

Abstract
Glycosylation is an important aspect of epigenetic regulation. Glycosyltransferase is a key enzyme in the biosynthesis of glycans, which glycosylates more than half of all proteins in eukaryotes and is involved in a wide range of biological processes. It has been suggested previously that homooligomerization in glycosyltransferases and other proteins might be crucial for their function. In this study, we explore functional homooligomeric states of glycosyltransferases in various organisms, trace their evolution and perform comparative analyses to find structural features which can mediate or disrupt the formation of different homooligomers. First we make a structure-based classification of the diverse superfamily of glycosyltransferases and confirm that the majority of the structures are indeed clustered into the GT-A or GT-B folds. We find that homooligomeric glycosyltransferases appear to be as ancient as monomeric glycosyltransferases and go back in evolution to the last universal common ancestor (LUCA). Moreover, we show that interface residues have significant bias to be gapped out or unaligned in the monomers implying that they might represent features crucial for oligomer formation. Structural analysis of these features reveals that the vast majority of them represent loops, terminal regions and helices indicating that these secondary structure elements mediate the formation of glycosyltransferases' homooligomers and directly contribute to the specific binding. We also observe relatively short protein regions which disrupt the homodimer interactions although such cases are rare. These results suggest that relatively small structural changes in the non-conserved regions may contribute to the formation of different functional oligomeric states and might be important in regulation of enzyme activity through homooligomerization.
Keywords: Glycosyltransferase, Homodimer, Homooligomerization, Interface, Protein structural evolution
Introduction
Post-translational modifications play an important role in regulating protein function and glycosylation is one of the most complex forms of post-translational modifications making a large contribution to phenotypic diversity. It has been established that more than 50% of proteins in eukaryotes are glycosylated1 and glycans are involved in a wide range of biological processes, such as immune response, cell–cell interaction, cellular regulation, tumor growth, and cell invasion.2-7 Glycosyltransferase (GT) is a key enzyme in the biosynthesis of glycans, which sequentially transfers a monosaccharide unit from a nucleotide sugar donor to acceptor substrates including protein residues. Understanding the function and evolution of glycosyltransferases is crucial in providing the insights into the relationships between glycan diversification, protein glycosylation and the phenotype – the main area of research in glycomics. It has been shown that knockouts of some glycosyltransferases may result in cellular dysfunction and even death.8 Moreover, in bacteria, the majority of GTs are involved in the synthesis of glycolipids, peptidoglycans, and lipopolysaccharides and can be suitable targets for drug development against bacterial pathogens. Much effort has been devoted to the identification of genes encoding glycosyltransferases and the characterization of the structures and functions of these enzymes.9,10 Currently, there are more than 100 glycosyltransferase genes found in humans and according to the CAZy database glycosyltransferases consist of approximately 90 very diverse families,11 and account for 1-2% of protein-coding genes in eukaryotic genomes (the number of genes is correlated with the number of coding genes).12
Many soluble and membrane-bound proteins form homooligomeric complexes in a cell, in fact majority of all proteins from Protein Data Bank are homooligomers. The activity of many proteins such as enzymes, ion channel proteins, receptors, and transcription factors are regulated through homooligomerization. Indeed, it has been suggested that large assemblies consisting of many identical subunits have advantageous regulatory properties as they can undergo sensitive phase transitions.13 Moreover, oligomerization can also provide sites for allosteric regulation, generate new binding sites at dimer interfaces, increase affinity through multivalent binding and enhance the diversity in the formation of regulatory complexes. The regulation of protein activity through the transitions between different oligomeric states has been experimentally demonstrated on several systems.14-17 Phosphorylation of a residue on a dimer interface or interaction with different ligands may destabilize the oligomeric structure and might shift the reaction equilibrium which might be important in regulation of apoptosis and tumor formation. In the case of low affinity interactions between proteins and glycans, it has been shown that they can be compensated through the multivalent interactions realized through homooligomerization.2 In our recent work we showed, for example, that homooligomeric interfaces in proteins from the galectin family are very well conserved among a diverse spectrum of species.18 Such homooligomeric structures allow for precise positioning of glycoligands on galectins and increases their binding affinity.
Several glycosyltransferases are similarly proven to form functional homooligomers, which play important roles in controlling enzyme activity and Golgi localization.19,20 For example, α2,6-sialyltransferase and GM2 synthase form very stable functional homodimers by disulfide bonds,21,22 In addition, a recent study indicated that glycosyltransferases may catalyze consecutive steps on the glycan biosynthesis pathways by interacting with each other and forming larger complexes.23 Indeed, there are some glycosyltransferases functioning as homooligomers while others function as monomers even though they can belong to the same protein fold class. In this paper we explore functional oligomeric states of these enzymes in various organisms and study their evolution. We also perform comparative analyses to find structural features which can mediate or disrupt the formation of biological homooligomers in glycosyltransferases. Detecting such features will help to understand the nature of regulation of enzyme activity through homooligomerization and sets the stage for prediction of functionally important oligomeric states for glycosyltransferases and other proteins.
Results
Identification of structurally conserved elements in glycosyltransferase structures
We obtained 68 non-redundant glycosyltransferase structures of 31 families from the CAZy database. Then we calculated the gapped structural alignment score (GSAS)24 and loop Hausdorff metric (LHM)25 as measures of structural similarity for clustering members of the GT-A and GT-B folds (Figure S1, see Methods). First we confirmed that the majority of the structures were correctly clustered into the GT-A or GT-B folds. Only three families GT42 (sialyltransferase), GT51 (peptidoglycan glycosyltransferases), and GT66 (oligosaccharyltransferase), had no significant similarity to any other families and were excluded from the analysis. We generated another score matrix to assess similarities among members of the GT-B fold at the domain level (Figure S2). The score matrix heat map representation shows similarities in the same domain as well as the similarities between the N-terminal and the C-terminal domains. There are some structures with very low similarity to others, such as GT44 (Toxin B) in GT-A and GT23 (fucosyltransferases) in the GT-B fold. It is not a trivial task to classify members of the extremely diverse GT superfamily but despite low similarity between the members of these folds (12% and 11% average sequence identity for GT-A and GT-B folds respectively) these structures still share the core elements as described below.
Previous studies have pointed out the structural similarities in the diverse glycosyltransferase families.26,27 Here we defined structurally conserved core elements by using explicit structure-structure alignments and deliberately chosen structural similarity metrics. The core structure of the GT-A fold consists of a seven-stranded beta-sheet, flanked by six helices, and a two-stranded beta-sheet (Figure S3). The D×D motif, often interacting with the divalent cation, lies at the junction of the two beta-sheets. The structure of the N-terminal half of the core, including the first five strands, three helices, and the D×D motif, is more conserved than the C-terminal half of the core, in which insertions of secondary structures are frequently observed.
GT-B fold enzymes are composed of two domains that are similar to each other. The core structure of the N-terminal domain consists of a seven-stranded beta-sheet, flanked by six helices (Figure. S3). This domain is involved in acceptor sugar molecule binding and is more variable consistent with the diversity of acceptor molecules. The core structure of the C-terminal domain consists of a six stranded beta-sheet, flanked by five helices, and the consecutive three helices at the C-terminal end. This domain involves the donor molecule binding (usually nucleotide-diphospho-sugar) and is more conserved consistent with the conserved nature of the substrates. An additional strand is observed only in the GT72 family, indicating the C-terminal domain is more conserved than the N-terminal domain at the secondary structure level.
Figure 1 presents the structural similarity dendrogram trees for the GT-A and GT-B folds which should reflect the major course of evolution of glycosyltransferases. They were constructed based on the GSAS similarity matrix (trees constructed based on the loop similarity metric can be found in Figure S4). As can be seen from these figures, in the vast majority of the cases the members from the same CAZy family are clustered together with the exception of the poorly conserved GT4 family. Glycosyltransferase families can be classified into two groups based on the mechanism of their glycosylation reactions: retaining and inverting. For the former the stereochemistry of the anomeric carbon is retained between the donor substrate and the product, for the latter it is inverted between them. The trees show that enzymes with retaining and inverting of the anomer correspond very well to two phylogenetic groups on the tree (with the exceptions of the GT27 families of the GT-A fold and the GT70, GT63 and GT10 families with the GT-B fold). This indicates that two types of glycosyltransferase reaction mechanisms diverged quite early in the evolution of the GT-A and GT-B folds and go back in evolution to the last universal common ancestor.
Figure 1.

The model phylogenetic tree for the GT-A (a) and GT-B (b) folds. Nodes are labeled with a family identifier, a PDB code, and an organism name. Branches are colored depending on their reaction mechanisms (red – inverting and blue – retaining). Homooligomeric and monomeric states are indicated by black and open circles respectively, and ancestral oligomeric states inferred by parsimony are shown by squares. External nodes with the same color indicate conserved binding. The asterisk corresponds to the exceptional case 2Z86, which contains two domains in the single chain in the GT-A fold.
Evolution of oligomeric states and binding modes in glycosyltransferases
We identified and verified homooligomeric states (monomers, dimers, tetramers, and octamers) for all family members that are classified into the GT-A or GT-B folds using PISA algorithm (Protein Interfaces, Surfaces and Assemblies) and additionally confirmed with IBIS (Inferred Biomolecular Interactions Server) algorithm.28 We mapped oligomeric states and their binding modes on the external nodes of the model phylogenetic trees for folds A and B and inferred ancestral states using maximum parsimony (see Methods). Several observations are evident from these trees. First, the higher order homooligomeric states (dimers, tetramers, and octamers) appear to be as ancient as the monomeric state for GT and go back in evolution to the last universal common ancestor. Indeed as can be seen from Figure S5 majority of glycosyltransferase families include all three major kingdoms of life (additional sequences without structures were purged from CAZy database to assign taxonomic diversity) and monomeric and oligomeric states existed before the family specifications. Moreover, there seem to be multiple gene duplications leading to a large number of paralogs with different GT specificities and homooligomeric states. There are three families in each fold which are consistently annotated as monomers (GT44, GT7, GT27 in GT-A and GT5, GT9 and GT80 in GT-B) while some families seem to function only as homodimers (GT81, GT43 in GT-A and GT35, GT23 in the GT-B fold, respectively). In addition, five families from both folds show mixed oligomeric states which suggests that their activity can be regulated through the transition between different oligomeric states. We also observed strikingly different binding arrangements in various homodimeric glycosyltransferases. There is only one homodimer binding mode from the GT35 family which is well-conserved in a number of very diverse species. Others are conserved among paralogs from the same species (example: binding modes from the GT43 and GT81 families) while the diverse families GT1 and GT4 can be characterized by multiple binding arrangements.
Identifying and characterizing protein features affecting the formation of homooligomers
There are some glycosyltransferases functioning as monomers while others function as homooligomers even though they can belong to the same fold class. This indicates that relatively small differences between structures may enable or disable some enzymes to form a homooligomer. We performed a comparative analysis of monomers and homooligomers to detect these structural differences. We first identified the interface residues of all homooligomers using the PISA algorithm, and then mapped these interfaces to the closest homologs in the monomeric state using structure-structure superpositions (the mapping of monomers to closest homooligomers produced similar results (Table S1). We call those regions “gapped” which are present in homooligomers and absent from the monomers (and vice versa) and “unaligned” those regions which include both gapped and misaligned residues (see Figure 2 for the illustration). We checked whether there is an association between the interaction interface and gapped/unaligned regions, and found that for the overall dataset the interface residues of the homooligomers correspond to both gapped and unaligned regions (Fisher exact test with Bonferroni's correction p-values ≪ 0.01). Table 1 shows a list of the individual homooligomers, their corresponding monomers, and p-values calculated for each case. In 22 out of 30 homooligomers (73%) the interface residues have a significant bias toward unaligned regions. In a smaller but significant fraction of cases (13 out of 30, 43%) the interface residues have a significant bias to be gapped out in the monomers implying that they might represent features crucial for oligomer formation. Interestingly, for the GT-B fold this tendency is more pronounced and 9 out of 16 oligomers (56%) have interface features which are absent from monomers. We found that the presence of oligomer mediating (or disabling) features in a protein does not depend on the level of sequence and structural similarity between monomer and oligomer. This together with the observation that these features are preferentially located on the oligomer-forming interfaces implies that they cannot be attributed merely to the structural differences occurring as a result of evolutionary divergence.
Figure 2.
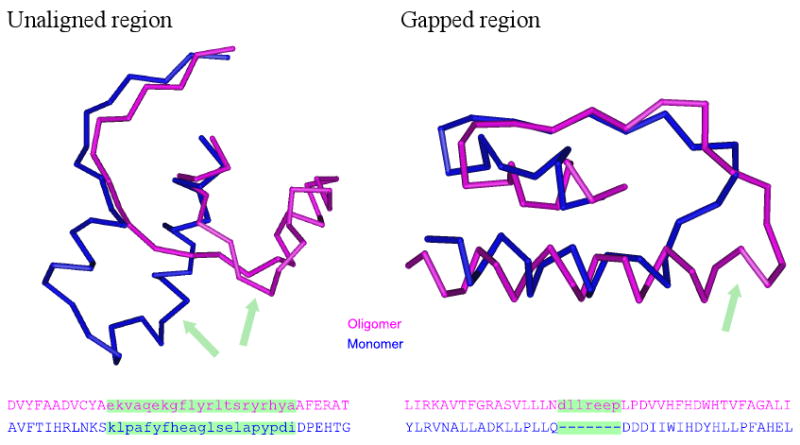
Examples of unaligned and gapped regions. Lower-case characters on the sequence alignments, shown by green, represent unaligned or gapped residues.
Table 1.
Statistical tests of independence between interface residues and unaligned or gapped residues
An asterisk indicates statistical significance (p-value < 0.01) after Bonferroni correction, the “pound sign” # indicates those cases where the monomer contains disabling regions and the symbol ‡ corresponds to those cases where the homooligomer is confirmed by IBIS.
| Fold | Oligomer | Monomer | % ID | Unaligned | Gapped |
|---|---|---|---|---|---|
| GT-A | 3BCV (GT2) | 1H7Q (GT2)# | 21% | 3.3E-04* | 1.4E-03* |
| 1LZJ (GT6)‡ | 1O7Q (GT6) | 45% | 1.8E-10* | 1.1E-02 | |
| 1LZI (GT6)‡ | 1O7Q (GT6) | 45% | 9.5E-10* | 1.0E-02 | |
| 1LL2 (GT8)‡ | 1G9R (GT8)# | 20% | 1.3E-03* | 3.5E-01 | |
| 2GAK (GT14) | 2D7I (GT27) | 7% | 1.0E-02 | 2.2E-08* | |
| 1S4P (GT15)‡ | 1G9R (GT8) | 13% | 7.1E-03* | 4.7E-01 | |
| 2J0B (GT31) | 2D7I (GT27) | 15% | 8.4E-01 | 1.0E-04* | |
| 3CU0 (GT43)‡ | 1H7Q (GT2) | 13% | 5.9E-06* | 4.4E-01 | |
| 1V84 (GT43)‡ | 1H7Q (GT2) | 14% | 1.7E-05* | 9.6E-01 | |
| 2D0J (GT43)‡ | 1XHB (GT27) | 13% | 5.7E-06* | 8.9E-01 | |
| 1OMZ (GT64) | 1XHB (GT27) | 10% | 2.8E-09* | 7.3E-01 | |
| 2BO8 (GT78) | 1XHB (GT27) | 15% | 2.7E-10* | 9.9E-01 | |
| 3E25 (GT81) | 2FFU (GT27) | 15% | 3.3E-09* | 1.1E-04* | |
| 3CKN (GT81) | 2FFU (GT27) | 14% | 1.3E-07* | 1.0E+00 | |
| GT-B | 3D0R (GT1)‡ | 2IYA (GT1) | 22% | 9.5E-01 | 8.6E-01 |
| 1IIR (GT1)‡ | 1RRV (GT1) | 57% | 1.6E-01 | 1.0E+00 | |
| 2P6P (GT1)‡ | 2IYA (GT1) | 21% | 9.7E-01 | 8.7E-01 | |
| 1PN3 (GT1)‡ | 1RRV (GT1) | 57% | 9.6E-01 | 1.0E+00 | |
| 3C48 (GT4)‡ | 2R60 (GT4) | 24% | 4.2E-01 | 1.0E+00 | |
| 2IUY (GT4)‡ | 2R60 (GT4)# | 15% | 8.1E-08* | 3.8E-06* | |
| 2JJM (GT4)‡ | 2GEK (GT4)# | 24% | 1.0E-03* | 2.9E-01 | |
| 1UQT (GT20) | 2BIS (GT5) | 15% | 9.2E-06* | 9.3E-05* | |
| 2HHC (GT23) | 2GT1 (GT9) | 13% | 6.0E-01 | 1.0E+00 | |
| 2DE0 (GT23) | 2GT1 (GT9) | 12% | 2.4E-11* | 2.8E-22* | |
| 3DDS (GT35)‡ | 2BIS (GT5) | 15% | 4.4E-06* | 3.7E-05* | |
| 2GJ4 (GT35)‡ | 2BIS (GT5) | 14% | 1.2E-08* | 1.1E-08* | |
| 1Z8D (GT35)‡ | 2BIS (GT5) | 15% | 1.1E-13* | 9.2E-15* | |
| 1L5W (GT35)‡ | 2BIS (GT5) | 14% | 1.9E-07* | 2.1E-06* | |
| 1YGP (GT35)‡ | 2BIS (GT5) | 15% | 3.3E-14* | 5.9E-12* | |
| 2C4M (GT35)‡ | 2BIS (GT5) | 13% | 9.1E-13* | 8.7E-10* | |
Next we analyzed the structural and sequence characteristics of those unaligned/gapped peptides which affect the homooligomerization. We identified secondary structures for 1534 interface residues in 30 homooligomers. Gapped loops, terminal regions and helices account for approximately 33% of all interface residues and unaligned loops, terminal regions and helices account for 53% of all interface residues, whereas beta strands are in a minority (Figure S6). There is a statistically significant preference for gapped loops, terminal regions and helices to be present on interface regions in comparison with noninterface regions (p-values < 0.01, Table S2), this tendency is especially evident for the GT-B fold. This indicates that these secondary structure elements play an important role in the formation of glycosyltransferase homooligomers. We have also calculated the amino acid composition of unaligned interface regions compared to aligned core regions but did not find any significant amino acid bias (Figure S7).
Structural and functional features mediating the formation of homooligomers
A typical example which illustrates how unaligned loops and helices can contribute to the formation of a homodimer is the glucuronyltransferase (GlcAT) from the GT43 family, which is involved in the biosynthesis of glycosaminoglycans. The C-terminal end of GlcAT, especially a long loop and the last helix, participates in the formation of a homodimer interface (marked by yellow in Figure 3a). Each subunit in a dimer possesses a substrate binding site near the interface, where a functionally important glutamine in one subunit interacts with a substrate of the other subunit.29 On the other hand, the C-terminal part (marked by yellow in Figure 3b) of SpsA protein, the closest monomer to the GlcAT dimer, is tied up by a disulfide bond (blue), cannot participate in any interactions and is located on the opposite side compared to the C-terminal region of the aligned dimer subunit.
Figure 3.
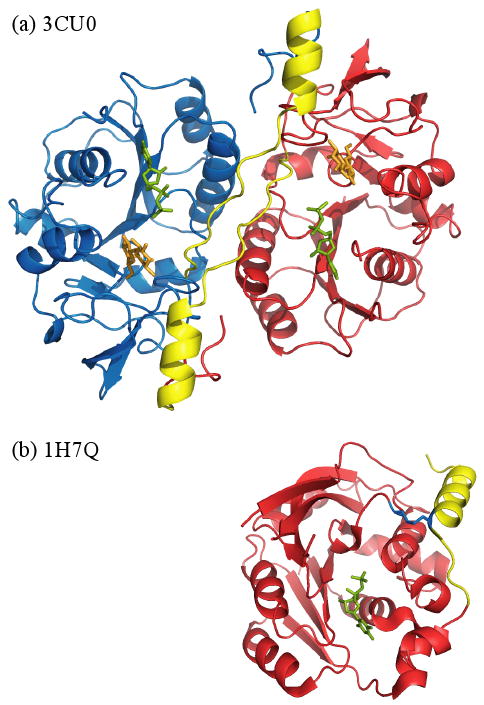
Structural comparison of homodimer glucuronyltransferase (GlcAT), 3CU0 (a) and a monomer SpsA,1H7Q (b). The C-terminus of 3CU0, shown by yellow, is located on the interface region, whereas the C-terminus of 1H7Q, also shown by yellow, is located on the opposite side.
Another example is glycogen phosphorylase from the GT35 family, which is an allosterically regulated enzyme, involved in the glycogen metabolism. The oligomeric states of all six members of this family, including four eukaryotes and two bacteria, are homodimers which is confirmed by previous experiments.30,31 Figure 4 shows a sequence alignment based on the pairwise structural alignment of six glycogen phosphorylases and three glycogen glucosyltransferases in the GT5 family, which were found to be the closest monomers. There are several alpha helices and loops in the glycogen phosphorylases that do not exist in the GT5 enzymes. Moreover, the majority of these structural elements are located on the homooligomeric interfaces. In particular, a long N-terminal tail containing many interface residues is distinctive because usually glycosyltransferases have only a short transmembrane domain at the N-terminal end. Interestingly, this N-terminal region also includes a phosphorylation and ligand binding sites, such as ATP and glucose. It has been shown that conformational changes upon phosphorylation occur at the dimer interface, involve ordering of the flexible N-terminal region and affect the enzymatic activity.32,33
Figure 4.

The multiple alignment of the GT35 and GT5 family enzymes at the N-terminal region. The alignment was constructed by superimposing eight pairwise structure alignments between 3DDS and the other structures. Blue characters represent structurally aligned residues, and red characters correspond to interface residues. Gray cylinders and arrows indicate alpha-helices and beta-strands of the 3DDS structure.
Structural and functional features disrupting the formation of homooligomers
The functional and structural features mentioned above are examples of specific regions that exist only in dimeric forms whereas we also found specific protein regions which are present only in a monomeric form and might disrupt the oligomerization. We tried to detect those residues in a monomer that are absent from a dimer and that are located between residues aligned to the dimer interface. We found four such monomers, they are indicated by the pound sign in Table 1. One of them represents a case of two GT8 structures: one is glycogenin glucosyltransferase from a eukaryote (1LL2), and the other is galactosyltransferase from bacteria (1G9R), which catalyzes a key step in the biosynthesis of lipooligosaccharide structure. Our results show that the eukaryotic enzyme is a homodimer and the bacterial enzyme is a monomer. In glycogenin glucosyltransferase from a eukaryote the interface of the homodimer has a cleft, which allows the formation of a stable complex (Figure 5a), whereas in galactosyltransferase from bacteria the residues corresponding to the interface are covered by other residues, thereby filling the cleft (Figure 5b). The alignment of these two structures reveals that the residues that cover the interface region are two loops not present in the homodimer and located close to the interface region as judged from the monomer-dimer alignment (Figure 5c). This result indicates that these two inserted loops might disrupt the formation of a homodimer and the deletion of the two loops enables the formation of the homodimer.
Figure 5.
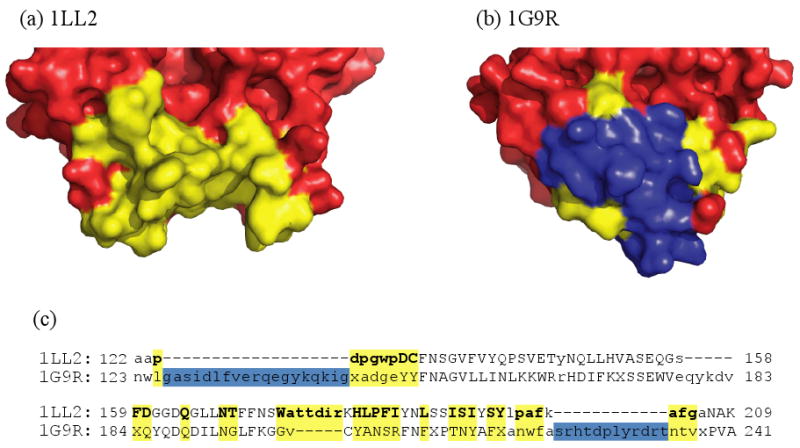
Structural comparison of homodimeric glycogenin glucosyltransferase, 1LL2 (a) and galactosyltransferase, 1G9R (b). Interface residues in 1LL2 and mapped interface residues in 1G9R are shown by yellow; and disabling loops are indicated by blue coloring. (c) The sequence alignment of the interface region is based on the structural superposition of 1LL2 and 1G9R.
Discussion
We analyzed the evolution, functional importance, and structural features which mediate or disable the formation of homooligomers in the glycosyltransferase family. Glycosyltransferases represent an extremely interesting and appropriate example to study these effects since this family is very ancient, encompassing all three kingdoms of life, showing a diversity of biological functions, substrates, binding selectivities, reaction mechanisms, and oligomeric states. The interactions between subunits as well as with other proteins are extremely important for the functionality of glycosyltransferases. Several roles of homooligomerization in the functionality of glycosyltransferases have been discussed previously. For example, binding affinity and selectivity might be regulated through oligomerization or namely through the bond formation between the substrate bound to one subunit in a dimer and the specific residues of another subunit.29 Oligomerization might also be required for stability and thermostability of some GTs (GT81 and GT78 families)34,35 while inter-subunit interactions in glycogenin dimers (GT8 family) are very important in self-glucosylation of glycogenin.36 At the same time glycogen phosphorylase from the GT35 family can undergo conformational changes upon phosphorylation at the dimer interface which involves ordering of the flexible N-terminal region 32 thus affecting and regulating the enzymatic activity.
Our phylogenetic analysis of the glycosyltransferases points to the ancient origin of higher order homooligomeric states which appear to be as ancient as the monomeric state and goes back in evolution as early as the time of divergence of prokaryotes and eukaryotes. Moreover, the examination of the trees for both the GT-A and GT-B folds shows multiple gene duplications leading to a large number of paralogs with different specificities. Although many different glycosyltransferase families form biological dimers, the dimer binding modes are surprisingly diverse among different families implying that paralogs might acquire different binding arrangements throughout evolution. Our recent study on a larger dataset of families exhibiting multiple binding arrangements showed that binding modes are conserved among homologous proteins from the same family sharing 50-70% identity or higher 18. It implies that oligomeric state, binding mode, and features affecting oligomerization can be reliably inferred only from close homologs.
As mentioned earlier, different oligomeric states may provide sites for allosteric regulation, generate new binding sites at dimer interfaces to increase specificity, increase affinity through multivalent binding, and provide the regulation through the transition between active and non-active forms which in turn depend on the protein oligomeric state. To understand such functional mechanisms and predict those features which might mediate/disrupt oligomerization and provide regulatory switches one might want to investigate the sequence and structural features which differentiate between different oligomeric states. In a recent landmark study this question was addressed on a set of fifty domain families.37 Manual inspection of these domains in monomeric and homooligomeric states revealed that for eleven domain families (22%) certain loop regions were responsible for enabling or disabling the oligomeric interfaces. In the current study we performed an automated analysis of different oligomeric forms from 31 families of two folds of glycosyltransferases and made several main observations. First, we have not observed any amino acid changes which would account for the difference between oligomeric states and even very remotely related glycosyltransferases with quite different amino acid usage on the homodimer interface regions have the same oligomeric state. We also have not observed examples of domain swapping which can be an important mechanism to form oligomeric proteins.38 Second, we found that there is a statistical bias for structurally unaligned and to a smaller degree for gapped regions (present in homooligomers, absent in the corresponding monomers) to occur on homodimer interfaces, which points to the importance of these regions in the formation of homooligomers. Finally, our further analysis showed that these unaligned/gapped interface regions for glycosyltransferases predominantly include loops and to a lesser extent alpha-helices. The observation that alpha-helices can affect the formation of specific homooligomeric interfaces is consistent with a recent study which showed that alpha-helices in some proteins from the p53 family might be essential for stabilizing p53 tetramers and can be lost in other species or acquire different functions.39 Although more flexible and variable than the core regions, loops are under strong evolutionary constraints to preserve their properties and might have pronounced functional significance for protein binding. It is evident from the correct GT classification provided by our loop similarity metric and consistent with the previous studies that loops might offer important clues to gauge remote evolutionary relatedness.25,40-42. Consistent with Akiva et al.37 we also show that loops play a crucial role in forming homooligomers and might regulate the binding affinity of homooligomeric interactions. Loops on interaction interfaces can be also used in a negative design strategy to protect proteins from non-specific aggregation.43 Consistent with the negative design strategy we also observed in some cases that inserted regions (with respect to the monomer) disrupt the homodimer interactions (so called “disabling” loops according to Akiva et al terminology) although such cases are rare compared to those regions which mediate the homooligomer formation. This points to the relative importance of enabling compared to disabling regions in the monomer-oligomer evolution of glycosyltransferases.
Materials and Methods
Calculating similarities between glycosyltransferases
First, we derived a list of glycosyltransferase structures from the CAZy database,11 which provides PDB codes of crystallographically determined glycosyltransferases. We then selected representative structures with the highest resolution in cases where the CAZy database lists multiple structures per a single protein. Structures of the GT-A fold with divalent metal ions were given priority since these ions are essential for their catalysis.10 The representative 73 non-redundant glycosyltransferase structures from 31 families are listed in Table S3. Additional information, such as function and reaction types of specific glycosyltransferases, was collected from CAZy, KEGG GLYCAN,44 and Conserved Domain Database (CDD).45
Glycosyltransferases are classified into two different folds: the so-called GT-A and GT-B folds. The GT-A fold consists of one domain while the GT-B fold comprises two similar domains. The diversity between glycosyltransferases is very high. Structure superpositions should provide additional insights into their classification. To achieve this goal we use all pairwise structure-structure alignments between glycosyltransferase representative structures, which are pre-calculated and stored in the PubVAST database.46 We calculate the gapped structural alignment score (GSAS)24 and loop Hausdorff metric (LHM)25 as measures of structural similarity for clustering members of the GT-A and GT-B folds. GSAS together with the LHM scores were shown to perform very well in ranking structurally similar proteins with respect to their homology.47 The GSAS score is defined as following24 (see Supplementary material for the LHM score definition):
Where Nmat is the number of aligned residues, Ngap is the number of gaps, and RMS is the root mean square deviation between the aligned pairs of α-carbon atoms. These two scores produced very similar cluster trees and therefore we will present only the tree obtained using the GSAS score (the LHM based tree is shown in Figure S4). We normalized the scores by dividing them by the maximum GSAS score among all pairwise alignments. To obtain more accurate alignments and similarity scores in the case of the GT-B fold, we separately aligned the N-terminal and C-terminal domains and calculated an average GSAS score over these two domains. If there were other domains present in the chains (SH3, TRP and others) we excluded them from the analysis. The domain boundaries were taken from the Molecular Modeling Database (MMDB).48 The dendrograms were calculated from GSAS distance matrices using the neighbor-joining method49 from PHYLIP version 3.68.50 It should be mentioned that very recently a new classification of glycosyltransferases from both folds has been released in CDD, which overall agrees with our structural classification with the exception of some entries from the diverse GT4 and GT1 families which are not very well resolved on both the CDD tree and our tree.
Annotating oligomeric states and interface residues
We annotated oligomeric states and interfaces of all the representative glycosyltransferases using the PISA algorithm (Protein Interfaces, Surfaces and Assemblies). PISA (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html) is based on chemical thermodynamics and estimates the stability of macromolecular assemblies in protein crystal structures 51. It should be mentioned that PISA can infer macromolecular assemblies and their interfaces using crystallographic symmetries even if such assemblies/interfaces are not present in the PDB asymmetric unit. To confirm the PISA results, we used IBIS (Inferred Biomolecular Interactions Server), a new server (http://www.ncbi.nlm.nih.gov/Structure/ibis/ibis.cgi), which reports interactions observed in experimentally determined protein structural complexes and infers conserved binding sites by inspecting protein complexes formed by close homologs.28 We then examined whether or not interfaces are conserved between homooligomers. Similarly to the conserved binding mode idea,52 if more than 50% of interface residues are structurally superimposed in different homooligomeric complexes, the interface is defined as a conserved binding mode.
Inferring ancestral oligomeric states
Oligomeric states were assigned to internal tree nodes using maximum parsimony. We implemented a modification of Fitch's algorithm,53 usually defined for a binary tree, to allow nodes to have any number of children. The first step of this algorithm applies the following rule recursively, starting from the leaves, to label every internal node: if a given oligomeric state (we distinguish here monomers and homooligomers) is present in more than, less than, or exactly half of labeled children, label the parent “present,” “absent,” or “unknown,” respectively. A second traversal of the tree from root to leaf removes unknown labels by assigning each node the same label as its parent. We break ties at balanced trees, i.e. trees with unknown root, by setting the root to “present”.
Comparison of glycosyltransferases in different oligomeric states
We compared glycosyltransferases in monomeric and homooligomeric states to determine the regions which are structurally different between the monomers and the oligomers. Two types of such regions were considered: “unaligned” and “gapped” regions. Here “gapped region” refers to the set of residues that are present in the oligomer and absent from the monomer (or vice versa), whereas “unaligned region” includes both gapped residues and structurally unaligned residues (Figure 2). First, we selected the most similar structure between monomers and oligomers using the GSAS similarity score. We then detected unaligned and gapped residues between an oligomer and its closest monomer using their VAST structure-structure alignment.46 The Fisher exact test was applied to find whether unaligned/gapped residues have a tendency to be among the interface residues. For each oligomer, for example, we counted the number of unaligned/aligned residues on the interface/non-interface, and generated a 2 × 2 contingency table (Table S4). The one-sided P-values were calculated and adjusted for multiple-testing using Bonferroni's correction.
Supplementary Material
Acknowledgments
We thank Yuri Wolf for careful reading of the manuscript. This work was supported by National Institutes of Health/DHHS (Intramural Research program of the National Library of Medicine). K.H. was supported by a JSPS Research Fellowship from the Japan Society for the Promotion of Science.
Abbreviations
- GT
Glycosyltransferase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Taniguchi N, Hancock W, Lubman DM, Rudd PM. The second golden age of glycomics: from functional glycomics to clinical applications. J Proteome Res. 2009;8:425–6. doi: 10.1021/pr801057j. [DOI] [PubMed] [Google Scholar]
- 2.Collins BE, Paulson JC. Cell surface biology mediated by low affinity multivalent protein-glycan interactions. Curr Opin Chem Biol. 2004;8:617–625. doi: 10.1016/j.cbpa.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 3.Mayer DC, Jiang L, Achur RN, Kakizaki I, Gowda DC, Miller LH. The glycophorin C N-linked glycan is a critical component of the ligand for the Plasmodium falciparum erythrocyte receptor BAEBL. Proc Natl Acad Sci USA. 2006;103:2358–2362. doi: 10.1073/pnas.0510648103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7:255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 5.van Kooyk Y, Rabinovich GA. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat Immunol. 2008;9:593–601. doi: 10.1038/ni.f.203. [DOI] [PubMed] [Google Scholar]
- 6.Jaeken J, Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu Rev Genomics Hum Genet. 2007;8:261–278. doi: 10.1146/annurev.genom.8.080706.092327. [DOI] [PubMed] [Google Scholar]
- 7.Nadanaka S, Kitagawa H. Heparan sulphate biosynthesis and disease. J Biochem. 2008;144:7–14. doi: 10.1093/jb/mvn040. [DOI] [PubMed] [Google Scholar]
- 8.Lowe JB, Marth JD. A genetic approach to Mammalian glycan function. Annu Rev Biochem. 2003;72:643–691. doi: 10.1146/annurev.biochem.72.121801.161809. [DOI] [PubMed] [Google Scholar]
- 9.Narimatsu H. Construction of a human glycogene library and comprehensive functional analysis. Glycoconj J. 2004;21:17–24. doi: 10.1023/B:GLYC.0000043742.99482.01. [DOI] [PubMed] [Google Scholar]
- 10.Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem. 2008;77:521–555. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 11.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hashimoto K, Tokimatsu T, Kawano S, Yoshizawa AC, Okuda S, Goto S, Kanehisa M. Comprehensive analysis of glycosyltransferases in eukaryotic genomes for structural and functional characterization of glycans. Carbohydr Res. 2009;344:881–887. doi: 10.1016/j.carres.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 13.Goodsell DS, Olson AJ. Structural symmetry and protein function. Annu Rev Biophys Biomol Struct. 2000;29:105–53. doi: 10.1146/annurev.biophys.29.1.105. [DOI] [PubMed] [Google Scholar]
- 14.Woodcock JM, Murphy J, Stomski FC, Berndt MC, Lopez AF. The dimeric versus monomeric status of 14-3-3zeta is controlled by phosphorylation of Ser58 at the dimer interface. J Biol Chem. 2003;278:36323–36327. doi: 10.1074/jbc.M304689200. [DOI] [PubMed] [Google Scholar]
- 15.Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005;15:300–8. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 16.Koike R, Kidera A, Ota M. Alteration of oligomeric state and domain architecture is essential for functional transformation between transferase and hydrolase with the same scaffold. Protein Sci. 2009;18:2060–6. doi: 10.1002/pro.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sigalov AB. Signaling chain homooligomerization (SCHOOL) model. Adv Exp Med Biol. 2008;640:121–63. doi: 10.1007/978-0-387-09789-3_12. [DOI] [PubMed] [Google Scholar]
- 18.Dayhoff JE, Shoemaker BA, Bryant SH, Panchenko AR. Evolution of Protein Binding Modes in Homooligomers. J Mol Biol. 2009;395:860–870. doi: 10.1016/j.jmb.2009.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCormick C, Duncan G, Goutsos KT, Tufaro F. The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc Natl Acad Sci USA. 2000;97:668–673. doi: 10.1073/pnas.97.2.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Meer G. What sugar next? Dimerization of sphingolipid glycosyltransferases. Proc Natl Acad Sci USA. 2001;98:1321–1323. doi: 10.1073/pnas.98.4.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Yen TY, Allende ML, Joshi RK, Cai J, Pierce WM, Jaskiewicz E, Darling DS, Macher BA, Young WW., Jr Disulfide bonds of GM2 synthase homodimers. Antiparallel orientation of the catalytic domains. J Biol Chem. 2000;275:41476–41486. doi: 10.1074/jbc.M007480200. [DOI] [PubMed] [Google Scholar]
- 22.Qian R, Chen C, Colley KJ. Location and mechanism of alpha 2,6-sialyltransferase dimer formation. Role of cysteine residues in enzyme dimerization, localization, activity, and processing. J Biol Chem. 2001;276:28641–28649. doi: 10.1074/jbc.M103664200. [DOI] [PubMed] [Google Scholar]
- 23.Noffz C, Keppler-Ross S, Dean N. Hetero-oligomeric interactions between early glycosyltransferases of the dolichol cycle. Glycobiology. 2009;19:472–478. doi: 10.1093/glycob/cwp001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kolodny R, Koehl P, Levitt M. Comprehensive evaluation of protein structure alignment methods: scoring by geometric measures. J Mol Biol. 2005;346:1173–1188. doi: 10.1016/j.jmb.2004.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Panchenko AR, Madej T. Analysis of protein homology by assessing the (dis)similarity in protein loop regions. Proteins. 2004;57:539–547. doi: 10.1002/prot.20237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu Y, Walker S. Remarkable structural similarities between diverse glycosyltransferases. Chem Biol. 2002;9:1287–1296. doi: 10.1016/s1074-5521(02)00295-8. [DOI] [PubMed] [Google Scholar]
- 27.Breton C, Snajdrova L, Jeanneau C, Koca J, Imberty A. Structures and mechanisms of glycosyltransferases. Glycobiology. 2006;16:29R–37R. doi: 10.1093/glycob/cwj016. [DOI] [PubMed] [Google Scholar]
- 28.Shoemaker BA, Zhang D, Thangudu RR, Tyagi M, Fong JH, Marchler-Bauer A, Bryant SH, Madej T, Panchenko AR. Inferred Biomolecular Interaction Server--a web server to analyze and predict protein interacting partners and binding sites. Nucleic Acids Res. 2009;38:D518–D524. doi: 10.1093/nar/gkp842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tone Y, Pedersen LC, Yamamoto T, Izumikawa T, Kitagawa H, Nishihara J, Tamura J, Negishi M, Sugahara K. 2-o-phosphorylation of xylose and 6-o-sulfation of galactose in the protein linkage region of glycosaminoglycans influence the glucuronyltransferase-I activity involved in the linkage region synthesis. J Biol Chem. 2008;283:16801–16807. doi: 10.1074/jbc.M709556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campagnolo M, Campa C, Zorzi RD, Wuerges J, Geremia S. X-ray studies on ternary complexes of maltodextrin phosphorylase. Arch Biochem Biophys. 2008;471:11–19. doi: 10.1016/j.abb.2007.11.023. [DOI] [PubMed] [Google Scholar]
- 31.Pautsch A, Stadler N, Wissdorf O, Langkopf E, Moreth W, Streicher R. Molecular recognition of the protein phosphatase 1 glycogen targeting subunit by glycogen phosphorylase. J Biol Chem. 2008;283:8913–8918. doi: 10.1074/jbc.M706612200. [DOI] [PubMed] [Google Scholar]
- 32.Barford D. Molecular mechanisms for the control of enzymic activity by protein phosphorylation. Biochim Biophys Acta. 1991;1133:55–62. doi: 10.1016/0167-4889(91)90241-o. [DOI] [PubMed] [Google Scholar]
- 33.Rath VL, Ammirati M, LeMotte PK, Fennell KF, Mansour MN, Danley DE, Hynes TR, Schulte GK, Wasilko DJ, Pandit J. Activation of human liver glycogen phosphorylase by alteration of the secondary structure and packing of the catalytic core. Mol Cell. 2000;6:139–148. [PubMed] [Google Scholar]
- 34.Flint J, Taylor E, Yang M, Bolam DN, Tailford LE, Martinez-Fleites C, Dodson EJ, Davis BG, Gilbert HJ, Davies GJ. Structural dissection and high-throughput screening of mannosylglycerate synthase. Nat Struct Mol Biol. 2005;12:608–614. doi: 10.1038/nsmb950. [DOI] [PubMed] [Google Scholar]
- 35.Fulton Z, McAlister A, Wilce MC, Brammananth R, Zaker-Tabrizi L, Perugini MA, Bottomley SP, Coppel RL, Crellin PK, Rossjohn J, Beddoe T. Crystal structure of a UDP-glucose-specific glycosyltransferase from a Mycobacterium species. J Biol Chem. 2008;283:27881–27890. doi: 10.1074/jbc.M801853200. [DOI] [PubMed] [Google Scholar]
- 36.Lin A, Mu J, Yang J, Roach PJ. Self-glucosylation of glycogenin, the initiator of glycogen biosynthesis, involves an inter-subunit reaction. Arch Biochem Biophys. 1999;363:163–170. doi: 10.1006/abbi.1998.1073. [DOI] [PubMed] [Google Scholar]
- 37.Akiva E, Itzhaki Z, Margalit H. Built-in loops allow versatility in domain-domain interactions: lessons from self-interacting domains. Proc Natl Acad Sci USA. 2008;105:13292–13297. doi: 10.1073/pnas.0801207105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bennett MJ, Schlunegger MP, Eisenberg D. 3D domain swapping: a mechanism for oligomer assembly. Protein Sci. 1995;4:2455–68. doi: 10.1002/pro.5560041202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joerger AC, Rajagopalan S, Natan E, Veprintsev DB, Robinson CV, Fersht AR. Structural evolution of p53, p63, and p73: implication for heterotetramer formation. Proc Natl Acad Sci USA. 2009;106:17705–17710. doi: 10.1073/pnas.0905867106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berezovsky IN, Trifonov EN. Loop fold nature of globular proteins. Protein Eng. 2001;14:403–7. doi: 10.1093/protein/14.6.403. [DOI] [PubMed] [Google Scholar]
- 41.Trifonov EN, Berezovsky IN. Evolutionary aspects of protein structure and folding. Curr Opin Struct Biol. 2003;13:110–4. doi: 10.1016/s0959-440x(03)00005-8. [DOI] [PubMed] [Google Scholar]
- 42.Wolf Y, Madej T, Babenko V, Shoemaker B, Panchenko AR. Long-term trends in evolution of indels in protein sequences. BMC Evol Biol. 2007;7:19. doi: 10.1186/1471-2148-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richardson JS, Richardson DC. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci U S A. 2002;99:2754–9. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hashimoto K, Goto S, Kawano S, Aoki-Kinoshita KF, Ueda N, Hamajima M, Kawasaki T, Kanehisa M. KEGG as a glycome informatics resource. Glycobiology. 2006;16:63R–70R. doi: 10.1093/glycob/cwj010. [DOI] [PubMed] [Google Scholar]
- 45.Marchler-Bauer A, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH, Mullokandov M, Song JS, Tasneem A, Thanki N, Yamashita RA, Zhang D, Zhang N, Bryant SH. CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009;37:D205–D210. doi: 10.1093/nar/gkn845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gibrat JF, Madej T, Bryant SH. Surprising similarities in structure comparison. Curr Opin Struct Biol. 1996;6:377–385. doi: 10.1016/s0959-440x(96)80058-3. [DOI] [PubMed] [Google Scholar]
- 47.Madej T, Panchenko AR, Chen J, Bryant SH. Protein homologous cores and loops: important clues to evolutionary relationships between structurally similar proteins. BMC Struct Biol. 2007;7:23. doi: 10.1186/1472-6807-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y, Addess KJ, Chen J, Geer LY, He J, He S, Lu S, Madej T, Marchler-Bauer A, Thiessen PA, Zhang N, Bryant SH. MMDB: annotating protein sequences with Entrez's 3D-structure database. Nucleic Acids Res. 2007;35:D298–D300. doi: 10.1093/nar/gkl952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 50.Felsenstein J. PHYLIP - phylogeny inference package. Cladistics. 1989;5:164–166. [Google Scholar]
- 51.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 52.Shoemaker BA, Panchenko AR, Bryant SH. Finding biologically relevant protein domain interactions: conserved binding mode analysis. Protein Sci. 2006;15:352–361. doi: 10.1110/ps.051760806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fitch WM. Toward defining the course of evolution: minimum change for a specific tree topology. Systematic zoology. 1971;20:406–416. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.