the OEC of PSII analogous to the `3+1 Mn tetramer' model of the OEC of PSII.8 Here, we extend these earlier studies to analyze the PCET reaction in complex 1. Our investigations are based on free energy calculations of pKa's and redox potentials, according to the thermodynamic cycle formalism in conjunction with a continuum solvation model.24 Gas-phase free energies are first calculated and then their values are corrected to account for solvation effects by using a dielectric continuum model. Such a standard computational procedure is one of the simplest approaches available to study redox and acid-base reactions in solution.25–31
Several studies have explored the capabilities of DFT methods for predictions of redox potentials of transition metal complexes.24–41 Most of these earlier studies investigated functionals based on the generalized gradient approximation (GGA) such as BLYP,42 BP8643 and PBE44 as well as hybrid functionals (e.g., B3LYP12,13) originally developed and parametrized without including transition metal compounds in the reference data set. However, the description of redox potentials of oxomanganese complexes and the regulatory effect of oxidation state transitions on the pKa's of oxo-ligands bridging the Mn centers remain to be investigated. Exploring the capabilities and limitations of these methods is crucial to gain insights on PCET mechanisms and to establish the validity of currently available computational tools for the rational design of transition metal catalysts.
The paper is organized as follows. Section II, outlines the computational methods applied for calculations of pKa's and redox potentials. Section III, presents our computational results and direct comparisons with experimental measurements. Concluding remarks and future research directions are outlined in Section IV.
II. Computational Methods
All electronic structure calculations were carried out using the Jaguar suite of electronic structure programs.45 Minimum energy configurations are obtained, as previously reported,8 in broken symmetry (BS) states where the α and β electronic densities are localized on different metal centers. The B3LYP exchange-correlation functional with unrestricted Kohn-Sham wave functions (UB3LYP) yields ground state configurations for the reduced and oxidized forms of complex 1 with antiferromagnetically coupled high-spin manganese centers. Minimum energy configurations were obtained by using a mixed basis set, including the LACVP basis set to account for a non-relativistic description of electron-core-potentials (ECP's) for the Mn4+ and Mn3+ centers, the 6-31G (d) and 6-31G (2df) basis sets for bridging O2− ions to include polarization functions for μ-oxo species, and the 6-31G basis sets for the rest of the atoms. All optimizations were followed by UB3LYP single point energy calculations based on Dunning's correlation-consistent triple-ζ basis set46–48 cc-pVTZ(−f), including a double set of polarization functions. We have also tested the cc-pVTZ(−f)++ basis set for which excellent agreement between calculated and experimental redox potentials was previously reported for other systems.28 Both basis set gave very comparable results.
Half-cell standard reduction potentials were obtained by computing the Gibbs free energy change ΔGred(aq) due to reduction of 1 in aqueous solution, as follows:
| (2) |
with the Faraday constant F=23.06 kcal mol−1 V−1 and n=1 the number of electrons involved in the redox couple [(bpy)2MnIII(μ-O)2MnIV(bpy)2]3+/[(bpy)2MnIII(μ-O)(μ-OH)MnIII(bpy)2]3+. The values of ΔGred(aq) were computed by using the half-reaction of the Born-Haber cycle, depicted in Figure 2, as follows:
| (3) |
Here, ΔGred(g) = ΔHred(g) − TΔSred(g) is the free energy change due to reduction of 1 in the gas phase, with ΔHred(g) = ΔHEA(DFT) + ΔHZPE + ΔHT. ΔHEA(DFT) is the electron attachment enthalpy, obtained at the DFT level for the complex in the gas phase, while the changes in the zero point energy ΔHZPE and corrections for molecular entropy changes ΔSred(g) were based on vibrational frequency calculations. The solvation free energies of 1 in the oxidized and reduced forms, ΔGsol(III,IV) and ΔGsol(III,III), were computed by using the standard self-consistent reaction field (SCRF) approach,49,50 based on accurate solutions of the Poisson-Boltzmann equation. Calculations were carried out for gas-phase geometries employing the dielectric constant of ε = 80.37 (water) for the solvating continuum medium with a solvent radii of 1.40 Å. The effect of hydrogen bonding with solvent molecules or coordination of buffer (phosphate) ligands to the metal centers is beyond the scope of this first study and will be addressed in a follow up publication. Corrections due to changes in the thermal enthalpy ΔHT were neglected.28 All redox potentials are reported as relative potentials referenced to Sliver Chloride electrode (Ag/AgCl). The Ag/AgCl potential is 0.199 V more positive than that of the standard hydrogen electrode (SHE). Considering that the absolute potential of the SHE has been determined experimentally to be 4.43 eV,51 we have subtracted 4.23 V from the absolute potentials to make direct comparisons to experimental data referenced to the Ag/AgCl.18
Figure 2.

Born-Haber thermodynamic cycle for free energy calculations of redox. potentials.
Our calculations of pKa's were based on the following equation:
| (4) |
where β = (kBT)−1 corresponds to room temperature T=298.15 K and kB is the Boltzmann constant. The free energy change ΔGa(aq) due to deprotonation of a μ-OH bridge for 1 in aqueous solutions was computed by using the half-reaction of the Born-Haber cycle, depicted in Figure 3, as follows:
| (5) |
where ΔGa(g) = ΔHa(g) − T ΔSa(g) is the free energy change due to deprotonation in the gas phase, with an enthalpy change ΔHa(g) = ΔHa(DFT) + ΔHZPE + ΔHT. Here, ΔHa(DFT) is the energy change computed at the DFT level, due to deprotonation of the complex in the gas phase. The solvation free energies associated with the deprotonated and protonated forms of the complex, ΔGsol(μ-O, μ-O) and ΔGsol(μ-O, μ-OH), were also computed according to the SCRF approach,49,50 as described above. We take the solvation free energy of a proton in water solvent to be ΔGsolv(H+) = −260 kcal mol−1, as widely adopted in the literature.51–53
Figure 3.

Born-Haber thermodynamic cycle used for free energy calculations of pKa's.
III. Results
Figure 4 shows the thermodynamic free energy diagram for PCET in complex 1 as obtained from DFT B3LYP/cc-pVTZ(−f) calculations. As described in Sec. II, the redox potentials and pKa's were obtained according to the Haber-Born cycle method, applied in conjunction with a continuum solvation model. Figure 4 shows that the reduced III,III state of complex 1 is expected to be protonated at pH<11.7, with most of its population in the oxo-hydroxo form [(bpy)2MnIII(μ-O)(μ-OH)MnIII(bpy)2]3+ 1red, while the 1 is protonated only at pH<2. In addition, Figure 4 shows that oxidation of 1red is thermodynamically much easier when the complex is deprotonated in the [(bpy)2MnIII(μ-O)2MnIII(bpy)2]3+ state (E0=0.31 V) than when it is protonated in the oxo-hydroxo state (E0=0.88 V).
Figure 4.

Thermodynamic energy diagram of PCET for complex 1 in aqueous solutions at pH=0, as described by DFT B3LYP/cc-pVTZ(−f) free energy calculations of redox potentials and pKa's based on the Haber-Born cycle method applied in conjunction with a continuum solvation model. Formal oxidation numbers are indicated as superscripts in Roman numbers and the spin populations obtained according to the Mulliken population analysis are indicated as subscripts in red.
As shown in Figure 4, the protonated species 1red can be oxidized via two possible pathways: (1) oxidation by a direct ionization process (red), requiring a rather high free energy of 0.88 eV; or (2) oxidation by a concerted removal of an electron from the complex and a proton from the μ -hydroxo bridge (green). The total energy requirement, thus, consists of two parts: 0.69–0.059*pH eV for the deprotonation step, and an extra 0.31 eV for subsequent oxidation of the deprotonated species. In an acidic environment, the oxidation related deprotonation steps are not spontaneous, but driven by the externally applied electric field in a CV experiment. Therefore, the CV peak position accounts for the free energy changes due to both deprotonation and oxidation. Because the electron ionization energy is constant, shifting of the potential for the redox reaction, thus, reflects the linear pH dependence of the associated deprotonation energy.
The results reported in Fig. 4 indicate that the oxidation of 1red to complex 1 is strongly coupled to deprotonation of the μ-OH bridge for a wide range of values of pH (i.e., pH=2.0–11.7). For 1red, the oxidation energy is constant at pH < 2.0 (oxidation takes the red path). It varies linearly at a rate of 59 mV/pH within the range of 2.0 < pH < 11.7 (green path with non-spontaneous deprotonation), as determined by the Nernst equation:
| (6) |
At pH > 11.7, the oxidation energy becomes a constant (the green path dominates with spontaneous deprotonation). Figure 5 shows the Pourbaix diagram, illustrating the pH dependence of E1/2, as computed at the DFT B3LYP/cc-pVTZ(−f) level of theory and directly compared to experimental data.18 The experimentally measured redox potentials for oxidation of 1 under different pH conditions are presented as circles in Figure 5(b). The measured data points in the range of 4 < pH < 9 (the filled circles) exhibit linear pH dependence with a slope of ~59 mV/pH (the blue solid line). This particular value of the slope corresponds exactly with the slope expected for a system that loses one proton for each electron removed. Therefore, this linear relationship indicates a PCET in the range of 4 < pH < 9. The measured potentials at pH = 1 and pH = 14 (hollowed circles) obviously deviate from the above linear relation. At such strong acidic/basic solution environment, it is expected that the pH-independent (non-PCET) process dominates the oxidation of 1, leading to the horizontal lines around pH = 1 and pH = 14 (dashed black lines). Therefore, the crossover points of pH-independent and pH-dependent lines correspond to the pKa's of the redox system.
Figure 5.

Pourbaix diagram for complex 1 in aqueous solutions, obtained (a) from free energy calculations of redox potentials at the DFT B3LYP/cc-pVTZ(−f) level of theory, and (b) experimental data (the circles) from Ref. [18].
The computational construction of the Pourbaix diagram (Figure 5(a)) is based on the ab initio calculation of pKa's and redox potentials, and predict the redox potential of 1 for the entire pH range without relying on any kind of experimental data. According to Figure 4, the redox potential must be constant (0.88 V) at pH < 2.0 (oxidation takes the red path). It must vary linearly at a rate of 59 mV/pH within the range of 2.0 < pH < 11.7 (green path with non-spontaneous deprotonation), and it must be constant (0.31 V) at pH > 11.7 (the green path dominates with spontaneous deprotonation). The first crossover in Figure 5(a) corresponds to the conditions under which the red and green pathways, shown in Figure 4, are energetically identical. Therefore, the pH value of this crossover point provides the pKa value of Mn(III, IV). Analogously, the pH value of the subsequent crossover point provides the pKa value of Mn(III, III).
This comparison shows that there is a semiquantitative agreement between the calculated and experimental values of redox potentials throughout the whole range of pH.54 The estimated errors are approximately ±1 unit of pH and ±60 mV for calculations of pKa's and redox potential, respectively. These results, thus, suggest that the DFT B3LYP/cc-pVTZ(−f) level of theory could provide valuable descriptions of PCET processes in oxomanganese complexes, including other biomimetic catalysts and the OEC of PSII.
The molecular structure of the OEC of PSII has yet to be established.3 Several structural models have been proposed, including the `3+1 Mn tetramer' with oxo-bridged high-valent Mn ions and Ca2+ found to be partially consistent with mechanistic studies of water oxidation and high-resolution spectroscopy.2,7 However, it remains to be explored whether such a model is consistent with the well-known “redox leveling” effect by PCET. Such a regulatory mechanism is thought to avoid the buildup of charge in the cluster by deprotonation of water/hydroxo ligands, after oxidation state transitions, making all redox steps occur over a narrow range of potential during the accumulation of 4 oxidizing equivalents. A similar redox leveling process is observed here for the oxomanganese complex 1 for which the redox potential of the deprotonated state [(bpy)2MnIII(μ-O)2MnIII(bpy)2]3+ (E0 = 0.31 V) is significantly reduced as compared to the redox potential of the protonated state (E0=0.88 V) manifesting such a redox leveling effect in good agreement with experimental data. The reported results thus partially validate the DFT B3LYP/cc-pVTZ(−f) level of theory for systems with common structural features, such as the OEC of PSII, when applied by combining the thermodynamic cycle formalism in conjunction with a continuum solvation model.
IV. Conclusions
We conclude that DFT B3LYP/cc-pVTZ(−f) calculations of redox potentials and pKa's, obtained from standard gas-phase calculations and the subsequent correction for solvation effects by using a continuum solvation model, can provide valuable insights on the nature of PCET in oxomanganese complexes in aqueous environments. The reported computational results of redox potentials and pKa's and the favorable comparisons to experimental data from cyclic voltammogram measurements18 and pKa's determined by solution magnetic susceptibility and near-IR spectroscopy17 demonstrate the capabilities of current DFT techniques as applied to modeling PCET in oxomanganese complexes. Both the regulatory effect of oxidation state transitions on the pKa's of oxo ligands and the effect of deprotonation of hydroxo ligands on the redox potentials of metal centers can be properly modeled at the DFT B3LYP/cc-pVTZ(−f) level. Therefore, it is natural to expect that analogous redox-leveling processes could be modeled at the same level of theory for the OEC of PSII. Such calculations are currently underway in our group in an effort to establish structural and functional models of the OEC through rigorous comparisons to thermodynamic studies of water oxidation in PSII.
Supplementary Material
Acknowledgments
V.S. Batista acknowledges supercomputer time from NERSC and financial support from the grant NIH 1R01-GM-084267-01. G. W. Brudvig acknowledges support from the grant NIH GM32715.
Footnotes
Supporting Information Supporting information is available for this manuscript, including a description of the computational methods and the characterization of structural models in terms of the nuclear coordinates and computed thermodynamic data, and including solvatoin energies, entropies, the spin population analysis, and the effect of oxidation state transition coupled to protonation/deprotonation events on the electrostatic potential atomic charges. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- (1).McEvoy JP, Brudvig GW. Chem. Rev. 2006;106:4455–4483. doi: 10.1021/cr0204294. [DOI] [PubMed] [Google Scholar]
- (2).Sproviero EM, Gascon JA, McEvoy JP, Brudvig GW, Batista VS. J. Am. Chem. Soc. 2008;130:3428–3442. doi: 10.1021/ja076130q. [DOI] [PubMed] [Google Scholar]
- (3).Sproviero EM, Gascon JA, McEvoy JP, Brudvig GW, Batista VS. Coord. Chem. Rev. 2008;252:395–415. doi: 10.1016/j.ccr.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Sproviero EM, Gascon JA, McEvoy JP, Brudvig GW, Batista VS. J. Chem. Theory Comput. 2006;2:1119–1134. doi: 10.1021/ct060018l. [DOI] [PubMed] [Google Scholar]
- (5).Sproviero EM, Gascon JA, McEvoy JP, Brudvig GW, Batista VS. Curr. Opin. Struct. Biol. 2007;17:173–180. doi: 10.1016/j.sbi.2007.03.015. [DOI] [PubMed] [Google Scholar]
- (6).Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- (7).Sproviero EM, Gascon JA, McEvoy JP, Brudvig GW, Batista VS. J. Am. Chem. Soc. 2008;130:6728–6730. doi: 10.1021/ja801979n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sproviero EM, Gascon JA, McEvoy JP, Brudvig GW, Batista VS. J. Inorg. Biochem. 2006;100:786–800. doi: 10.1016/j.jinorgbio.2006.01.017. [DOI] [PubMed] [Google Scholar]
- (9).Li GH, Sproviero EM, Snoeberger RC, Iguchi N, Blakemore JD, Crabtree RH, Brudvig GW, Batista VS. Energy Environ. Sci. 2009;2:230–238. [Google Scholar]
- (10).McNamara WR, Snoeberger RC, Li G, Schleicher JM, Cady CW, Poyatos M, Schmuttenmaer CA, Crabtree RH, Brudvig GW, Batista VS. J. Am. Chem. Soc. 2008;130:14329–14338. doi: 10.1021/ja805498w. [DOI] [PubMed] [Google Scholar]
- (11).Abuabara SG, Cady CW, Baxter JB, Schmuttenmaer CA, Crabtree RH, Brudvig GW, Batista VS. J. Phys. Chem. C. 2007;111:11982–11990. [Google Scholar]
- (12).Becke AD. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- (13).Becke AD. J. Chem. Phys. 1993;98:1372–1377. [Google Scholar]
- (14).Cady CW, Crabtree RH, Brudvig GW. Coord. Chem. Rev. 2008;252:444–455. doi: 10.1016/j.ccr.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Mukhopadhyay S, Mandal SK, Bhaduri S, Armstrong WH. Chem. Rev. 2004;104:3981–4026. doi: 10.1021/cr0206014. [DOI] [PubMed] [Google Scholar]
- (16).Hagen KS, Westmoreland TD, Scott MJ, Armstrong WH. J. Am. Chem. Soc. 1989;111:1907–1909. [Google Scholar]
- (17).Cooper SR, Calvin M. J. Am. Chem. Soc. 1977;99:6623–6630. [Google Scholar]
- (18).Thorp HH, Sarneski JE, Brudvig GW, Crabtree RH. J. Am. Chem. Soc. 1989;111:9249–9250. [Google Scholar]
- (19).Sarneski JE, Thorp HH, Brudvig GW, Crabtree RH, Schulte GK. J. Am. Chem. Soc. 1990;112:7255–7260. [Google Scholar]
- (20).Nyholm RS, Turco A. Chem. Ind. (London) 1960:74–75. [Google Scholar]
- (21).Plaksin PM, Palenik GJ, Stoufer RC, Mathew M. J. Am. Chem. Soc. 1972;94:2121–2122. [Google Scholar]
- (22).Wang K, Mayer JM. J. Am. Chem. Soc. 1997;119:1470–1471. [Google Scholar]
- (23).Morrison MM, Sawyer DT. J. Am. Chem. Soc. 1977;99:257–258. [Google Scholar]
- (24).Li J, Fisher CL, Chen JL, Bashford D, Noodleman L. Inorg. Chem. 1996;35:4694–4702. [Google Scholar]
- (25).Roy LE, Batista ER, Hay PJ. Inorg. Chem. 2008;47:9228–9237. doi: 10.1021/ic800541w. [DOI] [PubMed] [Google Scholar]
- (26).Roy LE, Jakubikova E, Guthrie MG, Batista ER. J. Phys. Chem. A. 2009;113:6745–6750. doi: 10.1021/jp811388w. [DOI] [PubMed] [Google Scholar]
- (27).Wang T, Friesner RA. J. Phys. Chem. C. 2009;113:2553–2561. [Google Scholar]
- (28).Baik MH, Friesner RA. J. Phys. Chem. A. 2002;106:7407–7412. [Google Scholar]
- (29).Moens J, Geerlings P, Roos G. Chem. Eur. J. 2007;13:8174–8184. doi: 10.1002/chem.200601896. [DOI] [PubMed] [Google Scholar]
- (30).Moens J, Jaque P, De Proft F, Geerlings P. J. Phys. Chem. A. 2008;112:6023–6031. doi: 10.1021/jp711652a. [DOI] [PubMed] [Google Scholar]
- (31).Moens J, Roos G, Jaque P, Proft F, Geerlings P. Chem. Eur. J. 2007;13:9331–9343. doi: 10.1002/chem.200700547. [DOI] [PubMed] [Google Scholar]
- (32).Tsai MK, Rochford J, Polyansky DE, Wada T, Tanaka K, Fujita E, Muckerman JT. Inorg. Chem. 2009;48:4372–4383. doi: 10.1021/ic900057y. [DOI] [PubMed] [Google Scholar]
- (33).Cheng TY, Szalda DJ, Hanson JC, Muckerman JT, Bullock RM. Organometallics. 2008;27:3785–3795. [Google Scholar]
- (34).Muckerman JT, Fujita E, Hoff CD, Kubas GJ. J. Phys. Chem. B. 2007;111:6815–6821. doi: 10.1021/jp070153+. [DOI] [PubMed] [Google Scholar]
- (35).Fujita E, Brunschwig BS, Creutz C, Muckerman JT, Sutin N, Szalda D, van Eldik R. Inorg. Chem. 2006;45:1595–1603. doi: 10.1021/ic0515498. [DOI] [PubMed] [Google Scholar]
- (36).Hou H, Muckerman JT, Liu P, Rodriguez JA. J. Phys. Chem. A. 2003;107:9344–9356. [Google Scholar]
- (37).Uudsemaa M, Tamm T. J. Phys. Chem. A. 2003;107:9997–10003. [Google Scholar]
- (38).Yang X, Baik MH. J. Am. Chem. Soc. 2006;128:7476–7485. doi: 10.1021/ja053710j. [DOI] [PubMed] [Google Scholar]
- (39).Ayala R, Sprik M. J. Chem. Theory Comput. 2006;2:1403–1415. doi: 10.1021/ct600169e. [DOI] [PubMed] [Google Scholar]
- (40).Galstyan A, Knapp EW. J. Comput. Chem. 2009;30:203–211. doi: 10.1002/jcc.21029. [DOI] [PubMed] [Google Scholar]
- (41).De Groot MT, Koper MTM. Phys. Chem. Chem. Phys. 2008;10:1023–1031. doi: 10.1039/b714727a. [DOI] [PubMed] [Google Scholar]
- (42).Becke AD. Phys. Rev. A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- (43).Lee CT, Yang WT, Parr RG. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- (44).Perdew JP, Burke K, Ernzerhof M. Phys. Rev. Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- (45).Jaguar. 5.0 ed. Schrodinger, Inc; Portland, OR: 2000. [Google Scholar]
- (46).Dunning TH. J. Chem. Phys. 1989;90:1007–1023. [Google Scholar]
- (47).Kendall RA, Dunning TH, Harrison RJ. J. Chem. Phys. 1992;96:6796–6806. [Google Scholar]
- (48).Woon DE, Dunning TH. J. Chem. Phys. 1993;98:1358–1371. [Google Scholar]
- (49).Rashin AA, Honig B. J. Phys. Chem. 1985;89:5588–5593. [Google Scholar]
- (50).Marten B, Kim K, Cortis C, Friesner RA, Murphy RB, Ringnalda MN, Sitkoff D, Honig B. J. Phys. Chem. 1996;100:11775–11788. [Google Scholar]
- (51).Reiss H, Heller A. J. Phys. Chem. 1985;89:4207–4213. [Google Scholar]
- (52).Jang YH, Sowers LC, Cagin T, Goddard WA. J. Phys. Chem. A. 2001;105:274–280. [Google Scholar]
- (53).Lim C, Bashford D, Karplus M. J. Phys. Chem. 1991;95:5610–5620. [Google Scholar]
- (54).The Value of pKa may change by several pH units, if the buffer coordination is considered, see experiments by Meyer et al in Ref. 22.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.