

Abstract
Apoptosis has been shown to be induced by many agents, including the clinically useful Sorafenib and K vitamins (VKs). Since few agents have activity against pancreas cancer cell growth, we evaluated the role of naturally-occurring K vitamins and Sorafenib both independently and together on the growth in culture of pancreas adenocarcinoma cell lines, including PL-5, PANC-1 and MIA PaCa-2. We found that when a K vitamin was combined with Sorafenib, the dose of Sorafenib required for growth inhibition was substantially reduced. Furthermore, growth could be inhibited at doses of each VK plus Sorafenib in combination that were ineffective when used alone. This effect was seen using vitamins K1, K2 and K5. The combination of VK1 plus Sorafenib induced apoptosis, as determined by both FACS and TUNEL staining. Phospho-ERK and Bcl-2 levels were decreased, but not levels of other bcl-2 family members. Cleavage of caspases 3 and 8, PARP and Bid were all induced by this combination. Vitamin K1 plus Sorafenib combination also resulted in elevated levels of activated c-Jun N-terminal kinase (JNK) and its substrates c-Jun and FasL. JNK inhibition partly antagonized the induction of apoptosis. Thus, combination VK1 plus Sorafenib strongly induced growth inhibition and apoptosis in pancreas cancer cells, involving both inhibition of the RAF/MEK/ERK pathway as well as activation of the JNK, c-Jun and FasL apoptotic pathway. Since both agents are available for human use, the combination is attractive for evaluation against pancreas cancer growth in vivo.
Keywords: Sorafenib, vitamin K, cell growth inhibition, apoptosis
Introduction
Human pancreas cancer is one of the deadliest of malignancies, with a death: incidence ratio of approximately 0.85 (1). Diagnosis is usually made at a stage in the disease process that is not amenable to curative surgery. Moreover, chemotherapy, radiation therapy and immunotherapy only marginally affect the progression of the tumor or patient survival. Clearly, new treatments, based on a better understanding of pancreas cancer biology are needed.
Pancreas cancer displays the highest frequency (70–90%) of somatic activating mutations in RAS genes (mainly KRAS) (2–4) amongst human cancers. Ras has been shown to mediate activation of various downstream targets, including the family of the mitogen-activated protein kinase (MAPKs). The RAF/mitogen-activated protein kinase (MAPK) kinase 1/2 (MEK1/2)/extracellular signal-regulated kinase 1/2 (ERK1/2) or RAF/MEK/ERK pathway is frequently deregulated in neoplastic transformation (5–7). Because of the probable importance of the RAF→MEK→ERK pathway in the aberrant behavior of cancer cells, it has been the subject of intense study, in order both to understand its fundamental role in cancer cell biology and as a potential target for therapeutic intervention (8–10).
Sorafenib is a multi-kinase inhibitor that was originally developed as an inhibitor of Raf-1, but it was subsequently shown to inhibit multiple other kinases, including platelet-derived growth factor, vascular endothelial growth factor receptors 1 and 2, c-Kit, and FLT3 (11). Sorafenib shows broad activity against various tumor cell lines in vitro and in xenograft models (12). Anti-tumor effects of Sorafenib in renal cell carcinoma and in hepatoma have been ascribed to anti-angiogenic actions of this agent through inhibition of multiple growth factor receptors (13–15). Preliminary evidence of single agent activity has also been observed in malignant melanoma and hematological malignancies as well as against experimental pancreas cancer (16) and human trials in pancreas cancer have begun (17). As the clinical application of Sorafenib evolves, there is increasing interest in defining the mechanisms underlying its anti-proliferative activity as well as the effects of the agent in combination with other drugs.
Vitamins K (VK) are fat-soluble vitamins that are involved in blood coagulation and bone metabolism. In recent years, their anti-tumor effects have also been examined (18–19). They have been shown to suppress cancer growth and induce apoptosis and differentiation in various cancer cells, including leukemia and hepatocellular carcinoma (HCC) cells. There are several forms of VK: VK1 (phytonadione), which is produced by plants and is used to treat human coagulation disorders; VK2 (menaquinone), which is produced by certain bacteria and also occurs naturally and in the human gut and is used to treat osteoporosis and HCC (20–21); synthetic VK3 (menadione), which is a short chain chemically synthesized form that induces redox cycling and is toxic in humans; and VK5, another short chain synthetic analog with inhibitory actions on both cells and bacteria. The natural VK1 and VK2 are thought to be without toxicity in adult humans. It has been shown that synthetic VK3 can inhibit pancreas cancer cell line growth (22), but there was recent evidence that natural VKs can also do this (19). We reasoned that since both Sorafenib and VKs are available for human use, and since they each independently have been shown to both to induce apoptosis in tumor cells, then the combination might be expected to be better than either agent alone. In the current study, we show that combining Sorafenib with natural K vitamins more effectively inhibits pancreas cancer cell line growth than either agent alone. By therapeutically targeting the MAPK and caspase dependent pathways, these combined agents dramatically induce apoptosis. Furthermore, we show that combined Sorafenib plus vitamin K induces pancreas cancer cell death via activation of the extrinsic apoptotic pathway, involving activation of JNK, c-Jun and FasL, as well as via phospho-ERK inhibition. These results lay the ground work for possible future clinical trials, testing this combination for the treatment of pancreas cancer patients.
Materials and Methods
Cell lines
Human pancreas tumor cell lines (PL5 or CRL-2555, MIA PaCa-2 or CRL-1420 and PANC-1 or CRL-1469) were obtained from American Type Culture Collection (Rockville, MD, ATCC) and cultured in DMEM containing 10% fetal bovine serum (FBS) in 5% CO2 at 37°C. Unless otherwise indicated, cell culture reagents were obtained from Life Technologies, Inc. (Gaithersburg, MD).
Compounds
Sorafenib [N-(3-trifluoromethyl-4-chlorophenyl)-N'-(4-(2-methylcarbamoyl pyridin-4-yl) oxyphenyl) urea] was synthesized at Bayer Corporation (West Haven, CT). Sorafenib was dissolved in 100% DMSO (Sigma, St. Louis, MO) and diluted with MEM to the desired concentration with a final DMSO concentration of 0.1% for in vitro studies. Vitamin K1 was purchased from (Sigma-Aldrich Chemical Co.) and dissolved in 99.9% ethanol at a stock concentration of 50 mM and then diluted to appropriate concentrations with medium in using. U0126 and JNK inhibitor (SP600125) were purchased from Calbiochem (San Diego, CA). Compounds were diluted with DMEM to the desired concentration with final DMSO or ethanol concentration of 0.1% for in vitro studies. DMSO and ethanol were added to cultures at 0.1% (V/V) as a solvent control.
Cell viability
Cells were plated at a concentration of approximately 2 × 104 cells per well in 24-well plates. Twenty-four hours after plating, the medium was replaced with fresh Dulbecco’s Modification of Eagle’s Medium (DMEM) containing vitamin K, Sorafenib or a combination of the two agents at the indicated concentrations. Three days after the treatment, the medium was removed, and the plates were stored at −80 °C until the day of the assay. The cell number was estimated by a DNA fluorometric assay using the fluorochrome Hoechst 33258.
In situ end labeling of fragmented DNA (TUNEL)
Cells were cultured on chamber slides and treated with compounds as described above. After 48 h of treatment, cells were fixed with 10% buffered formalin at room temperature, washed with PBS and air-dried. Fragmented DNA was detected by an in situ end labeling kit (ApopTag kit, Oncor, Gaithersburg, MD) according to the manufacturer's protocol. Briefly, digoxigenin-labeled dUTP was incorporated at the 3’-OH ends of the fragmented DNA by terminal deoxynucleotidyl transferase, the anti-digoxigenin antibody conjugated with peroxidase was then applied, and the peroxidase activity was revealed by 3-amino-9-ethylcarbazol. Nuclei were then counterstained lightly with hematoxylin.
Immunoblot analysis
Pancreas cancer cells were plated in 100 mm tissue culture dish with a density of 5 × 106/dish overnight. The cells were treated in the next morning and harvested in different time. After harvest, the cells were washed twice with cold phosphate-buffered saline (PBS), then lysed in 100 µl lysis buffer (150 mM NaCl; 50 mM Tris-HCl, pH 8.0; 0.1% SDS; 1% Triton X-100; 1 mM orthovanadate; 1 mM phenylmethylsulfonyl fluoride; 10 mg/ml leupeptin; 10 mg/ml aprotinin). Whole cell extracts (20 µg) were resolved on a 10% SDS-PAGE and transferred onto Hybond-PVDF membranes (Amersham, Arlington Heights, IL). Membranes were blocked using Tris-buffered saline with Tween-20 (150 mM NaCl, 10 mM Tris-HCl, pH 8.0, and 0.05% Tween-20) containing 1% BSA for 1 h, then probed with primary antibody for 2 h. After washing four times with Tris-buffered saline with Tween-20 (TBST buffer), the membranes were probed with horseradish peroxidase-conjugated secondary antibody to allow detection of the appropriate bands using enhanced chemiluminescence (Amersham). The antibodies used in these experiments (PARP, Caspase-3, 8, 9, ERK1/2, c-Jun, JNK, FasL, FADD and Actin) from Santa Cruz Biotechnology, Santa Cruz, CA or Cell Signaling, Waltham, MA.
Apoptosis determination
Annexin V-FITC kit (Jingmei Biotech) was used to measure the percentage of apoptosis induced by vitamin k1 and Sorafenib. After treatment for 36h, cells were harvested and washed with PBS at 4 °C and then re-suspended in 100 µL binding buffer (1 × 106 cells/ml) containing 5 µl of Annexin V-FITC and 10 µl of PI. After incubation away from light for 10–15 min at room temperature, stained cells were then analyzed by flow cytometry.
Results
Inhibition of pancreas cancer cell growth by Sorafenib plus vitamin K1
To assess the effect of combining Sorafenib with vitamin K1, 3 pancreas cancer cell lines (PL5, MIA PaCa-2 and PANC-1) were treated with the study agents either individually or in combination, and were then examined by DNA fluorometric assay. Cell viability was measured at 24, 48 and 72 hr after treatment (Fig. 1A). At the concentrations tested, neither Sorafenib nor vitamin K1 caused growth inhibition as a single agents, compared with vehicle control (DMSO or Ethanol), but the combination significantly inhibited cell proliferation compared with the single agent treatments (p<0.01, Two-way ANOVA when compared with vitamin K1 or Sorafenib alone). We also tested whether other K vitamins plus Sorafenib could also inhibit pancreas cancer cell line growth (Fig.1B). Vitamins K2 and K5 were similarly able to add to Sorafenib-mediated cell growth inhibition (p<0.01, with respect to control cells treated with either k or Sorafenib alone).
Figure 1. Sorafenib plus vitamin K inhibits pancreas cancer cell growth.
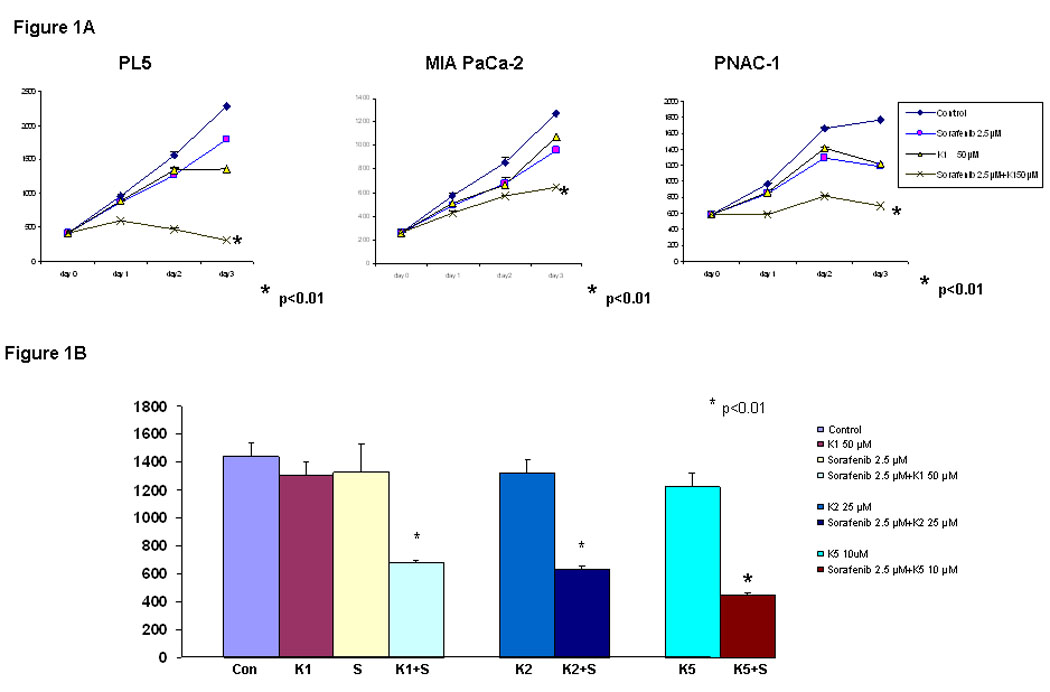
A, PL5, MIA PaCa-2 and PANC-1 cells were treated with K1 50 µM, Sorafenib 2.5 µM or K1 plus Sorafenib and the cell number was estimated by a DNA fluorometric assay. B. PL5 cells were treated separately with 50 µM K1, 25 µM K2, 10 µM K5, 2.5 µM Sorafenib or combination Sorafenib with Ks. Cell number was counted on day 3. Each experiment was run in triplet, repeated three times and expressed mean SD. P value is determined by student’s t test.
Induction of caspase-mediated apoptosis by Sorafenib plus vitamin K1
Since combination Sorafenib plus vitamin K1 caused a significant reduction in cell proliferation, the underlying mechanisms were investigated. First, TUNEL staining of treated cells showed the presence of apoptosis following this combination treatment, compared with either agent alone (Fig.2A). Pre-treatment with ZVAD, a pan-caspase inhibitor, significantly blocked the induced apoptosis, as measured by TUNEL staining. To confirm the induction of apoptosis by this combination, PL5 cells were treated with the agents individually or in combination and examined by Annexin V/propidium iodide staining (Fig.2B). At the concentrations tested, neither Sorafenib nor vitamin K1 elicited significant apoptosis as single agents, but the combination induced apoptosis in 50% of the cells. Pan-inhibition of caspase activity using ZVAD significantly reduced the cell death ratio. These results show that combination Sorafenib plus vitamin K1 caused apoptosis, which was inhibited by a caspase antagonist.
Figure 2. Combination Sorafenib and vitamin K1 induces apoptosis in pancreas cancer cell.
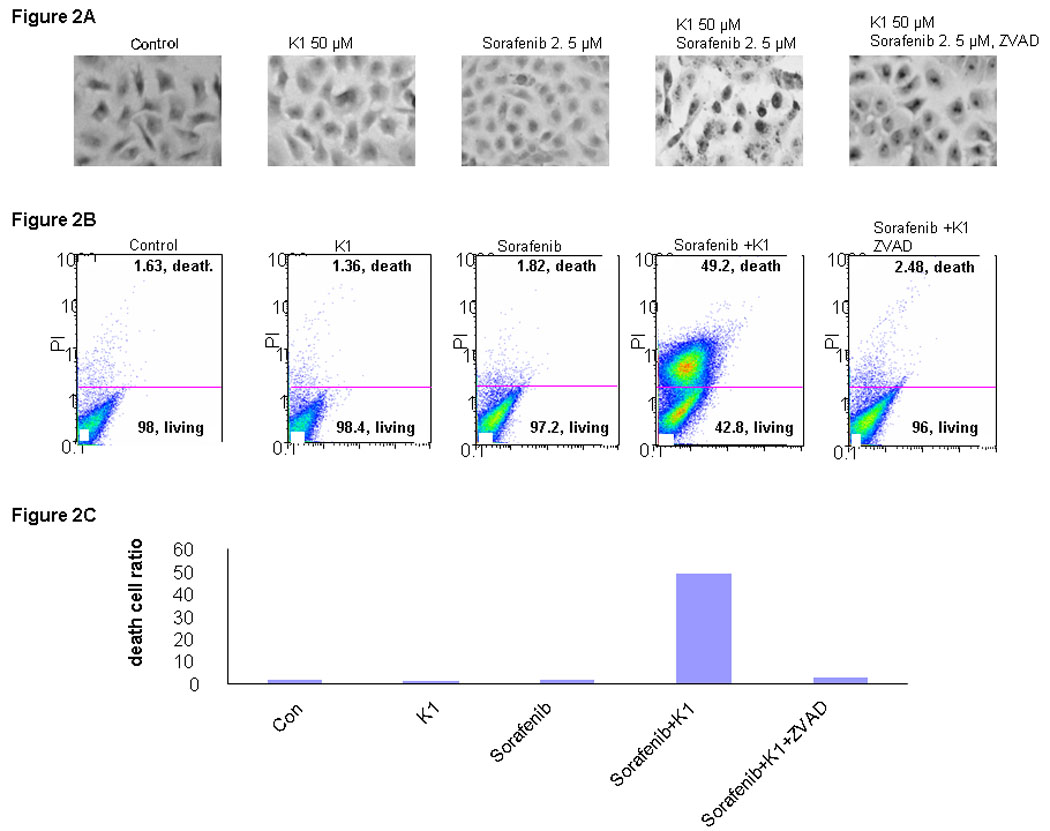
A: TUNEL staining. PL5 cells were treated with vitamin k1 (50 µM), Sorafenib (2.5 µM), combination vitamin k1 (50 µM) and Sorafenib (2.5 µM), or pretreated with casepases inhibitor 2 hours then incubated with vitamin k1 plus Sorafenib. TUNEL staining cells were observed at 40X magnification. B. PL5 cells were treated as the same condition in Fig. A. Floating and adherent cells were harvested at 36 hours and analyzed by flow cytometry. C: Quantitative of death cells in B.
Inhibition of RAF/MEK/ERK signaling and caspase-8 activation by combination Sorafenib plus vitamin K1
RAF kinases are key regulators of the MEK/ERK cascade, and up-regulated signaling through this pathway has an important role in pancreas cancer cell growth (16). Since Sorafenib was synthesized as a RAF inhibitor, we examined the phosphorylation levels of key proteins in the pathway by Western blot in cells treated with various doses of Sorafenib and vitamin K1. Sorafenib or vitamin K1 alone could inhibit ERK phosphorylation, but only at high concentrations (Fig. 3A, B). Neither Sorafenib (2.5 µM) nor vitamin K1 (50µM) alone inhibited MEK or ERK phosphorylation at the lower concentrations that were used in the combination, but the combination of these two agents at these lower doses dramatically reduced both MEK and ERK phosphorylation (Fig. 3C). Total MEK and ERK levels were unchanged. To examine whether the decrease in phospho-MEK and phospho-ERK levels could result in apoptosis, we employed UO126, a specific inhibitor of MEK. We found UO126 alone inhibited p-ERK levels, as expected, and also induced apoptosis, which was associated with caspase-8 cleavage (Fig.3D).
Figure 3. Inhibition of p-ERK increases caspase-8 activation.
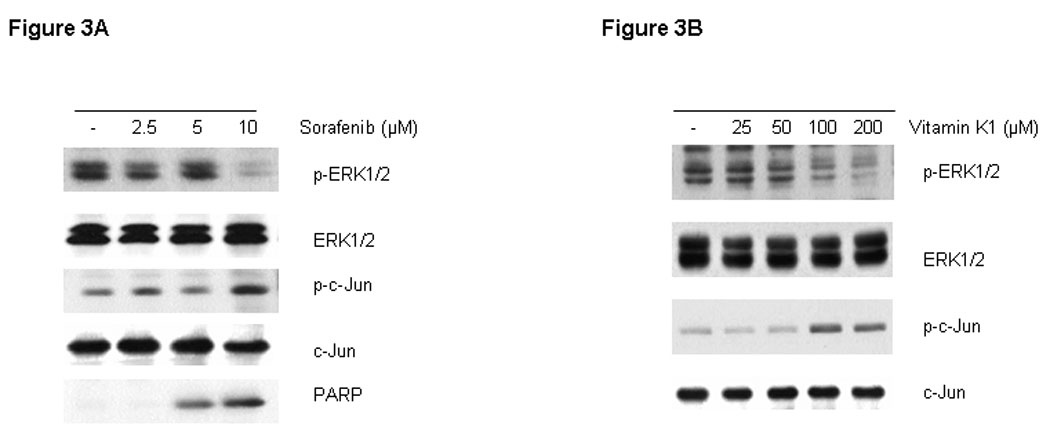
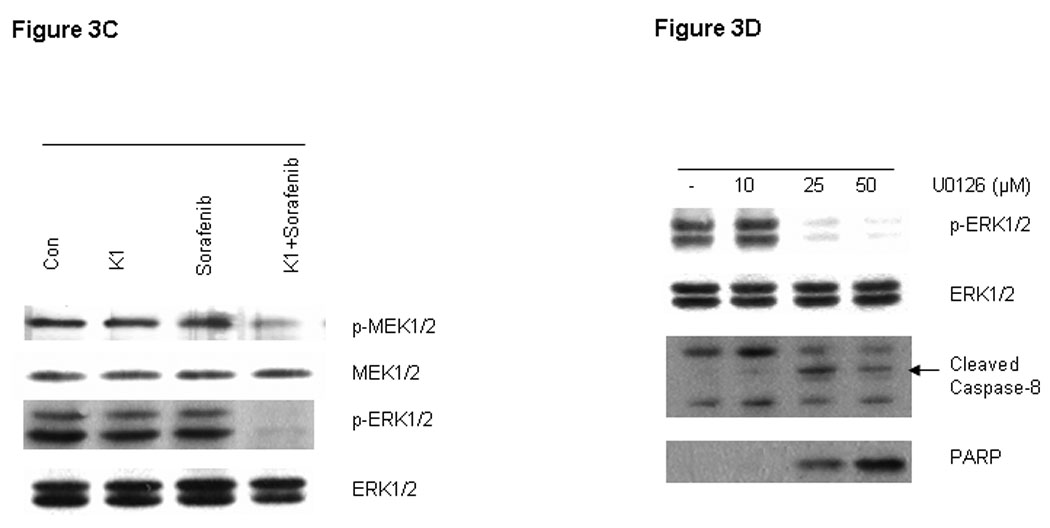
Sorafenib or vitamin k1 inhibits p-ERK in does dependent in PL5 cells (A, B). C. Lower concentration of Vitamin k1 (50 µM) plus Sorafenib (2.5 µM) inhibits RAF/MEK/ERK pathway in PL5 cells. D. PL5 cells were treated with various does of U0126 for 36 hours. Cells were lysed and 20 µg of soluble protein was separated by electrophoresis on a SDS-PAGE gel. Protein phosphorylation was detected by Western blot analysis.
Involvement of the extrinsic pathway in Sorafenib plus vitamin K1 mediated apoptosis
To further examine the processes of cell death induced by this combination, we analyzed cell extracts for expression of biological markers of apoptosis. The combination drug treatment resulted in marked cleavage of pro-caspase-3 and poly (ADP-ribose) polymerase (PARP) induction, whereas the individual agents did not (Fig.4A). In order to distinguish the extrinsic from the intrinsic apoptosis pathways, the upstream caspases of caspase-3 were examined. Treatment with the combination of Sorafenib plus vitamin K1 activated caspase-8 activity and subsequently cleaved BID, but neither pro-caspase-9 activity, nor cytochrome C release could be detected in the same treated samples (Fig.4 B, C). These findings suggest that caspase-8 signaling and the extrinsic pathway were involved in the combination-induced apoptosis. Since Bcl-2 molecule appears to be a possible likely target of the MEK/ERK pathway and the Bcl-2 expression level is known to be up-regulated by ERK (23), we next examined how combined exposure to Sorafenib plus vitamin K1 might affect the expression of Bcl-2 family members. Decreased levels of anti-apoptotic Bcl-2 was observed after exposure to the combined agents, but not after exposure to either Sorafenib or vitamin K1 alone. There was also no discernible change in the expression of other Bcl-2 family members, Bcl-xL, or Bax (Fig. 5A).
Figure 4. Combination vitamin k1 and Sorafenib activates the extrinsic caspase death pathway.
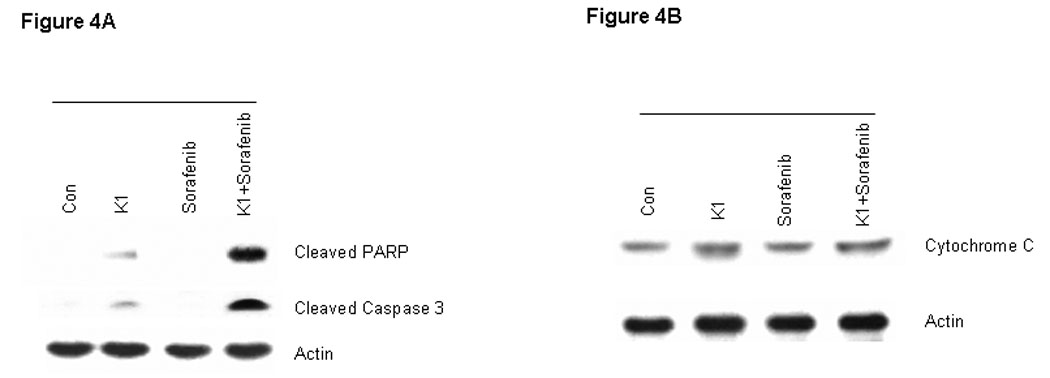
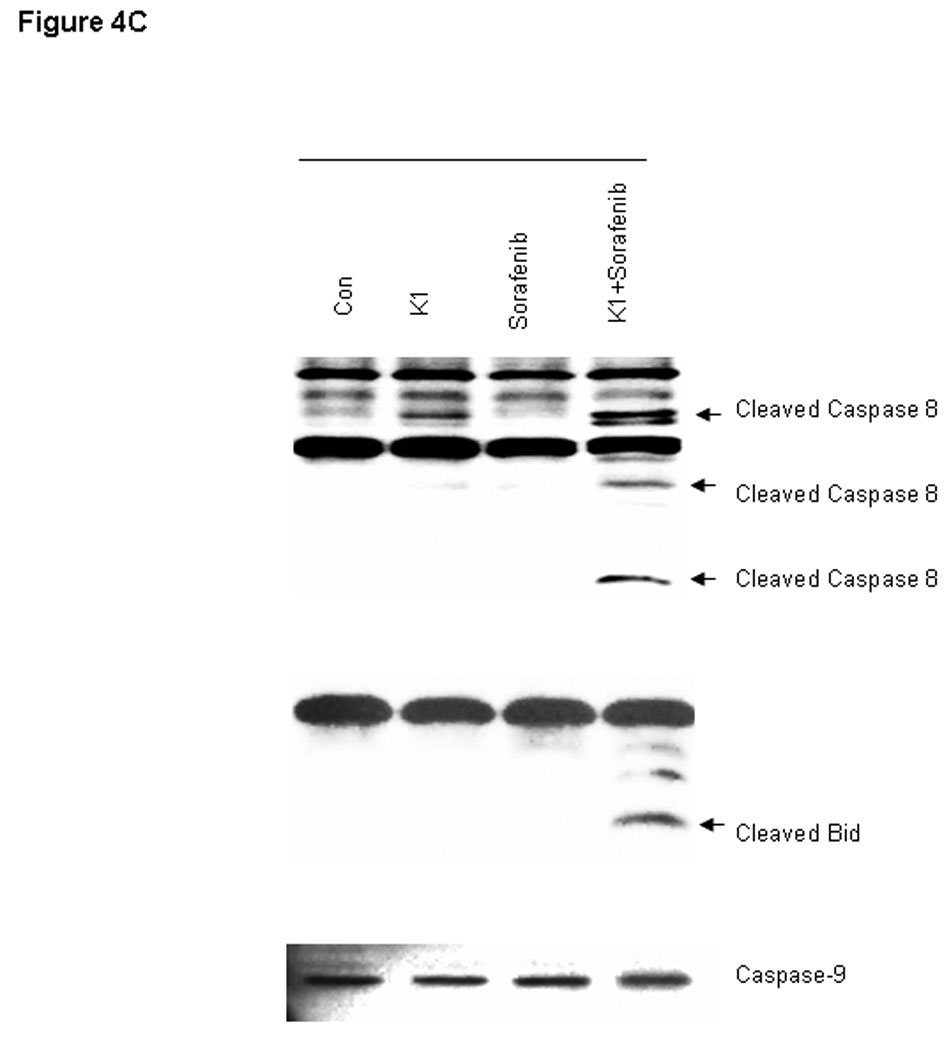
A: Cleaved PARP and caspase-3 can be detected after treatment 36 hours with vitamin k1(50 µM) plus Sorafenib (2.5 µM) in PL5 cells; B: Combination treatment with Sorafenib and vitamin k1 does not affect the cytochrome C release from the mitochondria in PL5 cells. C: Procaspase-8 was activated and its substrate Bid was cleaved after treatment with vitamin k1 and Sorafenib in PL5 cells, but no caspase-9 activity was detected in the same lysates. Cells were lysed and 20 µg of soluble protein was separated by electrophoresis on a SDS-PAGE gel. Protein levels were detected by Western blot analysis. Actin was used as loading control.
Figure 5. Sorafenib plus vitamin k1 modulates the expression of Bcl-2 family and FasL in PL5 cells.
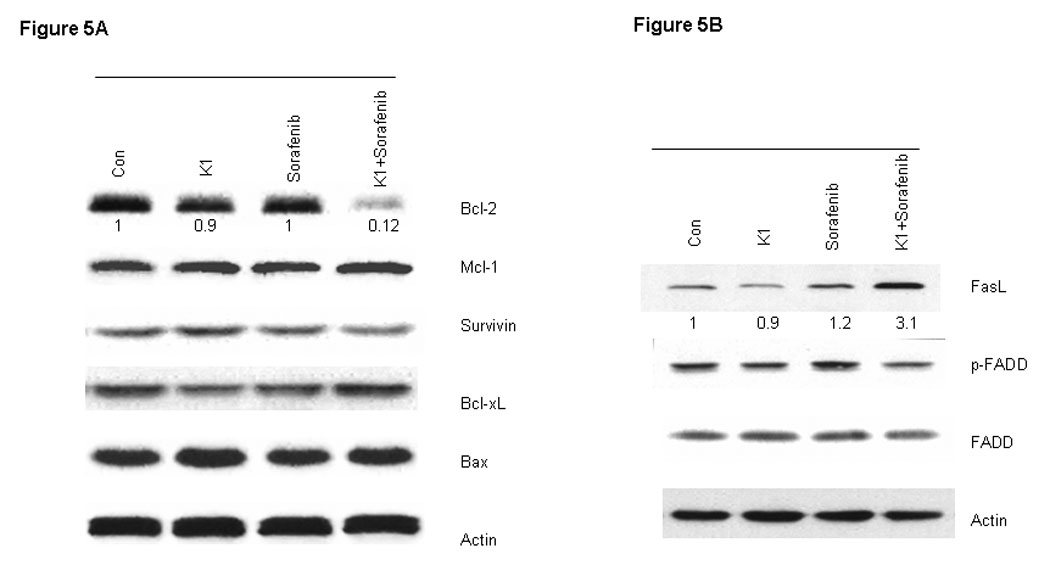
PL5 cells were plated and treated for 36 hours with vehicle, vitamin k1 (50 µM), Sorafenib (2.5 µM), and both vitamin k1 and Sorafenib. Cells were isolated and subjected to SDS-PAGE followed by immunobloting to determine the expression of Bcl-2, Mcl-1, Survivin, Bcl-xL and Bax (A); FasL, FADD (B).
Combination Sorafenib plus vitamin K1 activation of the death receptor (extrinsic) apoptosis pathway
The extrinsic apoptosis pathway involves binding of a ligand (FasL) to one of the tumor necrosis factor family of death receptors, and then activation procaspase-8 to induce apoptosis. To investigate if Fas signaling was involved in combination Sorafenib plus vitamin K1 induced apoptosis, PL5 cells were treated with Sorafenib, vitamin K1 or the combination of the two agents. Following 36 hours of treatment, cells were harvested and the lysates were analyzed in 10% SDS-PAGE gel. As shown in figure in Fig 5B, combination Sorafenib plus vitamin K1 enhanced expression of FasL, but there was little change in FADD levels.
Contribution of JNK to apoptosis induction by Sorafenib plus vitamin K1
Activated JNK can cause cancer cells to undergo apoptosis through phosphorylation and activation of c-Jun, an important member of the AP-1 family of transcription factors (24). We assessed the role of this signal by exposing cells to the Sorafenib plus vitamin K1 combination. Levels of cell phospho-JNK, but not total JNK were found to be elevated following this exposure (Fig.6A). Levels of phospho-c-Jun, a key substrate for phospho-JNK, were also increased in parallel with the elevated phospho-JNK levels, but total JNK and total c-Jun levels were not changed. In order to further to assess the involvement of JNK in the apoptosis induction, PL5 cells were pre-treated with the JNK inhibitor SB600125 and then subjected to Sorafenib plus vitamin K1 treatment. The JNK inhibitor decreased the elevated levels of induced phospho-JNK, as well as the levels of phospho-c-Jun and FasL, evidence of blocked JNK activity (Fig. 6B). Furthermore, the JNK inhibitor also partially antagonized the apoptosis induced by combination Sorafenib plus vitamin K1 (Fig.6C).
Figure 6. JNK and c-Jun activation are involved in vitamin k1 plus Sorafenib induced apoptosis in PL5 cells.
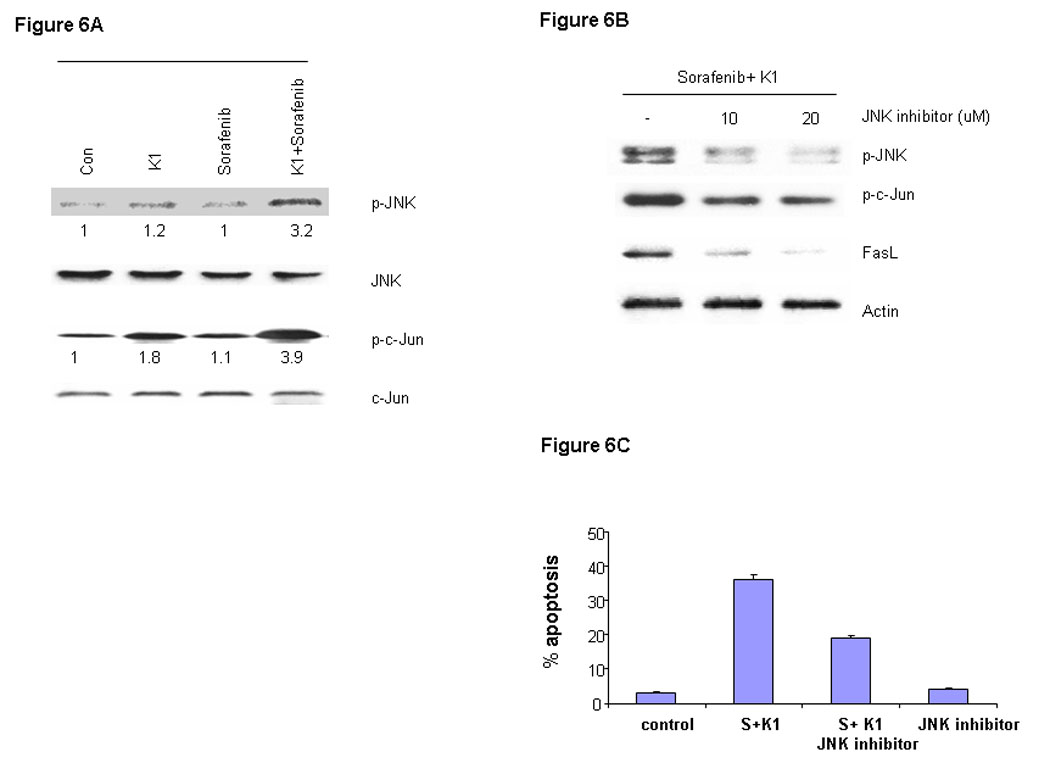
A. PL5 cells were treated with 50 µM vitamin k1 and 2.5 µM Sorafenib for 36 hours and levels of phospho-JNK and its key substrate phospho-c-Jun were analyzed by Western blot from whole-cell lysates. B. PL5 cells were treated with 50 µM vitamin k1 and 2.5 µM Sorafenib for 36 hours in the presence or absence of JNK inhibitor. After treatment, cells were lysed and 20 µg proteins were separated on SDS-PAGE gel or the percentage of apoptotic cells were determined by flow cytometry (C).
DISCUSSION
Pancreas cancer is often a systemic disease by virtue of its metastases, which respond poorly to conventional cytotoxic chemotherapy. This has led to a search for novel approaches to therapy, including the targeting of the EGF receptor (Erlotinib) and the RAF/MEK/ERK pathways by new therapeutic agents. The RAF/MEK/ERK cascade, one of the principal RAS-regulated pathways, is hyper-stimulated in pancreas cancers with activating K-ras mutations. Sorafenib, with its molecular targeting on RAF in this vital pathway, has been shown to have antitumor activity against renal cell cancer (25–26) and HCC (27–28). However, Sorafenib is less effective for treating other types of cancer, although preliminary data have shown some activity for Sorafenib in pancreas cancer also (16). This was the stimulus to our search for agents that could be combined with Sorafenib to enhance its pancreas cancer growth-inhibitory actions.
We previously showed that combination of vitamin K1 with Sorafenib could sensitize HCC cell lines to Sorafenib actions and enhance Sorafenib-mediated apoptosis in vitro and in vivo (29). The present studies attempted to determine whether Sorafenib plus vitamin K1 could also result in enhanced cell growth inhibition in pancreas cancer cell lines and, if so, to elucidate the mechanism(s) responsible for this phenomenon. We show here for the first time, that combination of vitamin K1 with low and clinically relevant concentrations of Sorafenib inhibited growth and induced apoptosis in pancreas cancer cells in vitro. This finding has clinical implications, as it suggests that the combination might be a candidate for therapy in pancreas cancer patients, especially since each component is currently used in patients in other settings.
Prior studies have shown that Sorafenib in the 10 µmol/L range, which is at pharmacologically achievable concentrations, induced cell death in human leukemic cells (30–31), HCC cells (28) and pancreatic cancer cells (32). The results of the present study show that low concentrations of Sorafenib (2.5 µM) or vitamin K1 (50 µM) when used as a single agent, did not induce growth inhibition or apoptosis or inhibit phosphorylation of MEK/ERK, although high doses of either can inhibit phospho-ERK levels and increase phospho-c-Jun levels (Fig. 3A and B). However, treatment of pancreas cancer cells with low concentrations of both Sorafenib (2.5 µM) plus vitamin K1 (50 µM) resulted in cell growth inhibition and apoptosis (Fig. 1 and 2), as well as significantly inhibiting the phosphorylation of MEK and ERK (Fig 3C). Therefore, vitamin K1 seems to add to Sorafenib in inhibiting the MEK/ERK pathway. The apoptosis induced by Sorafenib plus vitamin K1 was caspase-dependent, since pre-treatment with pan-caspase inhibitor could dramatically block the apoptosis induced by Sorafenib plus vitamin K1. The apoptotic signaling pathways are generally divided into two types: the extrinsic, or death receptor pathway and the intrinsic or mitochondrial pathway (33). The extrinsic pathway involves cell surface death receptors, such as tumor necrosis factor or Fas, which upon binding of their ligands, initiate signaling to activate caspase-8, which cleaves caspase-3 directly to induce apoptosis. The intrinsic pathway involves mitochondrial changes and triggers the release of cytochrome C, which in turn activates caspase-9 and then caspase-3 (34). Our findings suggest that one central mechanism of Sorafenib plus vitamin K1 mediated apoptosis in pancreas cancer cells, involves activation of the extrinsic apoptosis pathway.
Fas signaling plays a fundamental role in induction of extrinsic apoptosis in a wide variety of different types of cancer cell (35–36). Increased expression of FasL signal transduction activates caspase-8 and triggers apoptosis. Fas ligand has been shown to be a target gene for c-Jun, primarily in lymphoid and neuronal cell types (37) and there are recent reports on c-Jun transcriptional regulation of Fas gene expression (38–39). Another interesting aspect of our studies is that we show the importance of JNK, c-Jun and Fas signaling in Sorafenib plus vitamin K induced apoptosis. C-Jun NH2-terminal kinase (JNK) belongs to the superfamily of MAP kinases that are involved in the regulation of cell proliferation, differentiation and apoptosis. The activation of JNK leads to apoptosis, which is dependent on the stimuli and cell type involved in such activation (40–41). Prolonged activation of JNK has been observed in human breast, gastric, prostate and ovarian cancer cells with apoptosis-inducing compounds. JNK has also been recognized as a key mediator of a variety of cell fates (42) and was initially identified as a protein kinase involved in the transactivation of c-Jun by phosphorylation (43) and activated apoptotic signaling through the up-regulation of pro-apoptotic genes through the transactivation of specific transcription factors such as c-Jun. FasL has been shown to be a target gene for c-Jun primarily in lymphoid neuronal cell type (36). Similarly, we found that Sorafenib plus vitamin K1 activated JNK and increased phosphorylation of c-Jun and increased FasL levels. Transcriptional regulation of Fas ligand has also been observed in ovarian cancer cells undergoing apoptosis following cisplatin treatment (44). Furthermore, our data showed that a JNK inhibitor reduced the phosphorylation levels of JNK and c-Jun and decreased the levels of FasL that were induced by Sorafenib plus vitamin K1 (Fig 6B). The apoptosis induced by Sorafenib plus vitamin K1 was also partially antagonized by the JNK inhibitor, supporting the idea that the increase in the JNK levels that were induced by our combination, were involved in their growth-inhibitory mechanisms. On the other hand, other signaling pathways also might contribute to the cell growth inhibition and apoptosis which were induced Sorafenib plus vitamin K1.
Recently, it has been found that FasL mediated death signal is abrogated by the mitogen-activated protein kinase (MAPK/ERK)-mediated signals (45). In the FasL-mediated apoptosis process, ERK activity inhibits the caspase-8 cleavage (46). This strongly suggests that MEK/ERK-mediated signals can suppress apoptosis and the source of this anti-apoptotic activity may exist at the level of caspase-8 activation. Here, we demonstrated that Sorafenib or vitamin K1 alone can inhibit phospho-MEK and phosphor-ERK levels, when used at high concentrations, but vitamin K1 at low concentrations significantly added to Sorafenib-mediated inhibition of the MEK/ERK pathway and induction of apoptosis via the extrinsic pathway. After treatment with the MEK inhibitor U0126 alone, we found the expected inhibition of phospho-ERK levels, but also found augmented activity of caspase-8 (Fig. 3D). Thus, the combination of Sorafenib plus vitamin K1 resulted in the simultaneous decrease in levels of phospho-MEK and phospho-ERK MAPKs, while increasing the levels of phospho-JNK MAPK and its downstream targets, phospho-c-Jun and FasL. Simultaneous and opposite effects on different MAPKs have been previously reported with different agents (47–48). The mechanisms underlying that selectivity were not elucidated. These opposite effects, which both result in enhanced apoptosis, are summarized in Fig 7.
Figure 7.
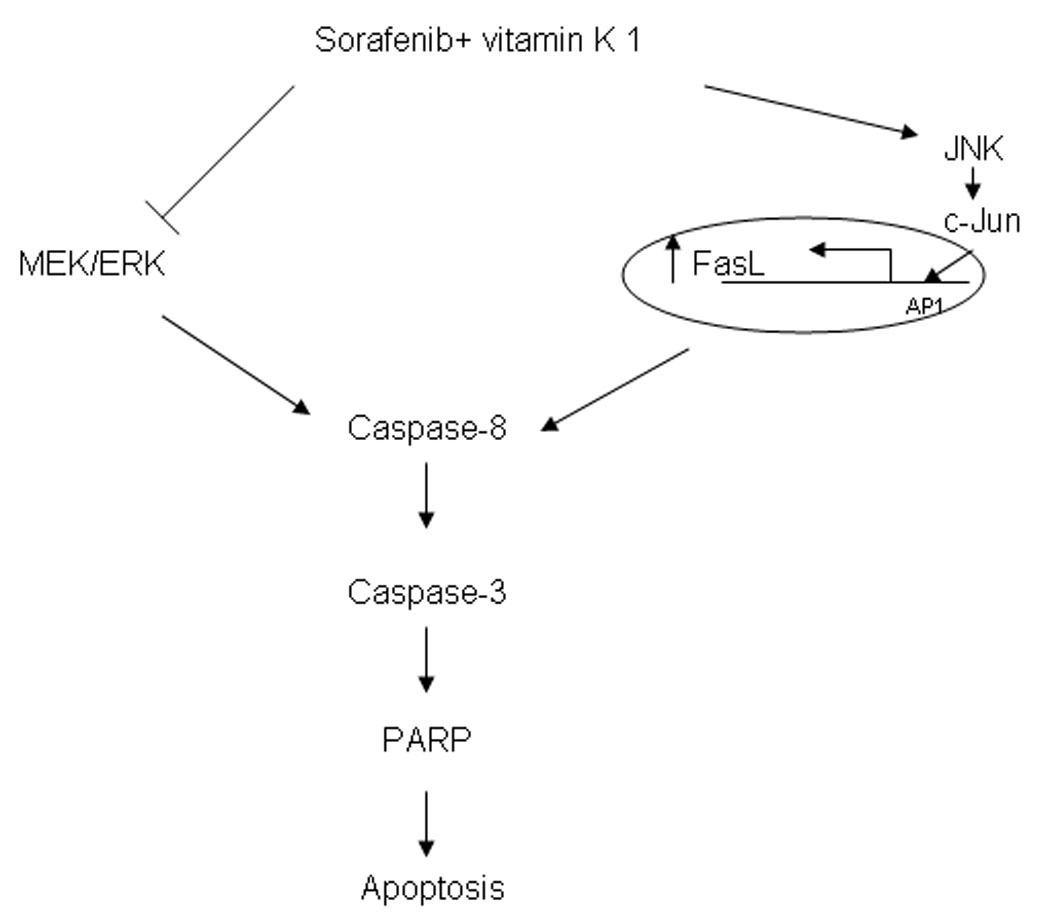
The mechanism of combination Sorafenib and vitamin k1 seems to involve inhibition of MEK/ERK activity and activation of JNK/c-Jun. Modulation of both of these pathways results in activation of caspase-8, ultimately leading to apoptotic cell death.
In conclusion, our data indicates that: 1) combination Sorafenib plus vitamin K1 can decrease the concentrations of Sorafenib that were needed to inhibit pancreas cancer cell growth and induce apoptosis; 2) vitamin K1 seems to be additive with Sorafenib in inhibiting the MEK/ERK pathway and activating caspase activity; 3) the possible mechanisms involved in Sorafenib plus vitamin K1 induced apoptosis of pancreas cancer cells involve both caspase-dependent extrinsic, as well as JNK/c-Jun-dependent upregulation of FasL-mediated apoptosis.
Supplementary Material
Acknowledgments
This work is supported in part by NIH grant CA 82723 (B.I.C)
Abbreviations
- MEK
mitogen-activated protein kinase kinase
- ERK
extracellular signal-regulated kinase
- JNK
c-Jun NH2-terminal kinase
Footnotes
Our research proposes a novel combination therapy involving vitamin K and Sorafenib for pancreas cancer that has the potential for immediate testing in patients, since each component is already approved for human use.
References
- 1.Devesa SS, Blot WJ, Stone BJ, Miller BA, Tarone RE, Fraumeni JF., Jr Recent cancer trends in the United States. J Natl Cancer Inst. 1995;87:175–182. doi: 10.1093/jnci/87.3.175. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 3.Grünewald K, Lyons J, Fröhlich A, Feichtinger H, Weger RA, Schwab G, Janssen JW, Bartram CR. High frequency of Ki-ras codon 127 mutations in pancreatic adenocarcinomas. Int J Cancer. 1989;43:1037–1041. doi: 10.1002/ijc.2910430614. [DOI] [PubMed] [Google Scholar]
- 4.Motojima K, Urano T, Nagata Y, Shiku H, Tsurifune T, Kanematsu T. Detection of point mutations in the Kirsten-ras oncogene provides evidence for the multicentricity of pancreatic carcinoma. Ann Surg. 1993;217:138–143. doi: 10.1097/00000658-199302000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishimura N, Yamasawa K, Karim Rumi MA, Kadowaki Y, Ishihara S, Amano Y, Nio Y, Higami T, Kinoshita Y. BRAF and K-ras gene mutations in human pancreatic cancers. Cancer Lett. 2003;199:169–173. doi: 10.1016/s0304-3835(03)00384-7. [DOI] [PubMed] [Google Scholar]
- 6.Dent P, Qiao L, Gilfor D, Birrer M, Grant S, Fisher PB. The regulation of tumor suppressor genes by oncogenes. Methods Mol Biol. 2003;222:269–292. doi: 10.1385/1-59259-328-3:269. [DOI] [PubMed] [Google Scholar]
- 7.Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK pathways in radiation responses. Oncogene. 2003;22:5885–5896. doi: 10.1038/sj.onc.1206701. [DOI] [PubMed] [Google Scholar]
- 8.Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, Cockerill M, Cartlidge S, Smith PD. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007;6:2209–2019. doi: 10.1158/1535-7163.MCT-07-0231. [DOI] [PubMed] [Google Scholar]
- 9.Milella M, Kornblau SM, Estrov Z, Carter BZ, Lapillonne H, Harris D, Konopleva M, Zhao S, Estey E, Andreeff M. Therapeutic targeting of the MEK/MAPK signal transduction module in acute myeloid leukemia. J Clin Invest. 2001;108:851–859. doi: 10.1172/JCI12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilhelm S, Chien DS. BAY 43-9006: preclinical data. Curr Pharm Des. 2002;8:2255–2257. doi: 10.2174/1381612023393026. [DOI] [PubMed] [Google Scholar]
- 11.Zhang W, Konopleva M, Shi YX, McQueen T, Harris D, Ling X, Estrov Z, Quintás-Cardama A, Small D, Cortes J, Andreeff M. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. J Natl Cancer Inst. 2008;10:184–198. doi: 10.1093/jnci/djm328. [DOI] [PubMed] [Google Scholar]
- 12.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 13.Flaherty KT. Sorafenib: delivering a targeted drug to the right targets. Expert Rev Anticancer Ther. 2007;7:617–626. doi: 10.1586/14737140.7.5.617. [DOI] [PubMed] [Google Scholar]
- 14.Rini BI. Sorafenib. Expert Opin Pharmacother. 2006;7:453–461. doi: 10.1517/14656566.7.4.453. [DOI] [PubMed] [Google Scholar]
- 15.Strumberg D. Preclinical and clinical development of the oral multikinase inhibitor sorafenib in cancer treatment. Drugs Today (Barc) 2005;41:773–784. doi: 10.1358/dot.2005.41.12.937959. [DOI] [PubMed] [Google Scholar]
- 16.Burris H, 3rd, Rocha-Lima C. New therapeutic directions for advanced pancreatic cancer: targeting the epidermal growth factor and vascular endothelial growth factor pathways. Oncologist. 2008;13:289–298. doi: 10.1634/theoncologist.2007-0134. [DOI] [PubMed] [Google Scholar]
- 17.Siu LL, Awada A, Takimoto CH, Piccart M, Schwartz B, Giannaris T, Lathia C, Petrenciuc O, Moore MJ. Phase I trial of sorafenib and gemcitabine in advanced solid tumors with an expanded cohort in advanced pancreatic cancer. Clin Cancer Res. 2006;12:144–151. doi: 10.1158/1078-0432.CCR-05-1571. [DOI] [PubMed] [Google Scholar]
- 18.Osada S, Tomita H, Tanaka Y, Tokuyama Y, Tanaka H, Sakashita F, Takahashi T. The utility of vitamin K3 (menadione) against pancreatic cancer. Anticancer Res. 2008;28:45–50. [PubMed] [Google Scholar]
- 19.Shibayama-Imazu T, Sakairi S, Watanabe A, Aiuchi T, Nakajo S, Nakaya K. Vitamin K(2) selectively induced apoptosis in ovarian TYK-nu and pancreatic MIA PaCa-2 cells out of eight solid tumor cell lines through a mechanism different from geranylgeraniol. J Cancer Res Clin Oncol. 2003;129:1–11. doi: 10.1007/s00432-002-0393-7. [DOI] [PubMed] [Google Scholar]
- 20.Shiraki M, Shiraki Y, Aoki C, Miura M. Vitamin K2 (menatetrenone) effectively prevents fractures and sustains lumbar bone mineral density in osteoporosis. Journal of Bone and Mineral Research. 2000;15:515–521. doi: 10.1359/jbmr.2000.15.3.515. [DOI] [PubMed] [Google Scholar]
- 21.Otsuka M, Kato N, Shao RX, Hoshida Y, Ijichi H, Koike Y, Taniguchi H, Moriyama M, Shiratori Y, Kawabe T, Omata M. Vitamin K2 inhibits the growth and invasiveness of hepatocellular carcinoma cells via protein kinase A activation. Hepatology. 2004;40:243–251. doi: 10.1002/hep.20260. [DOI] [PubMed] [Google Scholar]
- 22.Sata N, Klonowski-Stumpe H, Han B, Häussinger D, Niederau C. Menadione induces both necrosis and apoptosis in rat pancreatic acinar AR4-2J cells. Free Radic Biol Med. 1997;23:844–850. doi: 10.1016/s0891-5849(97)00064-6. [DOI] [PubMed] [Google Scholar]
- 23.Boucher MJ, Morisset J, Vachon PH, Reed JC, Lainé J, Rivard N. MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of human pancreatic cancer cells. J Cell Biochem. 2000;79:355–369. [PubMed] [Google Scholar]
- 24.Lasham A, Lindridge E, Rudert F, Onrust R, Watson J. Regulation of the human fas promoter by YB-1, Puralpha and AP-1 transcription factors. Gene. 2000;252:1–13. doi: 10.1016/s0378-1119(00)00220-1. [DOI] [PubMed] [Google Scholar]
- 25.Kane RC, Farrell AT, Saber H, Tang S, Williams G, Jee JM, Liang C, Booth B, Chidambaram N, Morse D, Sridhara R, Garvey P, et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin Cancer Res. 2006;12:7271–7278. doi: 10.1158/1078-0432.CCR-06-1249. [DOI] [PubMed] [Google Scholar]
- 26.Ratain MJ, Eisen T, Stadler WM, Flaherty KT, Kaye SB, Rosner GL, Gore M, Desai AA, Patnaik A, Xiong HQ, Rowinsky E, Abbruzzese JL, et al. Phase II placebo-controlled randomized discontinuation trial of sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2006;24:2505–2512. doi: 10.1200/JCO.2005.03.6723. [DOI] [PubMed] [Google Scholar]
- 27.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 28.Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851–11858. doi: 10.1158/0008-5472.CAN-06-1377. [DOI] [PubMed] [Google Scholar]
- 29.Brian I. Carr, Gang Wei, Meifang Wang. Vitamin k1 enhances Sorafenib effects on HCC and induces apoptosis: a non-toxic prevention strategy; AACR 100th Annual Meeting; 2009. p. 1319. [Google Scholar]
- 30.Zhang W, Konopleva M, Ruvolo VR, McQueen T, Evans RL, Bornmann WG, McCubrey J, Cortes J, Andreeff M. Sorafenib induces apoptosis of AML cells via Bim-mediated activation of the intrinsic apoptotic pathway. Leukemia. 2008;22:808–818. doi: 10.1038/sj.leu.2405098. [DOI] [PubMed] [Google Scholar]
- 31.Dasmahapatra G, Yerram N, Dai Y, Dent P, Grant S. Synergistic interactions between vorinostat and sorafenib in chronic myelogenous leukemia cells involve Mcl-1 and p21CIP1 down-regulation. Clin Cancer Res. 2007;13:4280–4290. doi: 10.1158/1078-0432.CCR-07-0835. [DOI] [PubMed] [Google Scholar]
- 32.Ulivi P, Arienti C, Amadori D, Fabbri F, Carloni S, Tesei A, Vannini I, Silvestrini R, Zoli W. Role of RAF/MEK/ERK pathway, p-STAT-3 and Mcl-1 in sorafenib activity in human pancreatic cancer cell lines. J Cell Physiol. 2009;220:214–221. doi: 10.1002/jcp.21753. [DOI] [PubMed] [Google Scholar]
- 33.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaufmann SH, Hengartner MO. Programmed cell death: alive and well in the new millennium. Trends Cell Biol. 2001;11:526–534. doi: 10.1016/s0962-8924(01)02173-0. [DOI] [PubMed] [Google Scholar]
- 35.Gillenwater AM, Zhong M, Lotan R. Histone deacetylase inhibitor suberoylanilide hydroxamic acid induces apoptosis through both mitochondrial and Fas (Cd95) signaling in head and neck squamous carcinoma cells. Mol Cancer Ther. 2007;6:2967–2975. doi: 10.1158/1535-7163.MCT-04-0344. [DOI] [PubMed] [Google Scholar]
- 36.Turley JM, Fu T, Ruscetti FW, Mikovits JA, Bertolette DC, Birchenall-Roberts MC. Vitamin E succinate induces Fas-mediated apoptosis in estrogen receptor-negative human breast cancer cells. Cancer Res. 1997;57:881–890. [PubMed] [Google Scholar]
- 37.Kavurma MM, Khachigian LM. Signaling and transcriptional control of Fas ligand gene expression. Cell Death Differ. 2003;10:36–44. doi: 10.1038/sj.cdd.4401179. [DOI] [PubMed] [Google Scholar]
- 38.Jia L, Yu W, Wang P, Li J, Sanders BG, Kline K. Critical roles for JNK, c-Jun, and Fas/FasL-Signaling in vitamin E analog-induced apoptosis in human prostate cancer cells. Prostate. 2008;68:427–441. doi: 10.1002/pros.20716. [DOI] [PubMed] [Google Scholar]
- 39.Karin M, Gallagher E. From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance IUBMB Life. 2005;57:283–295. doi: 10.1080/15216540500097111. [DOI] [PubMed] [Google Scholar]
- 40.Lin A, Dibling B. The true face of JNK activation in apoptosis. Aging Cell. 2002;1:112–116. doi: 10.1046/j.1474-9728.2002.00014.x. [DOI] [PubMed] [Google Scholar]
- 41.Liu J, Lin A. Role of JNK activation in apoptosis: a double-edged sword. Cell Res. 2005;15:36–42. doi: 10.1038/sj.cr.7290262. [DOI] [PubMed] [Google Scholar]
- 42.Chen YR, Wang X, Templeton D, Davis RJ, Tan TH. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and gamma radiation. Duration of JNK activation may determine cell death and proliferation. J Biol Chem. 1996;271:31929–31936. doi: 10.1074/jbc.271.50.31929. [DOI] [PubMed] [Google Scholar]
- 43.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 44.Mansouri A, Ridgway LD, Korapati AL, Zhang Q, Tian L, Wang Y, Siddik ZH, Mills GB, Claret FX. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J Biol Chem. 2003;278:19245–19256. doi: 10.1074/jbc.M208134200. [DOI] [PubMed] [Google Scholar]
- 45.Wilson DJ, Alessandrini A, Budd RC. MEK1 activation rescues Jurkat T cells from Fas-induced apoptosis. Cell Immunol. 1999;194:67–77. doi: 10.1006/cimm.1999.1486. [DOI] [PubMed] [Google Scholar]
- 46.Holmström TH, Schmitz I, Söderström TS, Poukkula M, Johnson VL, Chow SC, Krammer PH, Eriksson JE. MAPK/ERK signaling in activated T cells inhibits CD95/Fas-mediated apoptosis downstream of DISC assembly. EMBO J. 2000;19:5418–5428. doi: 10.1093/emboj/19.20.5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanimura S, Uchiyama A, Watanabe K, Yasunaga M, Inada Y, Kawabata T, Iwashita K, Noda S, Ozaki K, Kohno M. Blockade of constitutively activated ERK signaling enhances cytotoxicity of microtubule-destabilizing agents in tumor cells. Biochem Biophys Res Commun. 2009;378:650–655. doi: 10.1016/j.bbrc.2008.11.109. [DOI] [PubMed] [Google Scholar]
- 48.Kim YS, Park GB, Lee HK, Song H, Choi IH, Lee WJ, Hur DY. Cross-linking of B7-H1 on EBV-transformed B cells induces apoptosis through reactive oxygen species production, JNK signaling activation, and fasL expression. J Immunol. 2008;181:6158–6169. doi: 10.4049/jimmunol.181.9.6158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.