

Abstract
Cartilage oligomeric matrix protein/thrombospondin 5 (COMP/TSP5) is a major component of the extracellular matrix of the musculoskeletal system. Although COMP/TSP5 abnormalities are associated with several pathological conditions, its normal function remains unclear. This study was undertaken to delineate the function(s) of COMP/TSP5 in cartilage, especially regarding its interaction with chondrocytes. We show that COMP/TSP5 can support chondrocyte attachment and that the RGD sequence in COMP/TSP5 and the integrin receptors α5β1 and αVβ3 on the chondrocytes are involved in mediating this attachment. The interactions of COMP/TSP5 with the integrins are dependent on COMP/TSP5 conformation. Chondrocyte attachment to COMP/TSP5 in the calcium-replete conformation was inhibited by function-blocking integrin α5 and β1 antibodies, suggesting the involvement of the α5β1 integrin. Under this condition, a function-blocking antibody against αVβ3 did not have any effect on cell attachment. On the other hand, chondrocyte attachment to reduced COMP/TSP5 was instead sensitive to αVβ3 function-blocking antibodies, suggesting that COMP/TSP5 mediates attachment through chondrocyte αVβ3 integrin under this condition. Cell attachment to reduced COMP/TSP5 was not inhibited by β1 antibodies. These data indicate that COMP/TSP5 in different conformations can utilize different integrin receptors. These results are the first to demonstrate that COMP/TSP5 can mediate chondrocyte attachment through interactions with integrins. Through these interactions, COMP/TSP5 may be able to regulate cellular activities and respond to environment in the surrounding cartilage matrix.
Cartilage oligomeric matrix protein (COMP),2 also known as thrombospondin 5 (TSP5), is an abundant extracellular matrix protein that has been shown to exist in tissues of the musculoskeletal system including cartilage, tendon, ligament, and synovium (1–7). Immunohistochemistry studies of COMP/TSP5 distribution in cartilage have revealed specific temporal and spatial patterns, and COMP/TSP5 can be found in cartilage at sites both in proximity to chondrocytes in the pericellular matrix and away from the cells in the territorial and interterritorial matrix (8–10). In fetal cartilage, COMP/TSP5 exists in the pericellular matrix, especially in the growth cartilage adjacent to the primary ossification center. In adult articular cartilage, COMP/TSP5 is found at higher levels in the interterritorial matrix, with stronger staining in the deeper zone (8–10).
The importance of COMP/TSP5 is suggested by its association with several pathological conditions. Compared with normal cartilage, the levels and patterns of COMP/TSP5 expression were found to change in cartilage of osteoarthritis and rheumatoid arthritis patients, and COMP/TSP5 in serum and synovial fluid has been suggested as a biomarker for these conditions (11–13). The importance of COMP/TSP5 to cartilage structure and function is further underscored by findings that COMP/TSP5 mutations lead to human skeletal dysplasias, pseudoachondroplasia, and multiple epiphyseal dysplasia, including the Fairbanks and Ribbing types, MED/EDM1 (14, 15). We and others have shown previously that COMP/TSP5 is a calcium-binding protein and that COMP/TSP5 structure depends on its calcium binding ability (16, 17). Mutations in the type 3 calcium binding repeats affect the protein’s ability to fold correctly into the mature conformation. Thus, calcium binding is important for maintaining the proper structure of COMP/TSP5. Approximately 30% of the individuals with pseudoachondroplasia carry a mutation, delGAC-(1430–1444), that leads to the deletion of a single aspartic acid residue (15, 18). A recombinant version of this mutant COMP/TSP5, designated MUT3, exhibits decreased calcium binding, defective protein folding, and increased intracellular retention (5, 16, 17).
Based on sequence homology, COMP/TSP5 is also the fifth member of the thrombospondin (TSP) family. It has the coiled coil region responsible for pentamerization and interchain disulfide bond formation, the type 2 epidermal growth factor-like repeats, the type 3 calcium-binding repeats, and the COOH-terminal globular domain (19, 20). The presence of the type 2, type 3, and the COOH-terminal domain is the hallmark of the TSPs, and there is a high degree of sequence homology, with an average sequence identity of 60% among the TSPs in their type 3 calcium-binding repeats and the COOH-terminal globular domain (21, 22). Studies on TSP1 and TSP2 have shown that that they can function at the interface between the cell surface and the extracellular matrix (21, 23–26). Both TSP1 and TSP2 have been shown to be able to support cell attachment through interaction with various cellular receptors including the integrins, and their adhesive activities are subjected to regulation by the calcium binding of the proteins (27–30). The RGD sequence, located in the type 3 calcium-binding sequence of TSP1 and TSP2, is active in supporting cell attachment. In TSP1, this RGD sequence is part of a calcium-binding motif, and its accessibility for interaction with integrin is sensitive to the calcium loading as well as disulfide bonding patterns of the protein (30). There is an RGD sequence located in the type 3 calcium-binding region of COMP/TSP5 from human, bovine, and mouse, but not from rat. Therefore it is unclear if this RGD sequence is active.
Although immunohistochemical data and COMP/TSP5 mutations in pseudoachondroplasia and MED/EDM1 suggest that COMP/TSP5 is important in connective tissues, its precise functional role remains unclear. Of the major components of cartilage, it has been reported that COMP/TSP5 can interact with collagen in a zinc-dependent fashion via its COOH-terminal domain (17, 31, 32). Although sequence homology with TSP1 and TSP2 suggest that COMP/TSP5 may interact with cell surface receptors, COMP/TSP5 has not been found to be a good adhesive substrate for chondrocytes (19, 33, 34). Previous studies reported either that chondrocytes did not adhere to COMP/TSP5 (19, 33) or that appreciable attachment could be observed only at a very high COMP/TSP5 concentration (34). However, these studies utilized COMP isolated from the native tissues that required either strong denaturant or EDTA for purification. According to previous studies (16, 17), we postulate that these treatment may have rendered the purified COMP in a denatured conformation and thus may not reflect its functions. Therefore, we have decided to reassess COMP function using recombinant COMP that was expressed into a culture supernatant and purified under the native condition in the presence of calcium (16).
In this paper we intend to investigate whether COMP/TSP5 has the ability to interact with chondrocyte and, if so, determine the cell surface receptors that are responsible for the interaction. Our data show that COMP/TSP5 can function to bind chondrocytes to support cellular attachment through interaction with cell surface integrins. We further show that the conformation of COMP/TSP5 influences its adhesive activity as well as the integrin receptors it recognizes. Our results suggest that COMP/TSP5 can mediate chondrocyte-extracellular matrix interaction; through this interaction COMP/TSP5 has the potential to regulate chondrocyte cellular activities as well as its phenotypic development.
MATERIALS AND METHODS
Protein and Reagents
Human recombinant COMP/TSP5 or its mutant MUT3 were produced using a mammalian expression system and purified as reported previously (16). GRGDSP and GRGESP peptides were from Peninsula Laboratories, Inc. (Belmont, CA). Mouse IgG control was from Sigma. Monoclonal antibodies against the integrin α1 or α2 subunit were from Upstate Biotechnology, Inc. (Lake Placid, NY). The monoclonal antibody ASC-1 against α3 subunit was from Chemicon International, Inc. (Temecula, CA). The monoclonal antibody against α6 (4F10) was from Biodesign (Kennebunk, ME). The monoclonal antibodies against the α5 subunit were mAb16 from BD Biosciences and P1D6 from Chemicon. The monoclonal antibodies against the β1 subunit were P4C10 from Invitrogen, B3B11 from Chemicon, and 4B4 from Coulter Corporation (Miami, FL). The monoclonal antibody JBS5 against α5β1 was from Serotec Ltd. (Oxford, UK). Antibodies that contained azide when purchased were dialyzed against HEPES-buffered saline (HBS) with 1 mM CaCl2 (HBS/C) using a Slide-A-Lyzer MINI dialysis unit (Pierce).
Cells
Primary human juvenile costochondral chondrocytes were isolated and cultured as described previously (5). Immortalized human juvenile costochondral chondrocytes (T/C-28a4) were derived from the originally reported SV40-TAg-immortalized T/C-28a2 lines (35) after two additional passages through suspension culture over agarose. The T/C-28a4 chondrocyte cell line expresses type II collagen, aggrecan, and biglycan mRNAs, but low or undetectable levels of decorin and versican mRNAs (36). These cells continue to express sox9, indicating that they are chondrocytes in nature (38). However, expression of extracellular matrix proteins decreases after continuous culture in serum-containing medium compared with normal chondrocytes in primary monolayer culture (36–38). A clonal expansion of this cell line, TC1a, was used for the attachment assays described in this paper. Monolayer TC1a cells were cultured in 1:1 mixture of Dulbecco’s modified Eagle’s medium and Ham’s F-12 (BioWhittaker, Walkersville, MD) plus 10% fetal bovine serum.
Cell Attachment Assays
To prepare cells for attachment assays, monolayer TC1a or primary chondrocyte cells grown in 10-cm tissue culture plates were washed twice with HBS and treated with 1 ml of 0.1 mg/ml tosylphenylalanyl chloromethyl ketone-treated trypsin in HBS (Worthington, Lakewood, NJ) at 37 °C for 5 min. Trypsinization was stopped with 2 ml of 0.5 mg/ml soybean trypsin inhibitor (Sigma) in HBS/C. Cells were flushed off the plate as a single cell suspension, sedimented, and washed twice with trypsin inhibitor solution followed by two more washes with HBS/C. Subsequently, cells were resuspended in HBS/C containing 1% heat-inactivated bovine serum albumin at a final concentration of 2–2.5 × 105 cells/ml. Immulon II 96-well plates (Dynex, Chantilly, VA) were coated with COMP/TSP5 at various concentrations in HBS/C or HBS containing 5 mM EDTA at 4 °C overnight. The plates were then blocked with 1% heat-inactivated bovine serum albumin in HBS/C or HBS containing 5 mM EDTA, respectively, for 30 min at 37 °C. Where indicated, wells coated in EDTA were reduced with 20 mM dithiothreitol in HBS containing 5 mM EDTA for 30 min at room temperature. Wells were washed four times with HBS/C before the addition of 100 μl of cells to each well and incubated for 2 h at 37 °C. At the end of the incubation and after examination under phase-contrast microscopy, the plate was washed three times with HBS/C. The number of cells attached to each well was quantified using a CyQuant kit (Molecular Probes, Eugene, OR). In studies where various reagents including antibodies were used, the wells were coated with 5 μg/ml COMP/TSP5. The reagent was incubated with cells for 30 min at room temperature with mild agitation before transfer of cells to the wells. Attachment was carried out as described above.
Integrin Profiling
Primary chondrocyte cell surface proteins were surface-labeled with biotin using a protein biotinylation system per the manufacturer’s instruction (Amersham Biosciences). The cells were lysed in lysis buffer containing 0.15 M NaCl and 1% Triton X-100 in 50 mM Tris, pH 7.4, with a protease inhibitor mixture for 60 min at 4 °C. Detergent insoluble materials were removed by centrifugation at 10,000 × g for 20 min. After preclearing with mouse IgG and protein G-Sepharose, the cell lysate was incubated with specific integrin antibodies in the presence of protein G-Sepharose. The immunoprecipitated integrins were analyzed by SDS-PAGE followed by transfer to a piece of nitrocellulose membrane. The biotin-labeled integrins were visualized by horseradish peroxidase-conjugated streptavidin incubation followed by enhanced chemiluminescence detection.
RESULTS
We first used the chondrocyte-derived cell line TC1a to test whether COMP/TSP5 is an adhesive protein for chondrocytes. Chondrocytes grown in monolayer systems were used for short-term attachment assays. For the attachment assays, cells were allowed to adhere to 96-well plates that were coated with recombinant COMP/TSP5 or MUT3 at various concentrations. Based on our previous results showing that COMP/TSP5 is a calcium-binding protein and that calcium influences the conformation of COMP/TSP5 protein (16), we investigated whether the conformation of COMP/TSP5 also affected its ability to support the chondrocyte attachment. For this purpose, we coated COMP/TSP5 or MUT3 in the presence of either calcium or EDTA. All attachment assays were performed in the presence of 2 mM calcium ions regardless of the protein-coating conditions. Attached cells were quantified using CyQuant (Molecular Probes). In our preliminary studies we have shown that fluorescence readings were linearly proportional to the number of cells. Typically ~70% of the cells initially added to the wells remained at the end of the attachment assay under our conditions unless inhibitors were used. For TC1a cells grown in monolayer, COMP/TSP5 in a calcium-replete conformation supported cell attachment in a dose-dependent manner (Fig. 1A). At protein-coating concentrations of 5 μg/ml and higher the cells also spread on the protein-coated wells (Fig. 2). Treating cells 2 h before and during cell attachment assay with cycloheximide did not affect cell attachment, suggesting that in this short-term assay protein synthesis is not required for the attachment of cells to COMP/TSP5 (Fig. 3). Treating COMP/TSP5 with calcium or EDTA without reduction had no obvious effect on attachment of the TC1a cells grown in a monolayer (Fig. 1A). MUT3 also supported TC1a cell attachment, and treating MUT3 with EDTA before coating did not affect cell attachment significantly (Fig. 1B). These results suggest that the removal of calcium from wild type COMP/TSP5 (Fig. 1A) or MUT3 (Fig. 1B) does not affect the capacity to bind to chondrocytes cultured in a monolayer compared with the calcium-replete condition.
FIGURE 1. Attachment of chondrocyte-derived cell line TC1a cultured in monolayer to COMP/TSP5 (A) and MUT3 (B).

COMP/TSP5 or MUT3 in the presence of 1 mM CaCl2 (protein/C) or 5 mM EDTA (protein/E) or coated in the presence of 5 mM EDTA followed by reduction (protein/R) were diluted to the final concentrations as indicated and coated onto 96-well plates. Attachment assays were carried out as described under ”Materials and Methods“ with TC1a cells grown in a monolayer. Results are shown as mean ± S.D. of quadruplicate experiments.
FIGURE 2. Morphology of TC1a attached to COMP/TSP5.

TC1a cells were allowed to attach to COMP/TSP5 coated at the indicated concentrations in the presence of 1 mM CaCl2 (COMP/C) or 5 mM EDTA followed by reduction (COMP/R). FN coated at 10 μg/ml (FN) or heat-inactivated bovine serum albumin (BSA) coated at the same concentration was used as control. Attachment assays were carried out as described under ”Materials and Methods.“ At the end of the 2-h incubation, phase-contrast pictures were taken.
FIGURE 3. Effect of cycloheximide on attachment of chondrocyte-derived cell line TC1a.
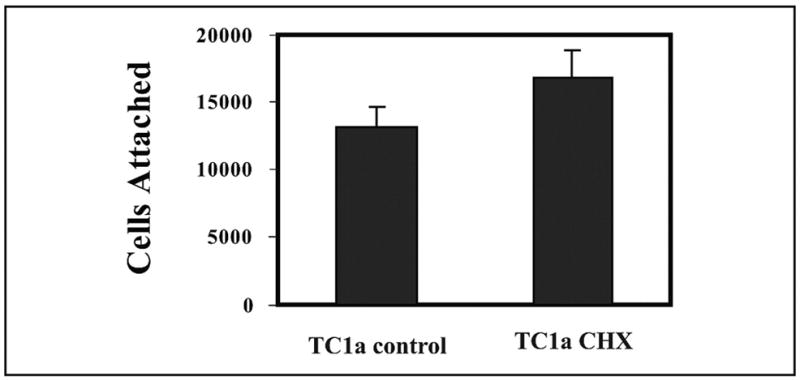
Attachment assays were carried out as described in the Fig. 1 legend with COMP/TSP5 coated in the presence of 1 mM CaCl2. TC1a cells were cultured in serum free Dulbecco’s modified Eagle’s medium containing 20 μg/ml cycloheximide 2 h before the attachment assay, and cycloheximide was included in all buffers for the attachment assay. Results are shown as mean ± S.D. of six data points.
It was shown previously with TSP1 and TSP2 that the availability of the RGD sequence, located in the calcium-binding type 3 repeats, is regulated by calcium binding as well as the complex intracellular disulfide bonds (28–30). Reduction of TSP1 and TSP2 helps to expose the RGD sequences that are possibly cryptic inside the complex intracellular disulfide bonds within the type 3 calcium-binding repeats (28, 29). We wished to determine whether this holds true for COMP/TSP5 because of the high degree of homology in the type 3 repeats among all TSPs and the presence of the RGD sequence in human COMP/TSP5. To test this hypothesis, we reduced the coated calcium-depleted COMP/TSP5 or MUT3 with the reducing reagent dithiothreitol before adding the cells for attachment. Indeed, reduction of either wild type COMP/TSP5 or MUT3 (protein/R in Fig. 1, A and B, respectively) after coating increased cell attachment, especially in wells coated with low concentrations, 1 μg/ml or less, of COMP/TSP5 or MUT3. Cells attached to and spread on dithiothreitol-treated COMP/TSP5 coated at 1 μg/ml (Fig. 2).
In addition to immortalized chondrocytes, we have also tested whether COMP/TSP5 can support the attachment of primary chondrocytes. For this purpose, juvenile costochondral chondrocytes were used for short-term attachment assays as above, and we observed similar results (data not shown).
To investigate the nature of the interaction between COMP/TSP5 and chondrocytes, sequences on COMP/TSP5 that mediate the attachment were studied. An RGD sequence is located in the type 3 calcium-binding repeats of COMP/TSP5. We assessed whether the RGD sequence is active. For this purpose, we included a peptide containing RGD in the attachment assay (GRGDSP). A peptide containing the RGE sequence (GRGESP) was used as control. COMP/TSP5 was coated either in the presence of calcium or in the presence of EDTA followed by reduction. Under both conditions, the RGD peptide was able to inhibit the attachment of primary chondrocytes to COMP/TSP5 in a RGD peptide concentration-dependent manner, whereas the RGE peptide had no significant effect (Fig. 4). These data suggest that COMP/TSP5 can support chondrocyte attachment, at least in part, via its RGD sequence.
FIGURE 4. Effect of RGD peptide on attachment of chondrocyte-derived cell line TC1a to COMP/TSP5.
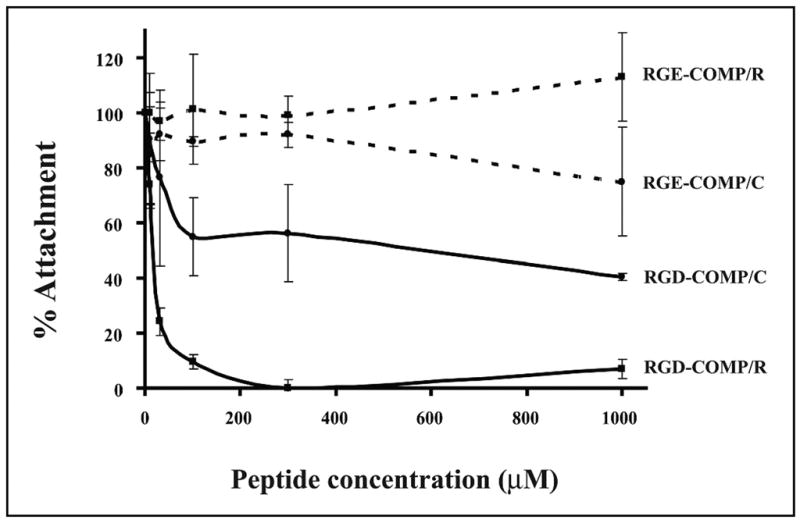
Attachment assays were carried out as described in the Fig. 1 legend with COMP/TSP5 coated in the presence of CaCl2 (COMP/C) or EDTA followed by reduction (COMP/R). Primary chondrocytes were used for this attachment assay. Cells were incubated with either the GRGDSP (RGD) or GRGESP (RGE) peptide for 30 min at the indicated concentrations before being added to the COMP/TSP5-coated wells.
The inhibitory effect of RGD suggests that the cells interact with COMP/TSP5 via cell surface integrins. To determine the integrin receptor expression on the primary human chondrocytes used in our experiments, we performed cell surface biotin labeling followed by immunoprecipitation with specific integrin antibodies. Our results showed that primary chondrocytes expressed integrins including α3β1 and α1β1 (not shown) as well as αVβ3 and those that contain α5 and α6 subunits in smaller amounts (Fig. 5). Similarly, integrins containing the β1 subunit and those that contain the α5 and α6 subunits, as well as α3β1 and αVβ3, were also found to be expressed by the TC1a cells (data not shown). The lack of any precipitated bands with control mouse IgG indicated the stringency of our immunoprecipitation studies. The existence of more than one band in a lane indicates the presence of both the α and the β subunits in the immunoprecipitated integrin as expected for these heterodimeric receptors and possibly other cell surface-associated proteins.
FIGURE 5. Integrin expression by the chondrocytes.

Primary chondrocyte cell surface proteins were labeled with biotin. The cells were lysed, and integrins were immunoprecipitated using specific integrin antibodies as indicated. The immunoprecipitated integrins were separated by SDS-PAGE followed by transfer to a piece of nitrocellulose membrane. The biotin-labeled integrins were visualized by horseradish peroxidase-conjugated streptavidin incubation followed by enhanced chemiluminescence detection. mIgG, mouse IgG.
To determine the integrin receptors on chondrocytes that mediate attachment to COMP/TSP5, various antibodies against integrin receptors were tested in attachment assays using chondrocytes. First, we examined the attachment of primary chondrocytes attached to calcium-replete COMP/TSP5, as shown in Fig. 6. A function-blocking monoclonal antibody against the β1 integrin subunit, mAb13, blocked the attachment, whereas another antibody without function-blocking activity against β1, B3B11, did not inhibit substantially the attachment of primary chondrocytes to calcium-replete COMP/TSP5. Monoclonal antibodies against either the α1 or α2 subunit, reported by the manufacturer to inhibit cell attachment to collagen at the concentrations used, did not have any obvious effect on attachment to COMP/TSP5. However, mAb16 against the α5 subunit was effective in inhibiting attachment (Fig. 6). A function-blocking antibody against αVβ3 did not block attachment of primary chondrocytes to COMP/TSP5 (see below and Fig. 7). These results suggest that chondrocyte attachment to calcium-replete COMP/TSP5 involves the cell surface integrin receptor α5β1.
FIGURE 6. Attachment of chondrocytes to COMP/TSP5/C in the presence of integrin antibodies.
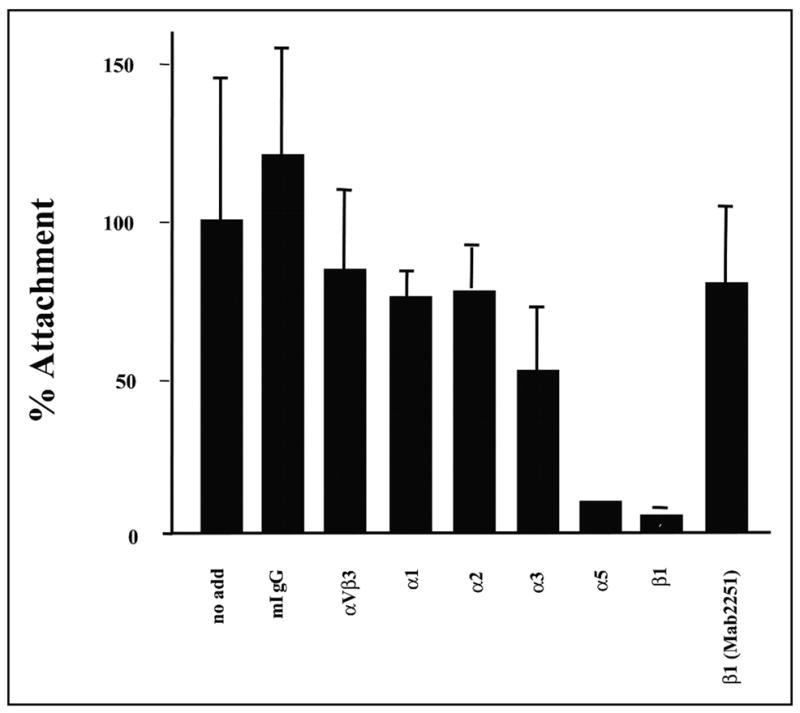
Attachment assays were carried out basically as described in the Fig. 1 legend. COMP/TSP5 in the presence of CaCl2 was coated on the wells. The primary chondrocytes used for this attachment assay were incubated with the various antibodies for 30 min before addition to the wells. Results are shown as mean ± S.D. of two triplicate experiments. The monoclonal antibodies used in this experiment and their final concentrations are as follows: mouse IgG (mIgG), 100 μg/ml; anti-α1, 6.3 μg/ml; anti-α2, 6 μg/ml; anti-α5,16.6 μg/ml; anti-α6, 16.6 μg/ml; anti-β1 (P4C10), 16.6 μg/ml; and anti-β1 (B3B11), 16.6 μg/ml.
FIGURE 7. Effect of COMP/TSP5 conformation on integrin usage by chondrocytes for attachment.

Attachment assays were carried out basically as described in the Fig. 1 legend. COMP/TSP5 was either coated in the presence of CaCl2 (COMP/C) or coated in the presence of EDTA followed by reduction (COMP/R) before the attachment assays. Chondrocytes were incubated with the different antibodies at the indicated concentrations for 30 min before addition to the wells. Shown here are the results using TC1a cells from a monolayer culture.
Similar to the primary chondrocytes, TC1a cell attachment to COMP/TSP5 also seemed to involve the α5β1 receptor. Attachment of the TC1a cells to calcium-replete COMP/TSP5 was inhibited by antibodies against either the α5 or β1 subunit. The antibodies tested were antibodies against the α5 subunit, P1D6 and JBS5, and antibodies against the β1 subunit, 4B4 and P4C10. Neither mouse IgG nor the B3B11 antibody without function-blocking activity against the β1 sub-unit inhibited attachment. Antibody LM609 against integrin αvβ3 did not have any inhibitory effect, indicating that αVβ3 integrin is not involved under this condition.
Because the different conformation induced by reduction of COMP/TSP5 seems to regulate its adhesive activity, we tested whether changing the conformations of COMP/TSP5 also affects its interaction with cell surface receptors. We showed above that chondrocyte attachment to calcium-replete COMP/TSP5 (COMP/C) was inhibited by antibodies against α5β1 but not against the αVβ3 integrin. Under this condition, antibody 4B4 against the β1 integrin subunit inhibited attachment in a concentration-dependent manner, whereas the antibody LM609 against αVβ3 integrin did not have any appreciable effect on the attachment (Fig. 7). When chondrocytes were allowed to attach to calcium-depleted and reduced COMP/TSP5 (COMP/R), attachment was not inhibited by the 4B4 antibody against the β1 subunit in all concentrations tested, including the concentration range at which 4B4 blocked these cells from attaching to COMP/C. Instead, the attachment was inhibited by antibody LM609 against αVβ3 in an antibody concentration-dependent fashion (Fig. 7). Under this condition, the attachment was also inhibited by the RGD peptide in a concentration-dependent manner but not inhibited by the RGE peptide (Fig. 2). These data suggest that in the calcium-depleted, reduced conformation, COMP/TSP5 can now recognize the cell surface αVβ3 receptor and interact with the chondrocyte.
DISCUSSION
COMP/TSP5 is an extracellular matrix protein that is abundant in musculoskeletal tissues including cartilage, tendon, ligament, and synovium. However, despite intensive studies on COMP/TSP5 and the fact that COMP/TSP5 is associated with several pathological conditions, its function in the connective tissue remains to be defined. To investigate the potential role of COMP/TSP5 in cartilage, in this paper we have studied the interaction of COMP/TSP5 with chondrocytes, the unique cell type found in cartilage.
Several important conclusions can be drawn from our current studies. First, we have shown that COMP/TSP5 can bind chondrocytes and serves as an adhesive factor for these cells using short-term attachment assays. This is in agreement with the distribution of COMP/TSP5 close to the chondrocytes in early development in cartilage pericellular and territorial matrix (7, 8). Through binding to the chondrocytes, COMP/TSP5 has the potential to influence cellular activities and cellular phenotype. Indeed, Kipnes et al. (39) reported that COMP/TSP5 plays a role in mesenchymal chondrogenesis in vitro. COMP/TSP5 was shown to be able to regulate cellular proliferation and differentiation of the mesenchymal stem cells under the influence of bone morphogenetic protein-2 (BMP-2). Thus, COMP/TSP5 can coordinate with BMP-2 to promote early stages of chondrogenic differentiation of the mesenchymal stem cells. Whether this process involves the interaction of COMP/TSP5 with integrin warrants further investigation.
Furthermore, we have investigated the sequences on COMP/TSP5 involved in the adhesive function of this molecule. It has been debated whether the RGD sequence in COMP/TSP5 is active because it is buried in the calcium-binding domain and is conserved in human, mouse, and bovine COMP/TSP5 but not in rat COMP/TSP5. Our data suggest that the RGD sequence does play a role in mediating the attachment of chondrocytes in human cartilage.
In addition, we have studied the influence of the calcium binding conformation of COMP on its cellular attachment activities. In our previous studies we have shown that COMP/TSP5 is a calcium-binding protein and that its conformation depends on the amount of calcium bound by the protein (16). Because COMP/TSP5 under physiological conditions exists in an environment where the concentrations of calcium are in the millimolar range, we purified COMP/TSP5 in a calcium-replete conformation. We then asked if chelation of calcium ions from COMP/TSP5 affects its function and, furthermore, if mutations in COMP/TSP5 that render the protein defective in calcium binding also affect its function. Using TC1a cells cultured in a monolayer, we found that the calcium binding conformation of COMP/TSP5 does not have substantial influence on the adhesiveness of the protein. In agreement, MUT3 also supports adhesion of these cells as well as wild type COMP/TSP5. These results suggest that calcium binding conformation of COMP/TSP5 did not have any major influence on its ability to support chondrocyte attachment under our conditions.
Most importantly, our data also indicate that COMP/TSP5 can interact with α5β1 and αVβ3 under different conditions. Through interactions with different receptors, COMP/TSP5 can potentially mediate different cellular responses. Although α5β1 and αVβ3 are present on the cell surface of chondrocytes, they have different distribution patterns. Both integrins are expressed by articular and growth plate chondrocytes in fetal cartilage (40–42), and αVβ3 expression seems to be lower in the adolescent growth plate cartilage (43). It is possible that the ligation of the two integrins can confer different functions. For example, it has been shown that activation of either α5β1 or αVβ3 plays opposite roles in mediating the inflammatory response in cartilage in osteoarthritis (44). The question of whether COMP/TSP5 interaction with either of these two integrins also leads to different downstream effects warrants further studies.
Integrin receptors exist on chondrocytes and recognize various cartilage extracellular matrix proteins (45, 46). Both the α5β1 and αvβ3 integrins have been found on the chondrocyte cell surface along with the α1β1, α2β1, α3β1, α6β1, α10β1, and αvβ5 integrins (47, 48). The integrins are important for cartilage morphogenesis and differentiation. Inhibition of integrins using anti-β1 antibodies leads to the inhibition of chondrogenesis (49). In addition, it has been established that human articular chondrocytes use the α5β1 integrin as a mechanoreceptor. Mechanical stimulation of normal human knee joint chondrocytes initiates a signal cascade involving stretch-activated ion channels, the α5β1 integrin, and the interleukin-4 loop that results in membrane hyperpolarization, increased levels of aggrecan mRNA, and decreased levels of matrix metalloprotease-3 (50–53). Although an extracellular matrix-integrin interaction can result in anabolic responses in chondrocytes, catabolic consequences of this interaction have also been reported. Although the mechanoreceptor in osteoarthritis chondrocytes is also the α5β1 integrin, downstream signaling pathways may differ, resulting in membrane depolarization and no change in aggrecan or matrix metalloprotease-3 expression following mechanical stimulation (54–56). These responses may play a role in the phenotypic changes and increased cartilage breakdown in diseased cartilage. However, the extracellular matrix molecules that can interact with the integrin receptors, as well as the exact mechanism leading from extracellular matrix-integrin interactions to the changes in intracellular signaling events and downstream biosynthetic reactions, remain unclear at present and need further studies. Here, our results have shown that COMP/TSP5 can mediate the interaction of chondrocytes with the cartilage extracellular matrix through interaction with cell surface integrin receptors.
Fibronectin (FN) and COMP/TSP5 have been shown to interact with each other and to colocalize in some regions of cartilage (10). These observations suggest the possibility that some of the functions of COMP/TSP5 may be indirectly mediated by FN. However, several pieces of evidence suggest that this is not the case in our study. First, it has been shown that COMP/TSP5 exists in articular cartilage in much higher concentrations (up to 5 mg/ml) (57) than FN (~1.5 μg/ml) (58). Second, in our short-term attachment assays cycloheximide, which inhibits protein synthesis including FN synthesis, did not inhibit chondrocyte attachment to COMP/TSP5. This suggests that COMP/TSP5 can bind to chondrocyte integrin receptors directly instead of binding indirectly through FN. Further evaluations of the potential role of interactions between these two molecules may be explored in future studies. Likewise, the lack of effect of cycloheximide and the lack of the inhibitory effect of antibodies to α1 or α2 integrins, receptors for the collagens, also argues against the indirect involvement of collagen in our system.
For our studies, both immortalized and primary chondrocytes isolated from the costochondral cartilage in monolayer culture system were utilized. With these chondrocytes we have shown that COMP can interact with the cell surface integrin receptors, including α5β1 and αvβ3. Because of the extremely limited availability of healthy human articular chondrocytes, we were not able to test whether COMP can interact similarly with these freshly isolated cells. It has been known that chondrocytes dedifferentiate in an extensive monolayer culture. In addition, non-weight-bearing cartilage, such as costochondral cartilage, is different in its matrix organization as compared with the weight-bearing articular cartilage, e.g. COMP is present at a much higher concentration in articular cartilage than in costochondral cartilage (57). Therefore care needs to be taken when we interpret our data and extrapolate to articular cartilage in vivo. Because of the clinical importance of articular cartilage and its involvement in arthritis, further experiments investigating the interaction of COMP with articular cartilage is of great importance in the future.
In summary, we have shown that COMP/TSP5 supports chondrocyte attachment in vitro and that integrins are functional receptors on chondrocytes that mediate attachment to COMP/TSP5. We have further shown that the conformation of COMP/TSP5 influences the receptors on the chondrocyte cell surface utilized for attachment. Our studies would indicate that COMP/TSP5 may function in mediating cell-matrix interactions within cartilage and other musculoskeletal tissues in vivo and that through these interactions COMP/TSP5 may function to regulate chondrocyte cellular activities as well as its phenotypic development.
Acknowledgments
We thank Dr. Karen Yee for advice on the use of integrin antibodies, Mark Duquette for help with protein purification, and Lujian Tan for help with the analysis of proteoglycan expression.
Footnotes
The abbreviations used are: COMP, cartilage oligomeric matrix protein; FN, fibronectin; HBS, HEPES-buffered saline; HBS/C, HBS with CaCl2; MED/EDM1, multiple epiphyseal dysplasia/epiphyseal dysplasia, multiple, 1; MUT3, a mutant of COMP/TSP5 bearing a three-base pair GAC deletion in nucleotides 1430 –1444; TSP, thrombospondin.
This work was supported in part by NHLBI, National Institutes of Health Grant HL49081 (to J. L.), NIAMS, National Institutes of Health Grant AR45378 (to M. B. G.), NIA, National Institutes of Health Grant AG22021 (to M. B. G.), and Shriners Hospital for Children Grant 15955 (to J. T. H.).
References
- 1.DiCesare PE, Fang C, Leslie MP, Tulli H, Perris R, Carlson CS. J Orthop Res. 2000;18:713–720. doi: 10.1002/jor.1100180506. [DOI] [PubMed] [Google Scholar]
- 2.DiCesare PE, Carlson CS, Stollerman ES, Chen FS, Leslie M, Perris R. FEBS Lett. 1997;412:249–252. doi: 10.1016/s0014-5793(97)00789-8. [DOI] [PubMed] [Google Scholar]
- 3.DiCesare P, Hauser N, Lehman D, Pasumarti S, Paulsson M. FEBS Lett. 1994;354:237–240. doi: 10.1016/0014-5793(94)01134-6. [DOI] [PubMed] [Google Scholar]
- 4.Hedbom E, Antonsson P, Hjerpe A, Aeschlimann D, Paulsson M, Rosa-Pimentel E, Sommarin Y, Wendel M, Oldberg A, Heinegard D. J Biol Chem. 1992;267:6132–6136. [PubMed] [Google Scholar]
- 5.Hecht JT, Deere M, Putnam E, Cole W, Vertel B, Chen H, Lawler J. Matrix Biol. 1998;17:269–278. doi: 10.1016/s0945-053x(98)90080-4. [DOI] [PubMed] [Google Scholar]
- 6.Smith RK, Zunino L, Webbon PM, Heinegard D. Matrix Biol. 1997;16:255–271. doi: 10.1016/s0945-053x(97)90014-7. [DOI] [PubMed] [Google Scholar]
- 7.Dodge GR, Hawkins D, Boesler E, Sakai L, Jimenez SA. Osteoarth Cartil. 1998;6:435–440. doi: 10.1053/joca.1998.0147. [DOI] [PubMed] [Google Scholar]
- 8.DiCesare PE, Morgelin M, Carlson CS, Pasumarti S, Paulsson M. J Orthop Res. 1995;13:422–428. doi: 10.1002/jor.1100130316. [DOI] [PubMed] [Google Scholar]
- 9.Murphy JM, Heinegard R, McIntosh A, Sterchi D, Barry FP. Matrix Biol. 1999;18:487–497. doi: 10.1016/s0945-053x(99)00042-6. [DOI] [PubMed] [Google Scholar]
- 10.Di Cesare P, Chen F, Moergelin M, Carlson C, Leslie M, Perris R, Fang C. Matrix Biol. 2002;21:461–470. doi: 10.1016/s0945-053x(02)00015-x. [DOI] [PubMed] [Google Scholar]
- 11.DiCesare PE, Carlson CS, Stolerman ES, Hauser N, Tulli H, Paulsson M. J Orthop Res. 1996;14:946–955. doi: 10.1002/jor.1100140615. [DOI] [PubMed] [Google Scholar]
- 12.Neidhart M, Hauser N, Paulsson M, DiCesare PE, Michel BA, Hauselmann HJ. Br J Rheumatol. 1997;36:1151–1160. doi: 10.1093/rheumatology/36.11.1151. [DOI] [PubMed] [Google Scholar]
- 13.Petersson IF, Boegard T, Svensson B, Heinegard D, Saxne T. Br J Rheumatol. 1998;37:46–50. doi: 10.1093/rheumatology/37.1.46. [DOI] [PubMed] [Google Scholar]
- 14.Briggs MD, Hoffman SM, King LM, Olsen AS, Mohrenweiser H, Leroy JG, Mortier GR, Rimoin DL, Lachman RS, Gaines ES, Cekleniak JA, Knowlton RG, Cohn DH. Nat Genet. 1995;10:330–336. doi: 10.1038/ng0795-330. [DOI] [PubMed] [Google Scholar]
- 15.Hecht JT, Nelson LD, Crowder E, Wang Y, Elder FF, Harrison WR, Francomano CA, Prange CK, Lennon GG, Deere M, Lawler J. Nat Genet. 1995;10:325–329. doi: 10.1038/ng0795-325. [DOI] [PubMed] [Google Scholar]
- 16.Chen H, Deere M, Hecht JT, Lawler J. J Biol Chem. 2000;275:26538–26544. doi: 10.1074/jbc.M909780199. [DOI] [PubMed] [Google Scholar]
- 17.Thur J, Rosenberg K, Nitsche DP, Pihlajamaa T, Ala-Kokko L, Heinegard D, Paulsson M, Maurer P. J Biol Chem. 2001;276:6083–6092. doi: 10.1074/jbc.M009512200. [DOI] [PubMed] [Google Scholar]
- 18.Deere M, Sanford T, Ferguson HL, Daniels K, Hecht JT. Am J Med Genet. 1998;80:510–513. doi: 10.1002/(sici)1096-8628(19981228)80:5<510::aid-ajmg14>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 19.Oldberg A, Antonsson P, Lindblom K, Heinegard D. J Biol Chem. 1992;267:22346–22350. [PubMed] [Google Scholar]
- 20.Newton G, Weremowicz S, Morton CC, Copeland NG, Gilbert DJ, Jenkins NA, Lawler J. Genomics. 1994;24:435–439. doi: 10.1006/geno.1994.1649. [DOI] [PubMed] [Google Scholar]
- 21.Adams JC. Annu Rev Cell Dev Biol. 2001;17:25–51. doi: 10.1146/annurev.cellbio.17.1.25. [DOI] [PubMed] [Google Scholar]
- 22.Lawler J, Chen H. In: Encyclopedia of Molecular Medicine. Creighton T, editor. John Wiley & Sons, Inc; New York, NY: 2002. pp. 481–484. [Google Scholar]
- 23.Lawler J. Curr Opin Cell Biol. 2000;12:634–640. doi: 10.1016/s0955-0674(00)00143-5. [DOI] [PubMed] [Google Scholar]
- 24.Chen H, Herndon ME, Lawler J. Matrix Biol. 2000;19:597–614. doi: 10.1016/s0945-053x(00)00107-4. [DOI] [PubMed] [Google Scholar]
- 25.Bornstein P, Sage EH. Curr Opin Cell Biol. 2002;14:608–616. doi: 10.1016/s0955-0674(02)00361-7. [DOI] [PubMed] [Google Scholar]
- 26.Bornstein P. J Clin Investig. 2001;107:929–934. doi: 10.1172/JCI12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawler J, Weinstein R, Hynes RO. J Cell Biol. 1988;107:2351–2361. doi: 10.1083/jcb.107.6.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen H, Sottile J, O’Rourke KM, Dixit VM, Mosher DF. J Biol Chem. 1994;269:32226–32232. [PubMed] [Google Scholar]
- 29.Sun X, Skorstengaard K, Mosher DF. J Cell Biol. 1992;118:693–701. doi: 10.1083/jcb.118.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kvansakul M, Adams JC, Hohenester E. EMBO J. 2004;23:1223–1233. doi: 10.1038/sj.emboj.7600166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenberg K, Olsson H, Morgelin M, Heinegard D. J Biol Chem. 1998;273:20397–20403. doi: 10.1074/jbc.273.32.20397. [DOI] [PubMed] [Google Scholar]
- 32.Holden P, Meadows RS, Chapman KL, Grant ME, Kadler KE, Briggs MD. J Biol Chem. 2001;276:6046–6055. doi: 10.1074/jbc.M009507200. [DOI] [PubMed] [Google Scholar]
- 33.Sommarin Y, Larsson T, Heinegard D. Exp Cell Res. 1989;184:181–192. doi: 10.1016/0014-4827(89)90376-5. [DOI] [PubMed] [Google Scholar]
- 34.DiCesare PE, Morgelin M, Mann K, Paulsson M. Eur J Biochem. 1994;223:927–937. doi: 10.1111/j.1432-1033.1994.tb19070.x. [DOI] [PubMed] [Google Scholar]
- 35.Goldring MB, Birkhead JR, Suen LF, Yamin R, Mizuno S, Glowacki J, Arbiser JL, Apperley JF. J Clin Investig. 1994;94:2307–2316. doi: 10.1172/JCI117595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kokenyesi R, Tan L, Robbins JR, Goldring MB. Arch Biochem Biophys. 2000;383:79–90. doi: 10.1006/abbi.2000.2044. [DOI] [PubMed] [Google Scholar]
- 37.Loeser RF. Biorheology. 2000;37:109–116. [PubMed] [Google Scholar]
- 38.Finger F, Schorle C, Zien A, Gebhard P, Goldring MB, Aigner T. Arthritis Rheum. 2003;48:3395–3403. doi: 10.1002/art.11341. [DOI] [PubMed] [Google Scholar]
- 39.Kipnes J, Carlberg AL, Loredo GA, Lawler J, Tuan RS, Hall DJ. Osteoarth Cartil. 2003;11:442–454. doi: 10.1016/s1063-4584(03)00055-4. [DOI] [PubMed] [Google Scholar]
- 40.Durr J, Goodman S, Potocnik A, von der Mark H, von der Mark K. Exp Cell Res. 1993;207:235–244. doi: 10.1006/excr.1993.1189. [DOI] [PubMed] [Google Scholar]
- 41.Salter DM, Godolphin JL, Gourlay MS. J Histochem Cytochem. 1995;43:447–457. doi: 10.1177/43.4.7897185. [DOI] [PubMed] [Google Scholar]
- 42.Ustoa Ma S, Garciadiego-Cazares D, Vargas C, Chimal-Monroy J, Diaz de Leon L. Ann N Y Acad Sci. 1998;857:241–244. doi: 10.1111/j.1749-6632.1998.tb10122.x. [DOI] [PubMed] [Google Scholar]
- 43.Hausler G, Helmreich M, Marlovits S, Egerbacher M. Calcif Tissue Int. 2002;71:212–218. doi: 10.1007/s00223-001-2083-x. [DOI] [PubMed] [Google Scholar]
- 44.Attur MG, Dave MN, Clancy RM, Patel IR, Abramson SB, Amin AR. J Immunol. 2000;164:2684–2691. doi: 10.4049/jimmunol.164.5.2684. [DOI] [PubMed] [Google Scholar]
- 45.Loeser RF. Biorheology. 2002;39:119–124. [PubMed] [Google Scholar]
- 46.Loeser RF. Arthritis Rheum. 1993;36:1103–1110. doi: 10.1002/art.1780360811. [DOI] [PubMed] [Google Scholar]
- 47.Salter DM, Hughes DE, Simpson R, Gardner DL. Br J Rheumatol. 1992;31:231–234. doi: 10.1093/rheumatology/31.4.231. [DOI] [PubMed] [Google Scholar]
- 48.Woods VL, Jr, Schreck PJ, Gesink DS, Pacheco HO, Amiel D, Akeson WH, Lotz M. Arthritis Rheum. 1994;37:537–544. doi: 10.1002/art.1780370414. [DOI] [PubMed] [Google Scholar]
- 49.Shakibaei M. Exp Cell Res. 1998;240:95–106. doi: 10.1006/excr.1998.3933. [DOI] [PubMed] [Google Scholar]
- 50.Salter DM, Millward-Sadler SJ, Nuki G, Wright MO. Clin Orthop. 2001;391(suppl):S49–S60. doi: 10.1097/00003086-200110001-00006. [DOI] [PubMed] [Google Scholar]
- 51.Millward-Sadler SJ, Wright MO, Lee H, Nishida K, Caldwell H, Nuki G, Salter DM. J Cell Biol. 1999;145:183–189. doi: 10.1083/jcb.145.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wright MO, Nishida K, Bavington C, Godolphin JL, Dunne E, Walmsley S, Jobanputra P, Nuki G, Salter DM. J Orthop Res. 1997;15:742–747. doi: 10.1002/jor.1100150517. [DOI] [PubMed] [Google Scholar]
- 53.Lee HS, Millward-Sadler SJ, Wright MO, Nuki G, Salter DM. J Bone Miner Res. 2000;15:1501–1509. doi: 10.1359/jbmr.2000.15.8.1501. [DOI] [PubMed] [Google Scholar]
- 54.Millward-Sadler SJ, Wright MO, Davies LW, Nuki G, Salter DM. Arthritis Rheum. 2000;43:2091–2099. doi: 10.1002/1529-0131(200009)43:9<2091::AID-ANR21>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 55.Salter DM, Millward-Sadler SJ, Nuki G, Wright MO. Biorheology. 2002;39:97–108. [PubMed] [Google Scholar]
- 56.Millward-Sadler SJ, Wright MO, Lee H, Caldwell H, Nuki G, Salter DM. Osteoarth Cartil. 2000;8:272–278. doi: 10.1053/joca.1999.0301. [DOI] [PubMed] [Google Scholar]
- 57.Hauser N, Geiss J, Neidhart M, Paulsson M, Hauselmann HJ. Acta Orthop Scand. 1995;66(Suppl 266):71–79. [PubMed] [Google Scholar]
- 58.Clemmensen I, Holund B, Johansen N, Andersen RB. Histochemistry. 1982;76:51–56. doi: 10.1007/BF00493284. [DOI] [PubMed] [Google Scholar]