

Abstract
Leucine-rich repeat transmembrane neuronal proteins (LRRTMs) were recently found to instruct presynaptic and mediate postsynaptic glutamatergic differentiation. In a candidate screen, here we identify neurexin-1β lacking an insert at splice site 4 (−S4) as a ligand for LRRTM2. Neurexins bind LRRTM2 with a similar affinity but distinct code from the code for binding neuroligin-1 (the predominant form of neuroligin-1 at glutamate synapses, containing the B splice site insert). Whereas neuroligin-1 binds to neurexins 1, 2, and 3 β but not α variants, regardless of insert at splice site 4, LRRTM2 binds to neurexins 1, 2, and 3 α and β variants specifically lacking an insert at splice site 4. We further show that this binding code is conserved in LRRTM1, the family member linked to schizophrenia and handedness, and that the code is functional in a coculture hemisynapse formation assay. Mutagenesis of LRRTM2 to prevent binding to neurexins abolishes presynaptic inducing activity of LRRTM2. Remarkably, mutagenesis of neurexins shows that the binding face on neurexin-1β (−S4) is highly overlapping for the structurally distinct LRRTM2 and neuroligin-1 partners. Finally, we explore here the interplay of neuroligin-1 and LRRTM2 in synapse regulation. In neuron cultures, LRRTM2 is more potent than neuroligin-1 in promoting synaptic differentiation, and, most importantly, these two families of neurexin-binding partners cooperate in an additive or synergistic manner. Thus, we propose a synaptic code hypothesis suggesting that neurexins are master regulators of the cooperative activities of LRRTMs and neuroligins.
Introduction
Synapse development requires recruitment and precise alignment of synaptic vesicles and release apparatus in axons opposite appropriate neurotransmitter receptors and postsynaptic machinery on dendrites. Presynaptic neurexins and partner postsynaptic neuroligins are perhaps the best known synaptic organizing cell adhesion molecules that control this process (Craig and Kang, 2007). Copy number, frameshift truncating, and function altering missense variants in neurexins and neuroligins are linked to autism, schizophrenia, and mental retardation (Sudhof, 2008), emphasizing the importance of these genes for human brain development. Mimicking these alterations in mice can phenocopy the psychiatric disorder (Jamain et al., 2008).
Different isoforms of neurexins and neuroligins selectively contribute to glutamatergic or GABAergic synapse development. There are three neurexin genes, each producing longer α-neurexins and shorter β-neurexins in multiple splice forms (Missler et al., 1998). Neuroligins bind the laminin neurexin sex hormone-binding protein (LNS) domain common to α-and β-neurexins, which contains the alternatively spliced site 4 (S4) (Boucard et al., 2005). The major glutamatergic neuroligin is neuroligin-1 containing an insert at its B splice site (+B); it binds neurexin-1β but not neurexin-1α (Song et al., 1999; Boucard et al., 2005; Chih et al., 2006; Graf et al., 2006). The major GABAergic neuroligin is neuroligin-2, which appears to bind all neurexins (Graf et al., 2004; Varoqueaux et al., 2004; Boucard et al., 2005). Mouse knock-out (KO) studies indicate some redundancy among neurexins and neuroligins. Individual α-neurexin KO mice survive, whereas triple α-neurexin KO is lethal because of defective synaptic transmission (Missler et al., 2003) (β-neurexin KO has not yet been reported). Similarly, while individual neuroligin-1 or neuroligin-2 KOs survive but exhibit selective defects in glutamatergic or GABAergic synaptic development, respectively, triple neuroligin KO is perinatal lethal because of defective synaptic transmission (Varoqueaux et al., 2006; Chubykin et al., 2007; Poulopoulos et al., 2009). However, triple neuroligin knock-outs still form synapses, and multiple lines of evidence indicate that neurexins and neuroligins are not the only glutamatergic synaptic-organizing proteins (Akins and Biederer, 2006; Dalva et al., 2007).
We recently performed an unbiased screen to characterize the set of surface or secreted synaptic-organizing proteins, screening over 105 clones for ability to trigger presynaptic differentiation when presented to axons of cultured neurons in a coculture hemisynapse formation assay (Linhoff et al., 2009). We reisolated neuroligins and netrin-G ligands (NGLs) (Woo et al., 2009) and isolated the novel gene LRRTM1. LRRTM1 was recently linked to both schizophrenia and handedness (Francks et al., 2007; Ludwig et al., 2009). There are four related leucine-rich repeat transmembrane neuronal protein (LRRTM) genes in mammals, and all are enriched in brain and expressed from postnatal day one or earlier (Lauren et al., 2003). LRRTM1 and LRRTM2 are most potent at inducing presynaptic differentiation. Furthermore, in rodents in vivo, endogenous LRRTM2 concentrates at multiple classes of glutamate synapses, and LRRTM1 contributes to synaptic organization with targeted deletion resulting in altered distribution of the vesicular glutamate transporter in hippocampus (Linhoff et al., 2009). Thus, like neuroligins, LRRTMs are potent synaptic organizing molecules that contribute functionally to specific synaptic circuits.
One aspect required to understand how LRRTMs contribute to synapse development is to identify their presynaptic ligands. Here we show that LRRTMs bind neurexins, at an overlapping face but with a different isoform code than neuroligins. We explore further how these differential interactions among neurexins, neuroligins, and LRRTMs cooperate to promote glutamatergic synapse development.
Materials and Methods
DNA constructs.
LRRTM1-cyan fluorescent protein (CFP), LRRTM2-CFP, LRRTM3-CFP, LRRTM4-CFP, N-Cadherin-CFP, Amigo-CFP, LINGO1-CFP, LRTM1-CFP, LRTM2-CFP, LRR3B-CFP, and TPBG-CFP were described previously (Linhoff et al., 2009). Nrx1α(+S4)-CFP, Nrx2α(+S4)-CFP, Nrx3α(+S4)-CFP, Nrx1β(+S4)-CFP, Nrx2β′(+S4)-CFP, Nrx3β′(+S4)-CFP, and Nrx1β(−S4)-CFP were described previously (Graf et al., 2004; Kang et al., 2008). Nrx1α(−S4)-CFP, Nrx2α(−S4)-CFP, Nrx3α(−S4)-CFP, Nrx2β′(−S4)-CFP, and Nrx3β′(−S4)-CFP were generated from the above by the overlap PCR method. Thy1 (accession number BC054436) was obtained in an expression vector from Open Biosystems. Neuroplastin (accession number BC070947), IGSF8 (accession number BC092192), Nectin 1 (accession number BC104948), ROBO1 (accession number BC157861), ROBO2 (accession number BC055333), ROBO4 (accession number BC071193), CHL1 (accession number BC131670), and Neurofascin (accession number BC137013) were obtained from Open Biosystems and were cloned into enhanced CFP (ECFP)-N1 vector. EphrinB1 (accession number BC006797) was obtained from Open Biosystems and cloned into a modified pCDNA3 vector that coexpresses CFP under the SV40 promoter (Linhoff et al., 2009). Several candidate expression plasmids were kind gifts: LAR-CFP from Dr. Eunjoon Kim (Korea Advanced Institute of Science and Technology, Daejeon, South Korea); contactin-green fluorescent protein (GFP) from Dr. Stephen G. Waxman (Yale University, New Haven, CT); Myc-SALM1, Myc-SALM2, SALM3, Myc-SALM4, and SALM5 from Dr. Robert Wenthold (National Institutes of Health, Bethesda, MD); neural cell adhesion molecule (NCAM)-140-GFP and NCAM-180-GFP from Dr. Brigitte Schmitz (University of Bonn, Bonn, Germany); CD166-CFP from Dr. A. M. Carmo (Universidade do Porto, Portugal); SDFR1-flag from Dr. Martin E. Hemler (Harvard Medical School, Boston, MA); CD81-GFP from Dr. Francisco Sánchez-Madrid (Universidad Autónoma de Madrid, Madrid, Spain); and NgCAM-yellow fluorescent protein (YFP) from Dr. Peter Sonderegger (University of Zurich, Zurich, Switzerland), which was cloned into Clontech ECFP-N1 vector to generate NgCAM-CFP. CASPR1-HA, CASPR2-HA, CASPR3TM, and CASPR4 were kind gifts from Dr. Elior Peles (Weizmann Institute of Science, Rehovot, Israel) and were cloned into the Clontech ECFP-N1 vector to generate CASPR1-CFP, CASPR2-CFP, CASPR3-CFP, and CASPR4-CFP. CALEB was amplified from a rat cDNA library (Linhoff et al., 2009) and cloned into Clontech pECFP-N1.
All neuroligin-1 clones used in this paper contain the B splice insert. pNICE-HA-NLG1 (splice variant A2+ B+) and Nrx1β(−S4)-Fc were kind gifts from Dr. Peter Scheiffele (University of Basel, Basel, Switzerland) (Scheiffele et al., 2000); the HA-NLG1 insert here was expressed from the cytomegalovirus (CMV) promoter in pcDNA3 vector. HA-LRRTM1, HA-LRRTM2, Myc-LRRTM1, and Myc-LRRTM2 contain the signal sequence from rat NMDAR2B, then the HA or Myc tag, and then the mature sequence of LRRTM1 or LRRTM2, and express from the CMV promoter in the Clontech EGFP-N1 vector. Nrx1β(+S4)-Fc and Nrx1β(ΔLNS)-Fc were described previously (Graf et al., 2006). Mutations Y198A, L204A, I206A, S209R, and N153A were generated in Nrx1β(−S4)-Fc by site-directed mutagenesis; all other mutants in Figure 6 were described previously (Graf et al., 2006). LRRTM2-alkaline phosphatase (AP) has been described previously (Linhoff et al., 2009). NLG1-AP was generated by cloning the extracellular region of HA-NLG1, including the signal sequence, the acetylcholinesterase domain, and the region of disorder before the transmembrane region into the pCDNA4-PLAP-Myc-His vector from Linhoff et al. (2009); Myc was deleted from NLG1-AP used in Figure 5. For expression in neurons for coculture assays, the coding regions for YFP-NLG1 (Graf et al., 2004), YFP-LRRTM1, and YFP-LRRTM2 (Linhoff et al., 2009) were cloned under the neuron-specific synapsin promoter in pLentiLox3.7 vector (kind gift from Dr. Alaa El-Husseini, University of British Columbia, Vancouver, BC, Canada) (Huang et al., 2009). mCherry-LRRTM2 consists of the rat NMDAR2B signal sequence, then mCherry, then mature LRRTM2 sequence also in the pLentiLox3.7 synapsin promoter vector. For overexpression studies in neurons, YFP-NLG1 and YFP-LRRTM2 were cloned into a vector with the CAG chicken β-actin promoter (kind gift from Dr. Gary Banker, Oregon Health Sciences University, Portland, OR) (Niwa et al., 1991; Kaech and Banker, 2006). The YFP in YFP-LRRTM2 was replaced by CFP to generate β-actin driven CFP-LRRTM2. Mutations in CFP-LRRTM2 (T162A,H164A) and (D260A,T262A) were generated by site-directed mutagenesis or overlapping PCR.
Figure 6.

The binding face on neurexin-1 β (−S4) for LRRTM2 strongly overlaps that for neuroligin-1. Twenty-one point mutants of Nrx1β(−S4)-CFP altered at one to five residues were expressed in COS7 cells, and binding of recombinant LRRTM2-AP or NLG1-AP was measured. A, Representative examples of Nrx1β(−S4) mutants showing binding to recombinant LRRTM2-AP or NLG1-AP. Most mutants affected binding of LRRTM2 and NLG1 to a similar extent, but S209R differentially reduced binding to NLG1-AP, and β4β5 differentially reduced binding to LRRTM2-AP. Scale bar, 10 μm. B, Quantitation of the binding data (ANOVA, p < 0.0001; n = 15). Results are expressed as mean ± SEM. Mutations in Nrx1β(−S4) reduced binding of LRRTM2 and NLG1 to a similar extent for S107A, R109A, D137A, N153A, D158A, N184A, R202A, Q203A, L204A, T205A, I206A, N208A, β2β3 loop, β6β7 loop, and β10β11 loop. Binding was unaffected for both LRRTM2 and NLG1 for T156A, Y198A, and β3β4 loop. Mutation of Nrx1β(−S4) β4β5 loop did not affect binding of NLG1-AP but reduced binding of LRRTM2-AP, whereas mutation S209R did not affect binding of LRRTM2-AP but reduced binding of NLG1-AP. C, Diagram of crystal structure of Nrx1β(−S4) (Protein Data Bank ID#3BIW) showing the common binding face for neuroligin-1 and LRRTM2. The left structure shows point mutations at specific residues (green), including those involved in coordinating calcium (blue) that reduced binding of both LRRTM2 and NLG1 to similar levels. Mutation S209R (red), however, reduced binding of NLG 1 but not of LRRTM2 to Nrx1β(−S4). The right structure shows mutations in loops (green) that reduced the binding of LRRTM2 and NLG1 to similar levels. Mutations in the β4β5 loop (red) reduced binding of LRRTM2 but not of NLG1 to Nrx1β(−S4).
Figure 5.

LRRTM2 binding to neurexin-1β(−S4) is calcium dependent and competitive with neuroligin-1 binding. A, Recombinant LRRTM2-AP bound to COS7 cells expressing Nrx1β(−S4)-CFP in normal buffer but not in calcium-free buffer containing 10 mm EGTA. Also see Figure 6 for lack of LRRTM2-AP binding to Nrx1β(−S4) with mutations at known calcium coordination residues, D137 and N208. B, Recombinant NLG1-AP containing only an HA tag for detection bound to Nrx1β(−S4)-CFP expressed in COS7 cells. Preincubation of Nrx1β(−S4)-expressing COS7 cells with LRRTM2-AP reduced subsequent binding of NLG1-AP. C, Recombinant LRRTM2-AP containing only an Myc tag for detection bound to Nrx1β(−S4)-CFP expressed in COS7 cells. Preincubation of Nrx1β(−S4)-expressing COS7 cells with NLG1-AP reduced subsequent binding of LRRTM2-AP. D, Quantitation of the experiments in B and C (t test, *p < 0.0001; n = 20). Results are expressed as mean ± SEM. Scale bars: 10 μm.
Cell culture.
Dissociated primary hippocampal neuron cultures were prepared from embryonic day 18 rat embryos essentially as described previously (Goslin et al., 1998; Kaech and Banker, 2006). Neurons were plated at a final density of 300,000 cells per dish on poly-l-lysine-coated coverslips in 60 mm culture dishes inverted over a feeder layer of glia. After 2 d, cytosine arabinoside (5 μm) was added to neuron cultures to prevent the overgrowth of glia. For both overexpression and fibroblast-neuron coculture assays, neurons were transfected with 1–4 μg of DNA at day in vitro (DIV) 0 using electroporation (AMAXA Biosystems) and seeded at a density of 500,000 to 1 million per 60 mm dish. Neurons used for overexpression studies were fixed at 10–12 DIV.
COS7 and human embryonic kidney 293T (HEK293T) cells were cultured in DMEM-H supplemented with 10% fetal bovine serum. HEK293T and COS7 cells were transfected using Fugene 6 (Roche). Cocultures of COS7 cell with neurons were performed as described previously (Graf et al., 2004). Essentially, COS7 cells were harvested by trypsinization 18 to 24 h after transfection, seeded onto neuron coverslips pregrown for 9–11 DIV, supplemented with 100 μm APV (Research Biochemicals) to limit toxicity, and fixed 20–24 h later.
Protein binding assays.
Expression of the AP tagged fusion proteins or human Fc tagged fusion proteins was performed by transient transfection in HEK293T cells. Supernatant collected from protein expressing cells was concentrated using Centricon Plus-70 ultrafiltration units (30 kDa cutoff; Millipore). AP fusion proteins also contained Myc (except NLG1-AP for Fig. 5), and His tags and were purified using Ni-NTA agarose eluting with 200 mm imidazole. Imidazole was removed by overnight dialysis (Spectrapor). Purified AP fusion proteins were quantitated by SDS-PAGE relative to a BSA standard curve using Sypro Ruby gel stain (Invitrogen), UV illumination, and a Bio-Rad gel documentation system. Fc-tagged fusion proteins were purified with protein A columns from an ImmunoPure IgG Purification Kit (Pierce) and concentrated in PBS with Centricon filters (Millipore). Purified Fc fusion proteins were immunoblotted, visualized by chemiluminiscence, and quantitated by densitometry relative to a human IgG standard curve.
For the ligand screen in Figure 1, candidate molecules were transfected into COS7 cells and were allowed to express for 36 h. Cells were incubated for 1 h with LRRTM2-AP, subjected to heat inactivation (1 h 65°C) of native alkaline phosphatases, and then fixed and treated with NBT/BCIP reagent (Roche) for 15–20 min. Bound protein was detected in a bright field microscope. To determine LRRTM2-AP or NLG1-AP binding to neurexin-CFP variants, transfected cells were incubated with the fusion proteins live for 1 h at room temperature followed by anti-Myc antibodies for 30 min. Binding assays were done in buffer: 168 mm NaCl, 2.6 mm KCl, 10 mm HEPES, pH 7.2, 2 mm CaCl2, 2 mm MgCl2, 10 mm d-glucose, and 100 μg/ml BSA. For Figure 5B, transfected cells were stained live with LRRTM2-AP(Myc) or buffer for 45 min, then with NLG1-AP(HA) for 45 min followed by HA antibody for 30 min. For Figure 5C, transfected cells were stained live with NLG1-AP(HA) or buffer for 45 min, then with LRRTM2-AP(Myc) for 45 min followed by Myc antibody for 30 min. Cells were then fixed in prewarmed 4% formaldehyde/4% sucrose, blocked in 10% BSA in PBS, and incubated with Alexa-568-conjugated anti-mouse IgG to detect bound Myc or HA.
Figure 1.

Candidate screening identifies neurexin-1 β (−S4) as a ligand for LRRTM2. A, List of candidate molecules expressed in COS7 cells that were screened for binding of LRRTM2-AP ectodomain fusion protein. Only neurexin-1β(−S4) bound LRRTM2-AP. B, N-terminally HA-tagged LRRTM2, neuroligin 1(+B) (NLG1), or CD8 were expressed in COS7 cells and detected with surface anti-HA antibody. Cells were incubated with neurexin-1β ectodomain fused to human Fc (Nrx1β-Fc). Whereas HA-NLG1 readily bound both −S4 and +S4 variants, HA-LRRTM2 bound only −S4 and not +S4 splice variants or Nrx1β lacking the LNS domain (ΔLNS). The HA-CD8 negative control shows specificity of binding. Scale bar, 10 μm. C, D, Binding affinities of Nrx1β (−S4) and (+S4) Fc proteins to HA-LRRTM2 (C) and HA-NLG1 (D). Different concentrations of Nrx1β-Fc proteins were incubated with COS7 cells expressing HA-LRRTM2 or HA-NLG1. The means of bound Nrx/surface-expressed HA were plotted using nonlinear regression fit.
To determine Nrx1β (+S4), (−S4), and (ΔLNS) Fc fusion binding to HA-LRRTM2, HA-CD8, HA-NLG1, Myc-LRRTM1, Myc-LRRTM2, Myc-SALM2, and CFP-LRRTM2, the Fc fusion proteins were coincubated with HA, Myc, or GFP antibodies (1:500 for all) for 30 min at 4°C. Cells were then fixed in prewarmed 4% formaldehyde/4% sucrose, blocked in 10% BSA in PBS, and incubated with FITC-conjugated anti-human IgG to detect bound neurexin-Fc and with Alexa-568-conjugated anti-mouse or anti-rabbit IgG to detect surface HA, Myc, or CFP.
Immunocytochemistry.
Neurons, COS7 cells, and cocultures were fixed for 12–15 min with warm 4% formaldehyde and 4% sucrose in PBS, pH 7.4, followed by permeabilization with PBST (PBS plus 0.25% Triton X-100). Fixed and permeabilized cultures were blocked in 10% BSA in PBS for 30 min at 37°C and primary antibodies applied in 3% BSA in PBS. After overnight incubation at room temperature, the coverslips were washed with PBS and incubated in secondary antibodies in 3% BSA in PBS for 1 h at 37°C. The coverslips were then washed and mounted in elvanol (Tris-HCl, glycerol, and polyvinyl alcohol, with 2% 1,4-diazabicyclo[2,2,2]octane).
The following primary antibodies were used: rabbit anti-synapsin I (1:2000; Millipore; AB1543P), mouse anti-PSD-95 family (IgG2a; 1:500; clone 6G6-1C9; Thermo Scientific), mouse anti-bassoon (IgG2A; 1:1000; Stressgen; VAM-PS003), mouse anti-synaptophysin (IgG1; 1:1000; BD Biosciences; 611880). For labeling dendrites, we used anti-MAP2 (chicken polyclonal IgY; 1:2000; Abcam; ab5392). To determine surface localization of N-terminally Myc-tagged, HA-tagged, or CFP-tagged proteins, live staining was performed using mouse anti-Myc (9E10; IgG1; 1:500; Millipore), mouse anti-HA (IgG2b, 1:500; Roche), or rabbit anti-GFP (A11122; Invitrogen). Labeled secondary antibodies used were raised in goat against the appropriate species and monoclonal isotype, highly cross-adsorbed, and conjugated to Alexa-488, Alexa-568, and Alexa-647 dyes (1:500; Invitrogen). To visualize dendrites, we used AMCA (7-amino-4methylcoumarin-3-acetic acid)-conjugated anti-chicken IgY (donkey IgG; 1:200; Jackson ImmunoResearch; 703-155-155). Recombinant proteins tagged with human IgG Fc domain were stained with FITC-conjugated donkey anti-human IgG (L+H) (1:150 in live staining; Jackson ImmunoResearch).
Image analysis.
Images were acquired on a Zeiss Axioplan2 or Zeiss Axioskop microscope with a 63× 1.4 numerical aperture oil objective and Photometrics Sensys cooled CCD camera using MetaMorph imaging software (Molecular Devices) and customized filter sets. Images were acquired as grayscale and prepared for presentation using Adobe Photoshop (Adobe Systems). For quantitation, experimental sets were fixed and stained simultaneously and imaged with identical settings. All imaging and analysis were done blind, choosing similar fields among conditions by phase contrast and the transfected protein channel. Analysis was performed using MetaMorph (Molecular Devices), Kaleidagraph (Synergy Software), GraphPad Prism, and InStat3 Graph (GraphPad Software).
To determine the binding affinity of soluble proteins to surface-expressed proteins, regions were generated around the exact perimeter of each cell, and the average gray values of bound protein and expressed protein were measured within the region. The average gray value of the off-cell background was subtracted, and the corrected average gray values of bound protein and expressed protein were measured. For coculture scoring, coverslips were blinded and contacts between expressing neurons and CFP-positive COS7 cells counted. Clustering of the tagged protein in dendrites contacting expressing COS7 cells was counted as positive, whereas no clustering was counted as negative. To determine PSD-95 coclustering by YFP-LRRTM2 or YFP-NLG1 expressed in neurons at contacts with neurexin expressing COS7 cells, regions around the clustered YFP were generated and total area and average gray value, subtracted for off-cell background, were measured for YFP and PSD-95. To determine effects of YFP-LRRTM2, YFP-NLG1, and YFP-NLG 1 plus CFP-LRRTM2 expression in neurons, expressing neurons were chosen by YFP expression alone. Fields were also chosen to include expressing and nonexpressing dendrites with equivalent MAP2 staining. Regions around dendrites were drawn ensuring only single dendrites were chosen, and total area and total gray values corrected for off-cell background were measured for YFP, synaptophysin, and bassoon. Linescan was used to measure the length of the dendrites. Results are expressed as a ratio of expressing to nonexpressing dendrites.
Results
LRRTM2 binds α and β neurexins 1, 2, and 3 lacking the splice site 4 insert
To identify the trans-synaptic binding partner of LRRTMs, we generated a soluble ectodomain fusion protein with alkaline phosphatase (LRRTM2-AP) and assayed for binding to a series of candidate proteins expressed on COS7 cells (Fig. 1A). Candidates were chosen mainly on the basis of reported presynaptic or axonal surface localization and domain structure. Of 46 candidates tested, a single surprising positive result was found: LRRTM2-AP bound neurexin-1β(−S4). We then directly compared the binding of a neurexin ectodomain IgG Fc fusion protein [Nrx1β(−S4)-Fc] to HA-LRRTM2 with its previously identified major binding partner HA-neuroligin-1(+B). Neurexin-1β(−S4) interacted with both partners with an apparent high affinity in these assays of dimeric ligand binding to cell surface-expressed receptor [Nrx1β(−S4)-Fc to LRRTM2, Kd = 19.9 nm; Nrx1β(−S4)-Fc to NLG1, Kd = 9.6 nm] (Figure 1B–D). Whereas addition of the insert at splice site 4 reduced the affinity of neurexin for neuroligin-1 by 2.8-fold [Nrx1β(+S4)-Fc to NLG1, Kd = 26.9 nm], it completely abolished binding to LRRTM2 [no detectable binding of Nrx1β(+S4)-Fc to LRRTM2]. The LNS domain of neurexin-1β mediates binding to LRRTM2 (Fig. 1B) as it does for neuroligins.
We then compared the full set of 12 neurexins, 1, 2, and 3, α or β and −S4 or +S4, to determine the binding codes for LRRTM2 and NLG1 (Fig. 2) (neurexin-2β′ and neurexin-3β′ contain the functionally important LNS domain from 2β and 3β with the regions flanking the LNS domain derived from 1β). Each neurexin-CFP was expressed in COS cells, and binding of soluble LRRTM2-AP or NLG1-AP was assessed. LRRTM2 bound to all α and β-neurexins specifically lacking the splice site 4 insert. In contrast, NLG1 bound to all β-neurexins but not α-neurexins, regardless of splice site 4 insert, consistent with previous data on binding to neurexin-1 (Boucard et al., 2005; Chih et al., 2006; Graf et al., 2006). Thus, neurexins bind neuroligin-1 and LRRTM2 with a different code: β-neurexins(−S4) bind both neuroligin-1 and LRRTM2, β-neurexins(+S4) bind only neuroligin-1, α-neurexins(−S4) bind only LRRTM2, and α-neurexins(+S4) bind neither.
Figure 2.

LRRTM2 and neuroligin-1 bind neurexins with distinct binding codes. A, B, C-terminal CFP fusions of neurexins 1β, 2β′, 3β′, 1α, 2α, and 3α, (+S4) or (−S4), were expressed in COS7 cells and visualized by CFP fluorescence. Cells were incubated with ectodomain fusions. LRRTM2-AP (A) bound specifically to all (−S4) neurexin isoforms. NLG1-AP (B) bound to all β isoforms of neurexins, regardless of S4 variant, and did not bind to any α isoforms. C, Quantitation of the data in A, normalized to Nrxβ(−S4)-CFP (ANOVA, p < 0.0001; n = 60 cells each; Tukey's post hoc test, *p < 0.001 compared to control). Data for neurexins 1, 2, and 3 were pooled. D, Quantitation of the data in B, normalized to Nrxβ(−S4)-CFP [ANOVA, p < 0.0001; n = 30 cells each; Tukey's post hoc test, *p < 0.001 compared to control; p < 0.01 between Nrxβ (−S4) and (+S4)]. Scale bars: 10 μm. Results are expressed as mean ± SEM. Data for neurexins 1, 2, and 3 were pooled.
Neurexin isoforms differentially recruit LRRTM2 or neuroligin-1 in neuron coculture
We next determined whether these differential binding codes operate functionally in a COS7 cell and neuron coculture hemisynaptogenesis or artificial synapse formation assay (Biederer and Scheiffele, 2007). We expressed YFP-tagged LRRTM2 or neuroligin-1 in cultured hippocampal neurons at low expression levels such that the tagged proteins clustered at glutamate postsynaptic sites with low diffuse levels in dendrites (although under identical expression conditions, YFP-LRRTM2 clustered more strongly, whereas YFP-NLG1 showed higher diffuse dendrite levels) (Graf et al., 2004; Linhoff et al., 2009). Neurons were then cocultured with COS7 cells expressing neurexin-CFP, again all 12 major neurexin variants, neurexin-1, -2, and -3, α or β, and −S4 or +S4 (Fig. 3). Nrx1β, Nrx2β′, Nrx3β′, Nrx1α, Nrx2α, or Nrx3α (−S4) but not (+S4) induced clustering of LRRTM2 in dendrites at contacts with the expressing COS7 cells. Unlike bona fide synapses, these induced hemisynapses lack presynaptic components such as synapsin I. In contrast, Nrx1β, Nrx2β′, and Nrx3β′ (−S4) or (+S4), but not Nrx1α, Nrx2α, or Nrx3α (−S4) or (+S4) induced clustering of NLG1 in dendrites at contacts with the expressing COS7 cells (in agreement with Kang et al. (2008) as reported previously for the +S4 isoforms). Thus, the code for coculture recruitment to hemi-postsynaptic sites was the same as for binding: β-neurexins(−S4) recruited both neuroligin-1 and LRRTM2, β-neurexins(+S4) recruited only neuroligin-1, α-neurexins(−S4) recruited only LRRTM2, and α-neurexins(+S4) recruited neither.
Figure 3.

Hemis ynaptogenesis assay reveals distinct functional codes for neurexins acting on LRRTM2 or neuroligin-1. The coculture assay was modified such that YFP-LRRTM2 or YFP-NLG1(+B) were expressed in hippocampal neurons, and these were cocultured with COS7 cells expressing CFP-tagged neurexins 1β, 2β′, 3β′, 1α, 2α, and 3α, (−S4) or (+S4). Cocultures were immunostained for PSD-95 and synapsin I. A, Illustration of the assay showing YFP-LRRTM2 expressed in neurons clustering at contacts with COS7 cells expressing Nrx1β(−S4)-CFP. PSD-95 coclusters at these sites. The absence of synapsin I at these clusters indicates the absence of native synapses. B, Nrx1β(−S4) on COS7 cells clusters both YFP-LRRTM2 and YFP-NLG1 in neurons, Nrx-1β(+S4) clusters only YFP-NLG1, Nrx1α(−S4) clusters only YFP-LRRTM2, and Nrx 1α(+S4) does not cluster either of them. C, Table showing the clustering code for YFP-LRRTM2 and YFP-NLG1 with all 12 neurexin variants studied. Whereas YFP-LRRTM2 in neurons clustered at contacts with COS7 cells expressing all (−S4) neurexin variants, YFP-NLG1 clustered neurexin β variants and not α variants. (+++++, 90–95% contact sites were positive; +++, 60–80% contact sites were positive; −, 0–5% contact sites were positive; n > 30 contact sites). Scale bars: 10 μm.
PSD-95 was corecruited along with either LRRTM2 or neuroligin-1 (Fig. 3A). Interestingly, α-neurexins(−S4) recruited PSD-95 in neurons expressing low levels of YFP-LRRTM2 but not in neurons expressing YFP-NLG1 or nontransfected neurons (data not shown). Thus, endogenous levels of LRRTMs in hippocampal cultures are not sufficient to mediate such recruitment, consistent with previous data also indicating low expression levels in culture (Linhoff et al., 2009). This finding also shows that α-neurexins(−S4) can induce glutamatergic postsynaptic differentiation only through LRRTMs and not through neuroligins. PSD-95 was not recruited by α-neurexins(+S4), which bind neither LRRTM2 nor neuroligin-1, but can bind other neuroligins and recruit neuroligin-2 and gephyrin (Boucard et al., 2005; Kang et al., 2008).
LRRTM1 also binds and is recruited by α-and β-neurexins lacking the splice site 4 insert
Interest in LRRTMs stems in part from recent evidence for maternal imprinting and linkage of paternally derived LRRTM1 to schizophrenia and handedness (Francks et al., 2007; Ludwig et al., 2009). Furthermore, LRRTM1 and LRRTM2 both target to glutamate postsynaptic sites and have equally potent synapse-promoting activity in coculture, more so than LRRTM3 or LRRTM4 (Linhoff et al., 2009). Thus, we tested whether LRRTM1, like LRRTM2, binds neurexins. Indeed, Myc-LRRTM1, like Myc-LRRTM2, bound Nrx1β(−S4)-Fc but not Nrx1β(+S4)-Fc (Fig. 4 A, B). Binding was specific, as shown by the absence of Nrx1β(−S4)-Fc binding to Myc-SALM2. As for LRRTM2 and neuroligins, binding to LRRTM1 required the neurexin LNS domain.
Figure 4.

LRRTM1, the family member linked to schizophrenia, also binds and is recruited by (−S4) neurexins. A, Recombinant Nrx1β(−S4)-Fc, Nrx1β(+S4)-Fc, and Nrx1β(ΔLNS)-Fc were incubated with COS7 cells transfected with Myc-LRRTM1, Myc-LRRTM2, or Myc-SALM2. Both Myc-LRRTM1 and Myc-LRRTM2, detected by surface staining for Myc, bound Nrx1β(−S4)-Fc but not Nrx1β(+S4)-Fc or Nrx1β(ΔLNS)-Fc. Myc-SALM2 was the negative control for binding. Scale bar, 10 μm. B, Quantitation of the data in A (ANOVA, p < 0.0001; Tukey's post hoc test, *p < 0.001 compared to Myc-SALM2; n = 20; results are expressed as mean ± SEM). C, Hemisynaptogenesis assay as described in Figure 3. COS7 cells expressing Nrx1β(−S4)-CFP or Nrx1α(−S4)-CFP but not Nrx1β(+S4)-CFP or Nrx1α(+S4)-CFP clustered YFP-LRRTM1 expressed in neurons. Scale bar, 10 μm. D, The assay in C was also performed for +S4 and −S4 variants of neurexins 2β′, 3β′, 2α, and 3α. All (−S4) neurexin variants clustered YFP-LRRTM1 expressed in neurons. These clusters contained PSD-95 but not synapsin. (++++, 80–95% contact sites were positive; −, 0–5% contact sites were positive; n > 30 contact sites).
We next performed a neuron coculture hemisynapse recruitment assay for LRRTM1 with all 12 major neurexin variants, neurexin-1, -2, and -3, α or β, and −S4 or +S4. YFP-LRRTM1 expressed at low levels in neurons normally clusters at glutamatergic postsynaptic sites (Linhoff et al., 2009) but was recruited to dendrite contacts with COS7 cells expressing CFP-tagged α-or β-neurexin −S4 but not +S4 variants (Fig. 4C, D). PSD-95 was also corecruited along with LRRTM1 (data not shown). LRRTM1 traffics poorly to the cell surface in nonneuronal cells (Francks et al., 2007; Linhoff et al., 2009). Thus, we were unable to generate LRRTM1-AP ectodomain fusion protein for direct binding assays to all 12 neurexin variants (as we did for LRRTM2 in Fig. 2), but based on these coculture recruitment results, we predict that the binding code for LRRTM1 is the same as for LRRTM2, binding to all α and β −S4 but not +S4 neurexin variants.
LRRTM binding to neurexin is calcium dependent and competitive with neuroligin
Neurexin interaction with neuroligins is calcium dependent (Sudhof, 2008). The calcium ion at the interface is coordinated mainly by interactions with neurexin including side chain oxygens from D137 and N208 of neurexin-1β(−S4) (Fabrichny et al., 2007; Chen et al., 2008). We show here that neurexin interaction with LRRTM2 is also calcium dependent (Fig. 5A). LRRTM2-AP did not bind Nrx1β(−S4) in calcium-free buffer containing the calcium chelator EGTA [bound LRRTM2-AP intensity was reduced to 0.6 ± 3% of control binding in nominally calcium free conditions; t test, p < 0.0001; n = 20].
We next determined whether neurexin can simultaneously bind neuroligin-1 and LRRTM2 or whether binding is competitive. COS7 cells expressing Nrx1β(−S4)-CFP were first incubated in control buffer or with LRRTM2-AP, and subsequently, binding of NLG1-AP was assessed (in these experiments, NLG1-AP bears only an HA tag, and LRRTM2-AP bears only a Myc tag, allowing for separate detection). Preincubation with LRRTM2-AP significantly reduced the binding of NLG1-AP to Nrx1β(−S4) (Fig. 5B,D). Likewise, preincubation with NLG1-AP significantly reduced the binding of LRRTM2-AP (Fig. 5C,D), suggesting that neuroligins and LRRTMs bind to neurexins in a highly competitive manner.
LRRTM2 and neuroligin-1 interact with a highly overlapping surface of neurexin
We performed an extensive structure-function analysis of neurexin-1β(−S4) to compare residues required for interaction with LRRTM2 to those required for interaction with neuroligin-1 (Fig. 6). A series of 21 Nrx1β(−S4)-CFP mutants were expressed in COS7 cells and assessed quantitatively for binding of LRRTM2-AP or NLG1-AP. The neurexin-1β LNS domain is composed of two seven-stranded β sheets forming a jelly roll fold. Residues in the loops on one end of the β sandwich interact with neuroligins (Graf et al., 2006; Arac et al., 2007; Fabrichny et al., 2007; Chen et al., 2008). Here we find that mutations of three to five residues to Ala in loops β2β3 (107–109 STR), β6β7 (154–157 VGTD), and β10β11 (202–205 RQLT) of Nrx1β(−S4) disrupted binding to neuroligin-1, as expected (Graf et al., 2006), and also disrupted binding to LRRTM2 (Fig. 6). Mutations in loops β3β4 (119–122 TVQK) or β11β12 (218–222 KEQGQ) on the other side of the β sandwich did not disrupt binding to either neuroligin-1 or LRRTM2. Thus, LRRTM2 binds to the same face of neurexin as neuroligins. Interestingly, mutations in loop β4β5 (132–135 SSGL) disrupted binding to LRRTM2 but not to neuroligin-1, indicating some difference in the residues required for interaction.
We thus assayed single residue changes to Ala to assess how much overlap there is in the neurexin-1β(−S4) surface required for interacting with LRRTM2 compared with neuroligin-1 (Fig. 6). As expected based on the calcium dependence of interaction demonstrated above, Nrx1β(−S4) with mutations in key calcium coordinating side chains D137A or N208A bound neither LRRTM2 nor neuroligin-1 (numbering here refers to Nrx lacking the 30 residue S4 insert; thus, N208 here is referred to as N238 in some papers). Remarkably, 11 of the other 12 point mutations located within the β2β3, β6β7, β8β9, or β10β11 loops comprising the binding surface affected binding of LRRTM2 and neuroligin-1 in parallel. For example, Nrx1β(−S4) Y198A maintained wild-type binding to both partners, N184A reduced by approximately one-half, D158A reduced by approximately three-quarters, and R109A and L204A reduced to background levels binding to both partners. One exception was S209R in the β10β11 loop. As predicted from structural characterization (Fabrichny et al., 2007; Chen et al., 2008), S209R reduced the interaction between neurexin-1β(−S4) and neuroligin-1; however, S209R did not reduce interaction with LRRTM2. Thus, whereas the S209R and β4β5 loop mutations differentially affected binding to LRRTM2 or neuroligin-1, 19 of the 21 neurexin-1β mutants assayed affected binding to both partners in parallel, indicating a conserved mechanism of interaction despite the structurally disparate natures of LRRTMs and neuroligins.
LRRTM2 mutation that blocks binding to neurexin abolishes presynaptic inducing activity
Our data suggest that LRRTM2 triggers presynaptic differentiation via binding to neurexins on axons. A prediction of this idea is that point mutations in LRRTM2 that abolish binding to neurexins should abolish the ability of LRRTM2 to induce presynaptic differentiation in the coculture hemisynaptogenesis assay. To test this prediction, a structural model of LRRTM2 was generated based on the structure of the related leucine-rich repeat (LRR) protein Lingo-1 (Mosyak et al., 2006). Residues on the predicted concave face of the LRR domain were chosen for mutagenesis, in part based on the interactions of Listeria LRR protein internalin with E-cadherin (Schubert et al., 2002). Among the mutants generated, one abolished binding to neurexins, LRRTM2-D260A,T262A (Fig. 7 A, B). As predicted, this mutant was not able to induce presynaptic differentiation when presented to axons of cultured hippocampal neurons (Fig. 7C). Also consistent with the prediction, LRRTM2-T162A,H164A retained the ability to bind neurexin and retained the ability to trigger presynaptic differentiation. Thus, it is likely that LRRTMs signal through presynaptic neurexins.
Figure 7.
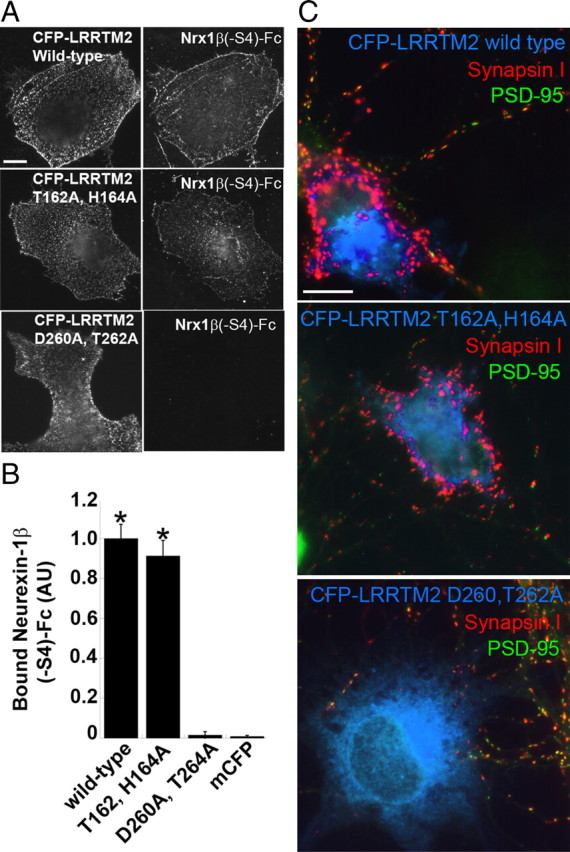
Mutation in LRRTM2 that prevents binding to neurexin abolishes its synaptogenic activity. A, Wild-type and point-mutation variants of LRRTM2 tagged at the N terminus with CFP to detect surface expression were expressed in COS7 cells and incubated with Nrx1β(−S4)-Fc. Nrx1β(−S4)-Fc had the same binding affinity to CFP-LRRTM2 T162A,H164A as to wild-type CFP-LRRTM2. However, Nrx1β(−S4)-Fc binding to D260A,T262A was abolished. B, Quantitation of the data in A [ANOVA, p < 0.0001; Tukey's post hoc test, *p < 0.001 compared to mCFP, (membrane associated CFP) n = 20]. Results are expressed as mean ± SEM. C, Coculture hemisynapse formation assay. COS7 cells expressing CFP-LRRTM2 wild-type and point-mutation variants were cocultured with hippocampal neurons. Wild-type CFP-LRRTM2 and CFP-LRRTM2 T162A,H164A induced clusters of synapsin I devoid of PSD-95 in contacting axons. CFP-LRRTM2 D260A,T262A expressing COS7 cells did not cluster synapsin I at axon contacts. Scale bars: 10 μm.
LRRTM2 and neuroligin-1 cooperate in synapse development
Given the mutually exclusive binding of LRRTMs and neuroligins to different subsets of neurexins, we began to explore how these multiple binding partners interact in synapse development. To test whether LRRTM2 and neuroligin-1 are recruited in separate domains or corecruited by a neurexin isoform that binds both partners, we expressed mCherry-LRRTM2 and YFP-NLG1 in neurons and cocultured these with COS7 cells expressing Nrx1β(−S4)-CFP (Fig. 8 A). Nrx1β(−S4) recruited both partners into a completely overlapping dendrite region comprising the induced hemi-postsynaptic site. LRRTMs and neuroligins could potentially link within the postsynaptic site via binding to a common intracellular partner, multimeric PSD-95 and related MAGUKs (Irie et al., 1997; Linhoff et al., 2009). To determine whether recruitment of LRRTM2 is sufficient to induce corecruitment of neuroligin-1, or vice versa, we cocultured the coexpressing neurons with COS7 cells expressing neurexin isoforms that bind only LRRTM2 or only neuroligin-1 (Fig. 8A). Nrx1β(+S4)-CFP that binds neuroligin-1 but not LRRTM2 recruited YFP-NLG1 but not mCherry-LRRTM2 to dendritic hemi-postsynaptic sites. Nrx1α(−S4)-CFP that binds LRRTM2 but not neuroligin-1 recruited mCherry-LRRTM2 but not YFP-NLG1 to dendritic hemi-postsynaptic sites. Thus, although different outcomes might be possible depending on the precise stoichiometry of all interacting partners, recruitment of LRRTM2 is not necessarily sufficient to recruit neuroligin-1 to a postsynaptic site, or vice versa.
Figure 8.

LRRTM2 and neuroligin-1 cooperate in excitatory synapse development. A, Coculture hemisynapse formation assay as described in Figure 3. mCherry-LRRTM2 and YFP-NLG1 were coexpressed in neurons, and these were cocultured with COS7 cells expressing neurexins. Nrx1β(−S4)-CFP in COS7 cells clustered both mCherry-LRRTM2 and YFP-NLG1, which were almost completely coclustered. In contrast, Nrx1β(+S4)-CFP clustered only YFP-NLG1, whereas COS7 cells expressing Nrx1α(−S4) clustered only mCherry-LRRTM2. B, LRRTM2 recruits PSD-95 more effectively than NLG1 at sites of contact between neurons expressing YFP-LRRTM2 or YFP-NLG1 and COS7 cells expressing Nrx1β(−S4). Similar results were obtained for (−S4) variants of neurexin-2β′ and neurexin-3β′. C, Quantitation of the data in B. In YFP-LRRTM2 or YFP-NLG1 clusters, the intensity ratio of PSD-95 to YFP was significantly higher for YFP-LRRTM2 than for YFP-NLG1. Measurements from neurexin-1β, neurexin-2β′, and neurexin-3β′ (−S4) variants were pooled (t test, p = 0.0005; n = 16 for YFP-LRRTM2; n = 19 for YFP-NLG1). D, Overexpression of YFP-LRRTM2 or YFP-NLG1 at similar levels in neurons increased both bassoon and synaptophysin clustering onto the transfected dendrites (arrows) compared to neighbor nontransfected dendrites (arrowheads). Coexpression of YFP-NLG1 and CFP-LRRTM2 appeared to have an additive or synergistic effect. E, Quantitation of the data in D. CFP-LRRTM2 and YFP-NLG1, when coexpressed, significantly increased apposed bassoon and synaptophysin intensities compared to expression of either YFP-NLG1 or YFP-LRRTM2 alone (ANOVA, p < 0.0001 for both Bassoon and Synaptophysin; Tukey's post hoc test, *p < 0.05; ***p < 0.001; n = 20). Results are expressed as mean ± SEM. Scale bars: 10 μm.
PSD-95 and other postsynaptic components are recruited along with LRRTM2 or neuroligin-1 to these induced hemi-postsynaptic sites. We assessed recruitment of endogenous PSD-95 and related MAGUKs (the antibody recognizes PSD-95, PSD-93, SAP102, and SAP97) at hemisynaptic sites induced by Nrx1β(−S4)-CFP contacting dendrites expressing YFP-LRRTM2 or YFP-NLG1 (Fig. 8B). The intensity of PSD-95 family proteins within the area of induced clusters was greater for YFP-LRRTM2 (PSD-95 average gray value, 552 ± 80; n = 16) than for YFP-NLG1 (PSD-95 average gray value, 176 ± 36; n = 19; t test, p < 0.0001). Furthermore, even normalizing to YFP intensity to control for differential recruitment of the two cell surface proteins, PSD-95 family proteins were more strongly recruited per YFP-LRRTM2 than per YFP-NLG1 (Fig. 8C). These results suggest that LRRTM2 may be more effective than neuroligin-1 at recruiting a glutamatergic postsynaptic scaffold.
Finally, to compare the efficacy and potential cooperation of LRRTM2 and neuroligin-1 for controlling synaptic differentiation in a more native system, we expressed these proteins in pure hippocampal neuron cultures (Fig. 8D,E). As reported previously, enhanced expression of either LRRTM2 or neuroligin-1 increased clustering of presynaptic antigens onto transfected dendrites (Prange et al., 2004; Linhoff et al., 2009). We report here that YFP-LRRTM2 was more effective at enhancing apposed clustering of both the synaptic vesicle component synaptophysin and the active zone component bassoon compared with YFP-NGL1 expressed from the same vector under the same conditions. Perhaps more importantly, coexpression of YFP-NLG1 with CFP-LRRTM2 in neurons had an additive or synergistic effect, resulting in significantly enhanced apposed clustering of synatophysin and bassoon relative to expression of YFP-NLG1 or YFP-LRRTM2 alone. Thus, LRRTM2 and neuroligin-1 can cooperate to promote glutamatergic synaptic development.
Discussion
We show here that LRRTM1 and LRRTM2 bind neurexins with a similar affinity but different binding code than neuroligin-1(+B). Whereas neuroligin-1 binds to neurexin-1, -2, and -3 β but not α variants regardless of insert at splice site 4, LRRTM2 binds to neurexin-1, -2, and -3 α and β variants specifically lacking an insert at splice site 4. This differential binding code operates functionally for recruitment of postsynaptic partners by neurexins in a coculture hemisynaptogenesis assay. Binding of LRRTM2 to neurexin is calcium-dependent and mutually exclusive with binding of neuroligin-1. Of 21 point mutants in neurexin-1β(−S4), only two differentially affected binding to LRRTM2 and neuroligin-1, whereas 15 reduced binding in parallel. Thus, despite the disparate nature of the LRR domain of LRRTM2 and the acetylcholinesterase-homologous domain of neuroligin-1, these partners bind to a highly overlapping face of neurexin-1β. A point mutation in LRRTM2 that abolished binding to neurexin also abolished synaptogenic activity in coculture, supporting the idea that the presynaptic-inducing activity of LRRTM2 is mediated by neurexins. Finally, we show that LRRTM2 is more potent than neuroligin-1 in recruiting postsynaptic PSD-95 in coculture and recruiting presynaptic vesicles and active zone marker in neuron overexpression, and most importantly, that LRRTM2 and neuroligin-1 work cooperatively in promoting presynaptic differentiation. Thus, neurexin isoforms might be considered master regulators of cooperative synaptic organization, with an S4 code controlling LRRTM1 and LRRTM2 contribution and mainly an α/β code controlling neuroligin-1 contribution. While this manuscript was in preparation, de Wit et al. (2009) and Ko et al. (2009) also reported interaction of LRRTM2 with neurexins. We discuss below important differences between the studies.
Interactions of LRRTMs and neuroligins with neurexins
The reports of de Wit et al. (2009) and Ko et al. (2009) generated a controversy over which neurexin isoforms bind LRRTM2: the former reported that only neurexin-1, α and β, but not neurexin-2 or neurexin-3, α or β, bind LRRTM2, whereas the latter reported that neurexin-1 α and β, −S4 but not +S4, bind LRRTM2 (the latter did not test binding of specific neurexin-2 or -3 isoforms). Here we present direct evidence for the complete binding code: LRRTM2 binds neurexins 1, 2, and 3 α and β, −S4 but not +S4. We demonstrate these interactions both in a COS cell surface binding assay (Fig. 2) and in a coculture hemisynaptogenesis assay (Fig. 3). For neurexin-2 and neurexin-3, de Wit et al. (2009) assayed +S4 constructs from Kang et al. (2008) corresponding to Nrx2α(+S4)-CFP, Nrx3α(+S4)-CFP, Nrx2β′(+S4)-CFP [called 2α1β by de Wit et al. (2009)], and Nrx3β′(+S4)-CFP (called 3α1β), and hence these isoforms do not bind to LRRTM2. Here we demonstrate binding when the S4 insert is removed from these constructs. The conclusion that LRRTM2 binds specific neurexin-2 and -3 isoforms explains the presence of peptides derived from neurexin-2 and neurexin-3 in material bound to an LRRTM2 affinity column (de Wit et al., 2009; Ko et al., 2009). All groups reported apparent high-affinity binding of LRRTM2 to neurexin-1β(−S4): estimated Kd = 19.9 nm (Fig. 1); Kd = ∼7 nm (de Wit et al., 2009), and Kd = 5.8 nm (Ko et al., 2009).
We show further here that LRRTM1, the family member linked paternally to schizophrenia and handedness (Francks et al., 2007; Ludwig et al., 2009), also binds neurexins with a similar affinity and identical binding code as LRRTM2 (Fig. 4). In a similar cell surface binding assay, we could detect no binding of Nrx1β(−S4)-Fc or Nrx1β(+S4)-Fc to LRRTM3 (data not shown). In contrast, Ko et al. (2009) report binding of all four LRRTMs to Nrx1β(−S4) in a pull-down assay. However, considering also the very weak activity of LRRTM3 for inducing hemi-presynaptic differentiation in coculture, more than ninefold less than that of LRRTM1 or LRRTM2 and only 1.6-fold above that of N-cadherin negative control (Linhoff et al., 2009), it seems unlikely that LRRTM3 binds neurexins with high affinity. Potential interaction of LRRTM4 with neurexins has yet to be examined in depth.
We show here that the code for neurexin binding and recruitment of the major glutmatergic neuroligin is different than that for LRRTMs: neuroligin-1(+B) binds and recruits neurexins 1, 2 and 3 β but not α regardless of S4 insert (Figs. 2, 3). Other neuroligin isoforms bind and are recruited by neurexin-1, α and β, −S4 and +S4 (Boucard et al., 2005; Chih et al., 2006; Graf et al., 2006), and likely all neurexins. Thus, of the neurexin variants, only α-neurexins(+S4) cannot bind either of the glutamatergic postsynaptic partners LRRTM1 or LRRTM2, or neuroligin-1(+B). This is consistent with the previous finding that α-neurexins(+S4) induce only GABAergic postsynaptic differentiation and recruit neuroligin-2 in coculture (Kang et al., 2008).
Numerous identical residues on the neurexin-1β(−S4) LNS domain are required for interacting with both LRRTM2 and neuroligin-1, including S107, R109, D137, N153, R202, Q203, L204, T205, I206, and two calcium coordinating residues, D158 and N208 (Fig. 6). This overlap in binding surface explains the competitive binding of LRRTM2 or neuroligin-1 to neurexin as shown here (Fig. 5) and by coimmunoprecipitation (Ko et al., 2009). Most of these residues are directly involved in hydrogen bonding, electrostatic, or hydrophobic interactions with neuroligin (Arac et al., 2007; Fabrichny et al., 2007; Chen et al., 2008). Only 2 of the 21 neurexin mutants tested differentiated between the partners; S209 was important only for neuroligin interaction, and residues 132SSGL135 in the β4β5 loop were important only for LRRTM2 interaction (Fig. 6). It is remarkable that these independently evolved structurally disparate LRRTM and neuroligin partners interact with such a highly overlapping surface of neurexins. For LRRTM2, we have only begun to identify residues important for neurexin interaction, D260,T262 (Fig. 7) in the center of the predicted concave face of the LRR domain.
Physiological roles of LRRTMs, neuroligins, and neurexins
The existence of LRRTMs with similar bioactivity as neuroligins (Linhoff et al., 2009) and now having overlapping binding partners in the neurexin family may explain the unexpectedly subtle phenotype of neuroligin KO with respect to glutamate synapses. Neuroligin-1 KO reduced NMDA-but not AMPA-mediated transmission in hippocampal slice (Chubykin et al., 2007), and triple neuroligin KO resulted in normal glutamatergic synapses in hippocampal culture and greater defects in inhibitory than excitatory synapse function in brainstem slice (Varoqueaux et al., 2006). The individual LRRTM1 KO phenotype also appears subtle, with altered VGluT1 immunofluorescence in hippocampal subfields expressing the lowest levels of LRRTM2 (Linhoff et al., 2009). Lentiviral LRRTM2 RNA interference was reported to reduce AMPA-and NMDA-mediated transmission in dentate granule cells compared to GFP-infected cells or uninfected neighbors (de Wit et al., 2009), suggesting that LRRTM2 KO, once generated, may have a significant effect. Although LRRTM2 can induce GABAergic presynaptic differentiation (Linhoff et al., 2009), whether it has a physiological role at GABAergic synapses, as do neurexins and neuroligins, is not yet clear. An important next step will be to assess the functions of these molecules individually and in combination in vivo.
Combinatorial effects can be addressed to some degree in culture systems. We show here that LRRTM2 is more potent than neuroligin-1(+B) at recruiting postsynaptic PSD-95 and at recruiting presynaptic synaptophysin and bassoon (Fig. 8). Furthermore, these two neurexin binding partners, LRRTM2 and neuroligin-1 (+B), have additive or synergistic effects at promoting synaptic differentiation when overexpressed in cultured neurons (Fig. 8). Cooperativity may occur by two mechanisms: (i) recruitment of the common binding partner β-neurexins(−S4) by both LRRTM2 and neuroligin-1(+B) increasing its total level, and (ii) recruitment of selective partners, α-neurexins (−S4) by LRRTM2 and β-neurexins (+S4) by neuroligin-1(+B), perhaps bringing new properties to the synapse (Fig. 9). In particular, selective recruitment of α-neurexins by LRRTM1 and LRRTM2 may mediate the recruitment of presynaptic calcium channels. The extracellular domain of α-neurexins is essential for presynaptic N-and P/Q-type calcium channel function, an activity not shared by β-neurexins (Zhang et al., 2005). Within the postsynaptic side, PSD-95 and related MAGUKs can link neuroligins and LRRTMs, via PSD-95 multimerization and via binding of neuroligins to PDZ3 (Irie et al., 1997), and likely LRRTMs via the -ECEV to PDZ1 and PDZ2 (Linhoff et al., 2009).
Figure 9.

Model of LRRTM, neurexin, and neuroligin interactions at a glutamate synapse. Diagram of a glutamate synapse containing the major postsynaptic LRRTMs (LRRTM1 and LRRTM2) and neuroligin (NLG1+B), and presynaptic neurexins. β-Neurexins(−S4) bind either LRRTMs or NLG1(+B), β-neurexins(+S4) bind only NLG1(+B), and α-neurexins(−S4) bind only LRRTMs. It is not clear whether α-neurexins(+S4) (not shown) are also present; these bind neither LRRTMs nor NLG1(+B). α-Neurexins are necessary to recruit presynaptic calcium channels. LRRTMs and NLG1 bind PSD-95 and related MAGUK scaffolds via different PDZ (PSD-95/Discs large/zona occludens-1) domains.
It is not yet clear precisely how LRRTMs, neuroligins, and neurexins contribute to synaptic development (Brose, 2009). The bulk of evidence indicates that these interacting complexes function in recruiting proteins to synapses, but defining specifically which components are recruited when and how these affect synaptic function requires further study. In addition, LRRTM–neurexin and neuroligin–neurexin complexes may also function in synaptic partner selection, initial adhesion, synaptic stabilization, and/or structural maturation. Altering the stoichiometry among the postsynaptic scaffolding partners and these synaptic organizing proteins can alter the excitatory/inhibitory balance (Levinson et al., 2005). Glial-derived factors can also modulate function of these synaptic organizing proteins; for example, thrombospondin interacts with neuroligin-1 to accelerate aspects of synapse development (Xu et al., 2010).
The complexities of these interactions among LRRTMs, neurexins, and neuroligins, and the expression of numerous isoforms with overlapping distributions in the brain, may begin to explain how these genes contribute to multiple psychiatric disorders. Perhaps it is not surprising that these genes controlling composition and function of synapses in cortical circuits involved in complex cognitive processing are linked to psychiatric disorders (Sudhof, 2008). Variants in LRRTM1, neurexin-1, and neuroligins 1, 3, and 4 are associated with schizophrenia, autism spectrum disorders, and mental retardation (Jamain et al., 2003; Francks et al., 2007; Szatmari et al., 2007; Sudhof, 2008; Glessner et al., 2009). These include copy number variants, promoter variants, and de novo microdeletions, truncating mutations, and coding region mutations altering protein function, thus constituting strong evidence of contribution to the disease. We face a challenge to unravel the in situ contributions of individual isoforms in specific synapses and circuits to understand how LRRTMs, neurexins, and neuroligins contribute to brain function and dysfunction.
Footnotes
This work was supported by Canadian Institutes of Health Research Grant MOP84241, National Institutes of Health Grant MH70860, the Mind Foundation of British Columbia, a Michael Smith Foundation for Health Research Postdoctoral Fellowship (T.J.S.), a Bluma Tischler Postdoctoral Fellowship (Y.K.), and the Canada Research Chair program (A.M.C.). We thank Xiling Zhou for excellent assistance with experiments, Dr. Filip Van Petegem for advice on LRRTM2 mutagenesis and generating a structural model, and Drs. Ethan R. Graf and Michael W. Linhoff for numerous constructs.
References
- Akins MR, Biederer T. Cell-cell interactions in synaptogenesis. Curr Opin Neurobiol. 2006;16:83–89. doi: 10.1016/j.conb.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Arac D, Boucard AA, Ozkan E, Strop P, Newell E, Sudhof TC, Brunger AT. Structures of neuroligin-1 and the neuroligin-1/neurexin-1 beta complex reveal specific protein-protein and protein-Ca2+ interactions. Neuron. 2007;56:992–1003. doi: 10.1016/j.neuron.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Biederer T, Scheiffele P. Mixed-culture assays for analyzing neuronal synapse formation. Nat Protoc. 2007;2:670–676. doi: 10.1038/nprot.2007.92. [DOI] [PubMed] [Google Scholar]
- Boucard AA, Chubykin AA, Comoletti D, Taylor P, Sudhof TC. A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha-and beta-neurexins. Neuron. 2005;48:229–236. doi: 10.1016/j.neuron.2005.08.026. [DOI] [PubMed] [Google Scholar]
- Brose N. Synaptogenic proteins and synaptic organizers: “many hands make light work.”. Neuron. 2009;61:650–652. doi: 10.1016/j.neuron.2009.02.014. [DOI] [PubMed] [Google Scholar]
- Chen X, Liu H, Shim AH, Focia PJ, He X. Structural basis for synaptic adhesion mediated by neuroligin-neurexin interactions. Nat Struct Mol Biol. 2008;15:50–56. doi: 10.1038/nsmb1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih B, Gollan L, Scheiffele P. Alternative splicing controls selective trans-synaptic interactions of the neuroligin-neurexin complex. Neuron. 2006;51:171–178. doi: 10.1016/j.neuron.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Chubykin AA, Atasoy D, Etherton MR, Brose N, Kavalali ET, Gibson JR, Sudhof TC. Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron. 2007;54:919–931. doi: 10.1016/j.neuron.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AM, Kang Y. Neurexin-neuroligin signaling in synapse development. Curr Opin Neurobiol. 2007;17:43–52. doi: 10.1016/j.conb.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalva MB, McClelland AC, Kayser MS. Cell adhesion molecules: signalling functions at the synapse. Nat Rev Neurosci. 2007;8:206–220. doi: 10.1038/nrn2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit J, Sylwestrak E, O'Sullivan ML, Otto S, Tiglio K, Savas JN, Yates JR, 3rd, Comoletti D, Taylor P, Ghosh A. LRRTM2 interacts with Neurexin1 and regulates excitatory synapse formation. Neuron. 2009;64:799–806. doi: 10.1016/j.neuron.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrichny IP, Leone P, Sulzenbacher G, Comoletti D, Miller MT, Taylor P, Bourne Y, Marchot P. Structural analysis of the synaptic protein neuroligin and its beta-neurexin complex: determinants for folding and cell adhesion. Neuron. 2007;56:979–991. doi: 10.1016/j.neuron.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francks C, Maegawa S, Laurén J, Abrahams BS, Velayos-Baeza A, Medland SE, Colella S, Groszer M, McAuley EZ, Caffrey TM, Timmusk T, Pruunsild P, Koppel I, Lind PA, Matsumoto-Itaba N, Nicod J, Xiong L, Joober R, Enard W, Krinsky B, et al. LRRTM1 on chromosome 2p12 is a maternally suppressed gene that is associated paternally with handedness and schizophrenia. Mol Psychiatry. 2007;12:1129–1139. doi: 10.1038/sj.mp.4002053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, Imielinski M, Frackelton EC, Reichert J, Crawford EL, Munson J, Sleiman PM, Chiavacci R, Annaiah K, Thomas K, Hou C, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslin K, Asmussen H, Banker G. Rat Hippocampal neurons in low-density culture. In: Banker G, Goslin K, editors. Culturing nerve cells. Ed 2. Cambridge, MA: MIT; 1998. pp. 339–370. [Google Scholar]
- Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 2004;119:1013–1026. doi: 10.1016/j.cell.2004.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf ER, Kang Y, Hauner AM, Craig AM. Structure function and splice site analysis of the synaptogenic activity of the neurexin-1 beta LNS domain. J Neurosci. 2006;26:4256–4265. doi: 10.1523/JNEUROSCI.1253-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K, Sanders S, Singaraja R, Orban P, Cijsouw T, Arstikaitis P, Yanai A, Hayden MR, El-Husseini A. Neuronal palmitoyl acyl transferases exhibit distinct substrate specificity. Faseb J. 2009;23:2605–2615. doi: 10.1096/fj.08-127399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie M, Hata Y, Takeuchi M, Ichtchenko K, Toyoda A, Hirao K, Takai Y, Rosahl TW, Sudhof TC. Binding of neuroligins to PSD-95. Science. 1997;277:1511–1515. doi: 10.1126/science.277.5331.1511. [DOI] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, Bourgeron T. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S, Radyushkin K, Hammerschmidt K, Granon S, Boretius S, Varoqueaux F, Ramanantsoa N, Gallego J, Ronnenberg A, Winter D, Frahm J, Fischer J, Bourgeron T, Ehrenreich H, Brose N. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc Natl Acad Sci U S A. 2008;105:1710–1715. doi: 10.1073/pnas.0711555105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kang Y, Zhang X, Dobie F, Wu H, Craig AM. Induction of GABAergic postsynaptic differentiation by alpha-neurexins. J Biol Chem. 2008;283:2323–2334. doi: 10.1074/jbc.M703957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Fuccillo MV, Malenka RC, Sudhof TC. LRRTM2 functions as a neurexin ligand in promoting excitatory synapse formation. Neuron. 2009;64:791–798. doi: 10.1016/j.neuron.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauren J, Airaksinen MS, Saarma M, Timmusk T. A novel gene family encoding leucine-rich repeat transmembrane proteins differentially expressed in the nervous system. Genomics. 2003;81:411–421. doi: 10.1016/s0888-7543(03)00030-2. [DOI] [PubMed] [Google Scholar]
- Levinson JN, Chery N, Huang K, Wong TP, Gerrow K, Kang R, Prange O, Wang YT, El-Husseini A. Neuroligins mediate excitatory and inhibitory synapse formation: involvement of PSD-95 and neurexin-1beta in neuroligin-induced synaptic specificity. J Biol Chem. 2005;280:17312–17319. doi: 10.1074/jbc.M413812200. [DOI] [PubMed] [Google Scholar]
- Linhoff MW, Lauren J, Cassidy RM, Dobie FA, Takahashi H, Nygaard HB, Airaksinen MS, Strittmatter SM, Craig AM. An unbiased expression screen for synaptogenic proteins identifies the LRRTM protein family as synaptic organizers. Neuron. 2009;61:734–749. doi: 10.1016/j.neuron.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig KU, Mattheisen M, Muhleisen TW, Roeske D, Schmal C, Breuer R, Schulte-Korne G, Muller-Myhsok B, Nothen MM, Hoffmann P, Rietschel M, Cichon S. Supporting evidence for LRRTM1 imprinting effects in schizophrenia. Mol Psychiatry. 2009;14:743–745. doi: 10.1038/mp.2009.28. [DOI] [PubMed] [Google Scholar]
- Missler M, Fernandez-Chacon R, Sudhof TC. The making of neurexins. J Neurochem. 1998;71:1339–1347. doi: 10.1046/j.1471-4159.1998.71041339.x. [DOI] [PubMed] [Google Scholar]
- Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K, Sudhof TC. Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature. 2003;424:939–948. doi: 10.1038/nature01755. [DOI] [PubMed] [Google Scholar]
- Mosyak L, Wood A, Dwyer B, Buddha M, Johnson M, Aulabaugh A, Zhong X, Presman E, Benard S, Kelleher K, Wilhelm J, Stahl ML, Kriz R, Gao Y, Cao Z, Ling HP, Pangalos MN, Walsh FS, Somers WS. The structure of the Lingo-1 ectodomain, a module implicated in central nervous system repair inhibition. J Biol Chem. 2006;281:36378–36390. doi: 10.1074/jbc.M607314200. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Poulopoulos A, Aramuni G, Meyer G, Soykan T, Hoon M, Papadopoulos T, Zhang M, Paarmann I, Fuchs C, Harvey K, Jedlicka P, Schwarzacher SW, Betz H, Harvey RJ, Brose N, Zhang W, Varoqueaux F. Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron. 2009;63:628–642. doi: 10.1016/j.neuron.2009.08.023. [DOI] [PubMed] [Google Scholar]
- Prange O, Wong TP, Gerrow K, Wang YT, El-Husseini A. A balance between excitatory and inhibitory synapses is controlled by PSD-95 and neuroligin. Proc Natl Acad Sci U S A. 2004;101:13915–13920. doi: 10.1073/pnas.0405939101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- Schubert WD, Urbanke C, Ziehm T, Beier V, Machner MP, Domann E, Wehland J, Chakraborty T, Heinz DW. Structure of internalin, a major invasion protein of Listeria monocytogenes, in complex with its human receptor E-cadherin. Cell. 2002;111:825–836. doi: 10.1016/s0092-8674(02)01136-4. [DOI] [PubMed] [Google Scholar]
- Song JY, Ichtchenko K, Sudhof TC, Brose N. Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc Natl Acad Sci U S A. 1999;96:1100–1105. doi: 10.1073/pnas.96.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, Vincent JB, Skaug JL, Thompson AP, Senman L, Feuk L, Qian C, Bryson SE, Jones MB, Marshall CR, Scherer SW, Vieland VJ, Bartlett C, Mangin LV, Goedken R, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Jamain S, Brose N. Neuroligin 2 is exclusively localized to inhibitory synapses. Eur J Cell Biol. 2004;83:449–456. doi: 10.1078/0171-9335-00410. [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Sudhof TC, Brose N. Neuroligins determine synapse maturation and function. Neuron. 2006;51:741–754. doi: 10.1016/j.neuron.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Woo J, Kwon SK, Kim E. The NGL family of leucine-rich repeat-containing synaptic adhesion molecules. Mol Cell Neurosci. 2009;42:1–10. doi: 10.1016/j.mcn.2009.05.008. [DOI] [PubMed] [Google Scholar]
- Xu J, Xiao N, Xia J. Thrombospondin 1 accelerates synaptogenesis in hippocampal neurons through neuroligin 1. Nat Neurosci. 2010;13:22–24. doi: 10.1038/nn.2459. [DOI] [PubMed] [Google Scholar]
- Zhang W, Rohlmann A, Sargsyan V, Aramuni G, Hammer RE, Sudhof TC, Missler M. Extracellular domains of alpha-neurexins participate in regulating synaptic transmission by selectively affecting N-and P/Q-type Ca2+ channels. J Neurosci. 2005;25:4330–4342. doi: 10.1523/JNEUROSCI.0497-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]