

Abstract
Lipoxygenases (LOs) comprise a class of substrate activating mononuclear non-heme iron enzymes which catalyze the hydroperoxidation of unsaturated fatty acids. A commonly proposed mechanism for LO catalysis involves H-atom abstraction by an FeIII-OH- site, best described as a proton coupled electron transfer (PCET) process, followed by direct reaction of O2 with the resulting substrate radical to yield product. An alternative mechanism that has also been discussed involves the abstraction of a proton from the substrate by the FeIII-OH leading to a σ-organoiron intermediate, where the subsequent σ bond insertion of dioxygen into the C-Fe bond completes the reaction. H-atom abstraction is favored by a high Eo of the FeII/FeIII couple and high pKa of water bound to the ferrous state, while an organoiron mechanism would be favored by a low Eo (to keep the site oxidized) and a high pKa of water bound to the ferric state (to deprotonate the substrate). A first coordination sphere mutant of soybean LO (N694C) has been prepared and characterized by near-infrared circular dichroism (CD) and variable-temperature, variable-field (VTVH) magnetic circular dichroism (MCD) spectroscopies (FeII site), as well as UV/Vis absorption, UV/Vis CD and electron paramagnetic resonance (EPR) spectroscopies (FeIII site). These studies suggest that N694C has a lowered Eo of the FeII/FeIII couple and a raised pKa of water bound to the ferric site relative to WT sLO-1 which would favor the organoiron mechanism. However, the observation in N694C of a significant deuterium isotope effect, anaerobic reduction of iron by substrate and a substantial decrease in kcat (~3000-fold) support H-atom abstraction as the relevant substrate-activation mechanism in sLO-1.
Introduction
The mononuclear non-heme iron enzymes comprise a large and expanding class catalyzing reactions of medical, pharmaceutical and environmental significance.1 These enzymes utilize dioxygen as a co-substrate and can be divided into two general groups: (1) dioxygen activating and (2) substrate activating enzymes.2 Oxygen activating mononuclear non-heme iron enzymes utilize a ferrous resting site which directly binds O2 to yield iron-oxygen intermediates which react with substrate to yield product. Substrate activating enzymes are characterized by their utilization of high-spin ferric sites to activate organic substrates for direct reaction with dioxygen. While numerous mononuclear non-heme enzymes exist, the majority are oxygen activating and differ only in the source of the additional reducing equivalents necessary for O2 activation (i.e. α-ketoglutarate-dependent, pterin dependent, extradiol dioxygenases, Rieske dioxygenases). In contrast, lipoxygenases along with the intradiol dioxygenases are mononuclear non-heme ferric enzymes which activate substrate.
Lipoxygenases catalyze the regio- and stereospecific hydroperoxidation of 1,4-Z,Z-pentadiene-containing polyunsaturated carboxylic acids in plants and animals.3-6 In humans, lipoxygenases produce leukotrienes and lipoxins, signaling molecules that mediate conditions such as asthma, atherosclerosis and psoriasis.7, 8 Current insight into the molecular mechanism of lipoxygenase derives from a variety of structural, kinetic and spectroscopic studies of soybean lipoxygenase (sLO-1) due to its ease of purification and stability. Crystallographic studies of ferrous sLO-1 have shown the active site iron to contain one C-terminal carboxylate ligand from Ile839, three histidine ligands (His690, His504 and His499), a water ligand and a distant side chain carbonyl of Asn694, at 2.87 Å.9-11 Circular dichroism (CD) and magnetic circular dichroism (MCD) studies of resting sLO-1 provided further insight into the active site structure having shown it to be a 40/60 % mixture of five-coordinate (5C) and six-coordinate (6C) ferrous species which becomes 6C upon substrate binding.12 The difference in coordination is due to the coordination flexibility of the Asn694 ligand which contributes to the reactivity of the site.
For activity, the enzyme must be oxidized to the ferric state. A commonly proposed mechanism for sLO-1 is shown in Scheme 1A. The catalytic cycle begins with hydrogen atom abstraction of the pro-S hydrogen from the 1,4-cis, cis-pentadiene fatty acid substrate by the FeIII-OH- species,13 shown to be a 5C species by electron paramagnetic resonance (EPR) with the signal being axial in a non-heme environment,14 to form a pentadienyl radical and the FeII-OH2 intermediate.6, 13, 15, 16 A large kinetic isotope effect (KIE) has been reported for sLO-1 (KIE ~ 80)17 and human 15-LO-1 (KIE ~ 50),18, 19 indicating that C-H cleavage contributes to the rate-determining step and its mechanism is via hydrogen tunneling. For this mechanism, the coordination flexibility (6C FeII → 5C FeIII), attributed to the Asn694 ligand, has been shown by site-directed mutagenesis to play an important role in tuning the active site for catalysis.20 Density functional theory calculations on H-atom abstraction by the FeIII-OH- site of sLO have suggested that the coordination flexibility contributes to the reaction energetics by stabilizing the FeII relative to the FeIII state, raising the redox potential and the pKa of the water ligand of the reduced site (to favor H-atom abstraction).21, 22 The transition state for H-atom abstraction is best described as a proton coupled electron transfer (PCET) process as the electron tunnels directly from the substrate to the iron through a superexchange pathway involving an Fe-O-H’-substrate bridge. Dioxygen would react directly with the substrate radical formed in the PCET process to yield a peroxyl radical which subsequently oxidizes the FeII-OH2 to give the active FeIII-OH- site and the hydroperoxide product.
Scheme 1.

Proposed Reaction Mechanisms for sLO-1: (A) H-atom abstraction and (B) Organoiron pathway.
Since both Eo of the FeII/FeIII couple and the pKa of water bound to the ferrous site are affected by the strength of the ligands to FeII, first coordination sphere mutants have been studied to gain further insight into the mechanism of catalysis in LOs. These mutant studies have focused on the Asn694 ligand responsible for the coordination flexibility of sLO-1 shown to be important in tuning the site in sLO-1 for catalysis. Two such mutants, N694H and N694G, were previously generated and characterized.23, 24 In contrast to WT sLO-1 which is 6C in the ferrous state with substrate analog bound and converts to a 5C site upon oxidation, CD and MCD studies showed that both N694H and N694G were 6C in both the reduced and oxidized forms. Furthermore, detailed analysis of both mutants indicated that they had a lowered Eo relative to WT sLO-1 due to the stronger donor ability of the bound ligand compared to Asn (His for N694H and hydroxide for N694G). The lowered Eo correlated with a decrease in kcat for the mutants, indicating that the Eo of the FeII/FeIII couple is a key driving force for H-atom abstraction.
To gain further molecular-level insight into the factors controlling catalysis in LO, we have extended our first sphere mutant studies of sLO-1 to a cysteine mutant of Asn694 (N694C). The thiolate ligand of cysteine in the mutant would have a higher affinity for the oxidized site which would lower the potential of the FeII/FeIII couple and the Cys is a strong donor ligand which would decrease the inductive effect of the FeIII on the coordinated water, raising the its pKa. Interestingly, this perturbation of the iron site would, in principle, favor an alternative, organoiron mechanism (Scheme 1B).25 In this mechanism, initial abstraction of a proton by the enzyme is facilitated by complexation of the carbanion to FeIII, yielding a σ-organoiron intermediate. The subsequent σ bond insertion of dioxygen in the C-FeIII bond would complete the reaction to yield product. Importantly, several aspects of this organoiron mechanism have been suggested to be feasible by model studies, where reaction of allylic organoiron intermediates with dioxygen was shown to be a facile process, able to compete with homolytic cleavage of the C-FeIII bond.26 In contrast to H-atom abstraction, the σ-organoiron mechanism would be favored by a low Eo to keep the site oxidized and high pKa for water bound to the ferric state to deprotonate the substrate. This organoiron mechanism has not been directly experimentally evaluated.
Near-infrared CD and variable-temperature variable-field (VTVH) MCD studies are utilized to determine the geometric and electronic structure of the ferrous form of N694C LO and UV/Vis absorption, UV/Vis CD and EPR spectroscopies are employed to study the ferric form of this variant. These studies suggest that the Eo of the FeII/FeIII couple and pKa of water bound to iron in N694C are shifted relative to WT sLO-1 to favor the organoiron mechanism and disfavor the H-atom abstraction mechanism. However, a large primary KIE, a 3000-fold decrease in kcat and anaerobic reduction of the iron site by substrate are observed providing further support of H-atom abstraction as the relevant substrate-activation mechanism in sLO-1.
Materials & Methods
Materials
LA was purchased from Aldrich Chemical Co. and per-deuterated linoleic acid (D-LA) was purified from a mixture of per-deuterated algal fatty acids from Cambridge Isotope Labs, as previously described.23 All commercial fatty acids were re-purified using a Higgins Semi-Prep Haisil (25 cm × 4.6 mm) C-18 column. Solution A was 99.9% MeOH and 0.1% acetic acid, while solution B was 99.9% H2O and 0.1% acetic acid. An isocratic elution of 85% A:15% B was used to purify all fatty acids and they were stored at −80 °C for a maximum of 6 months. All other chemicals were reagent grade or better and were used without further purification.
Mutagenesis, overexpression, and purification of N694C
Site-directed mutagenesis, overexpression, and purification of WT sLO-1 and N694C followed a protocol outlined previously 23. In brief, following expression of the protein in BL21-DE3 (Escherichia coli), the cells were harvested by centrifugation and their membranes were disrupted by sonication. Cell debris was pelleted and the supernatant was first dialyzed against 20 mM Bis-Tris buffer (pH 6.0) and then applied to a SP-Sephadex high-flow ion exchange column (Pharmacia), which was equilibrated with the same buffer. Eluted fractions containing lipoxygenase activity were pooled, concentrated, dialyzed against 20 mM Bis-Tris buffer (pH 6.0), and applied to a Macro-Prep 25-S ion exchange column (Bio-Rad). After concentration and buffer exchange into 0.1 M Borate (pH 9.2), the isolated enzyme was estimated to be greater than 90% pure (SDS-PAGE). Iron content of all lipoxygenase enzymes were determined on a Finnegan inductively coupled plasma mass spectrometer (ICP-MS) using internal standards of CoII-EDTA, and data were compared with standardized iron solutions.
Steady-State Kinetic Measurements
Steady-state kinetic values were determined by following the formation of the conjugated product at 234 nm (ε = 2.5 × 104 M-1 cm-1) with a Perkin-Elmer Lambda 40 spectrophotometer. No photo-degradation of the product was observed. All kinetic measurements were standardized to iron content. The assay samples were 2 mL in volume with substrate concentrations ranging from 1-80 μM (Borate buffer, pH 9.2, 25°C) under constant stirring with a rotating magnetic bar. The total ionic strength was adjusted to 50 mM by addition of NaCl. The temperature was controlled using a circulating water bath and jacketed cuvette system. Stock solution of LA was prepared in 95% ethanol and diluted into buffer so that the total ethanol concentration was less than 1.5%. Fatty acid concentrations were verified by allowing the enzyme reaction to proceed to completion and quantitating the conjugated product at 234 nm. Initial rates (up to the first 15% of the reaction) for each substrate were fitted to the Michaelis-Menten equation using the KaleidaGraph (Synergy) program and errors determined.
Competitive Kinetic Isotope Effect
The kineic isotope effect (KIE) reactions were performed as previously described.27 Briefly, a protio/per-deutero LA mixture, of known molar ratio, was reacted with LO in the kinetic buffer described above. The reaction was monitored at 234 nm and stopped with an acid quench at less than 5% total LA consumption. The acidified reaction mixture was extracted with methylene chloride, evaporated to dryness, reconstituted in methanol and injected onto an RP-HPLC column (Higgins Analytical, 250 × 4.6 mm). The products eluted at 1 mL/min (isochratic mobil phase 74.9% methnol, 25% water, 0.1% acetic acid). The molar protio/per-deutero HPOD ratios were equated to the corresponding peak area ratios and the KIE calculated as described previously.27
UV-vis Absorption and CD spectroscopies were carried out at 4° C. The UV-vis absorption spectra were recorded on a Perkin Elmer Lambda 40 monochromometer. Spectra were taken in 0.1 M Borate (pH 9.2) at 0.1 M ionic strength (adjusted with NaCl) at 4° C. CD spectra were obtained using an Aviv 60DS spectropolarimeter in 0.1 M Borate (pH 9.2). CD spectra were baseline corrected by subtracting the buffer and cell background from the raw data. Titrations were performed by the addition of 1 equiv of HPOD to 0.6 mL of lipoxygenase (20 mg/mL).
EPR spectroscopy
EPR measurements were performed on a Bruker ESP-500E spectrometer equipped with a dual mode X-band cavity and an Oxford Instruments ESR-900 helium flow cryostat. The program HAM (Dr. M. Hendrich, Carnegie Mellon University, Pittsburgh, PA) was used for data manipulation and EPR spectral simulation, as described previously 23. In brief, the signals were fit with an effective S’ = 1/2 model which uses a g-dependent linewidth that properly accounts for field-dependent transition probabilities. The temperature dependence was also fit with this program using the standard Boltzmann equation. The amount of EPR-active FeIII in the protein samples and the temperature dependence were determined relative to a FeIII-EDTA standard. A 100 μM Fe solution was made from a 1000 ppm iron standard (Fischer Scientific), with addition of 10-fold excess of EDTA in 25% glycerol. The EPR spectra were recorded at a modulation frequency of 100 kHz and at microwave frequencies of about 9.44 GHz. Precise microwave frequencies were recorded for individual spectra to insure accurate g-alignment. Other EPR parameters are specified in the figure legends.
Anaerobic Reactivity and Spectroscopic Properties of Ferric N694C
A ferrous sample of N694C (108 mg/ml in the kinetic buffer) was titrated anaerobically with one equivalent aliquots of HPOD until the ferric form of N694C was fully generated (as seen by the 762 nm band intensity, vida infra). An EPR sample was removed and determined to manifest a rhombic ferric signal. One equivalent of LA substrate was then added anaerobically and the decrease in signal intensity at 762 nm was measured. An EPR sample of the substrate oxidized species with LA added was removed and its change monitored with EPR.
Near-IR CD and MCD Spectrocopy
Near-infrared (600-2000 nm) CD and MCD data were recorded on a Jasco J-200D spectropolarimeter with a liquid N2-cooled InSb detector and equipped with an Oxford Instruments SM4000-7 Tesla (T) superconducting magnet/cryostat capable of fields up to 7T and temperatures down to 1.5 K. The N694C CD sample (~1.5 mM) was exchanged into deuterated CHES buffer (50 mM, pD 9.2) and kept at 5 °C during data collection with a circulating cooling bath attached to the sample holder. Glycerol-d3 was added at 60 % (v/v) to the N694C/FeII solution for preparation of the MCD sample and CD spectra were taken with and without glycerol addition to ensure that the site was unaffected by the glassing agent (Note: no change in the CD spectrum due to glycerol was observed). CD spectra were corrected for buffer and cell baselines by subtraction. Low temperature MCD data were obtained in custom made cells consisting of two Infrasil quartz disks separated by a 0.3 cm thick neoprene O-ring spacer into which the sample was injected and secured between two copper plates. The MCD spectrum was corrected for the natural CD and zero-field baseline effects caused by strain in the glasses by averaging the positive and negative field data at a given temperature (i.e (7T-(-)7T)/2). VTVH data were normalized to the maximum observed intensity over all isotherms for a given wavelength and the ground-state parameters were extracted by fitting in accordance with published procedures.28, 29
Results
Protein Purification and Kinetic Determinations
N694C and WT sLO-1 were purified with yields of 2-4 mg/L, with metal contents of 55 ± 10% and 80 ± 5%, respectively.
The steady-state parameters of N694C sLO, kcat (0.098 +/- 0.002 s-1),kcat/KM (0.026 +/- 0.002 s-1 μM-1 and KM (3.8 +/- 0.3 μM) (Table 1), were determined at 22 °C. The kcat value of N694C was ~ 2900 times lower than that of the WT sLO-1 and the kcat/KM value was ~ 730 times lower, however, the KM value is similar to that observed for WT sLO-1. The competitive kinetic isotope effect data (Dkcat/KM) for N694C were temperature independent, with values of 44 +/- 2 at 15°C, 48 +/- 3 at 22°C and 47 +/- 6 at 30°C (average value of approximately 46 +/- 2). In previous studies, it was shown that WT sLO-1 displayed a temperature-dependent, competitive Dkcat/KM at low substrate concentration (5μM).27 This temperature dependence is due to the multiple rate-limiting steps in the mechanism: substrate diffusion, hydrogen bond rearrangement and hydrogen bond abstraction (C-H bond cleavage).30 However, for N694C, the Dkcat/KM is temperature independent, indicating that the rate-determining step for N694C is solely limited by the C-H bond cleavage since tunneling mechanisms are not temperature dependent. The data for N694C are comparable to our previous data for N694H, which manifested a Dkcat/KM of 60 ± 9 at 30 °C.23 Although the magnitude of Dkcat/KM for N694C is less than that observed for N694H, the magnitude of the Dkcat/KM is much larger than semiclassical theory predicts (KIE ~ 7)31 and is consistent with a tunneling mechanism for hydrogen atom abstraction.32
Table 1.
Comparison of Kinetic Parameters for WT and N694C sLO-1a
| kcatb (s-1) | kcat/KMb (μM-1 s-1) | KM (μM) | |
|---|---|---|---|
| WT | 287(5) | 19(1) | 15(1) |
| N694C | 0.098(0.002) | 0.026(0.002) | 3.8(0.3) |
Numbers in parentheses represent errors as determined from replicate determinations.
Data were collected at 22 °C, in parallel.
Spectroscopy
(1) FeIII Site
Oxidation of N694C was accompanied by an increase in absorbance at 342 nm (ε = 2650 M-1 cm-1) and 762 nm (ε = 760 M-1 cm-1) (Figure 1, samples corrected for iron content). The latter transition is characteristic of a Cys to FeIII ligand-to-metal charge transfer band. It is lower in energy and has less intensity than the Cys to FeIII charge transfer observed, for example, in superoxide reductase.33, 34 UV/Vis CD spectroscopy manifests a band at 345 nm (Δε = 2.2 M-1 cm-1) and a shoulder at 420 nm (Figure 2). The Δε value was corrected for iron content and is comparable in magnitude to that of the WT sLO-1 (Δε = 1.45 M-1 cm-1), assigned as a His to FeIII charge transfer transition.
Figure 1.
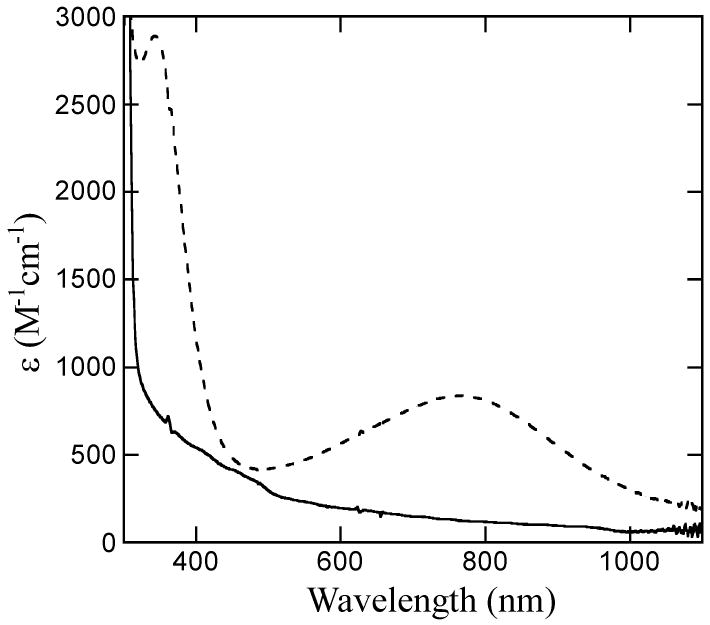
UV/Vis absorbance spectra of ferrous N694C (solid) and ferric N694C (dotted) at 4°C. Samples were corrected for iron content.
Figure 2.
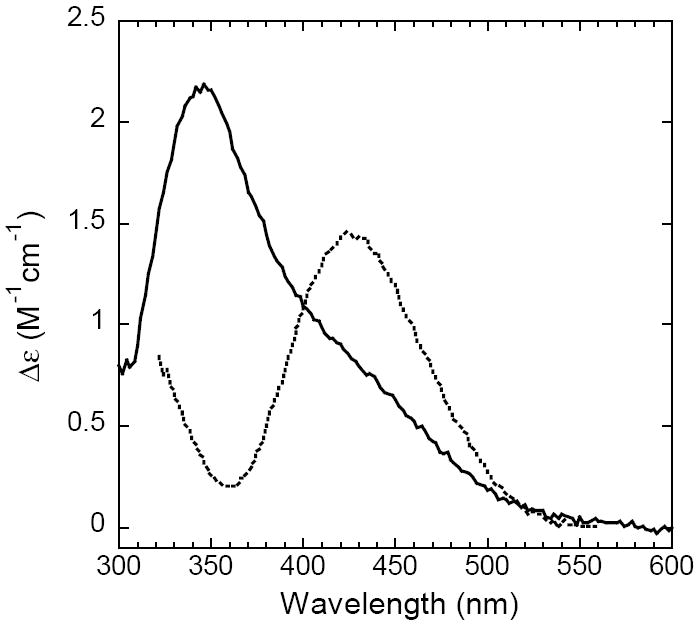
CD Spectra of ferric WT sLO-1 (dashed line) and ferric N694C (solid line) at 4°C. Samples were corrected for iron content.
The EPR spectrum of as-isolated N694C (60 μM iron) in frozen solution at 4 K, exhibited a weak signal at g ~ 4.3 (data not shown). The low intensity of this signal represents less than 5% of the total iron and indicates that the enzyme is mostly in its ferrous state. Addition of 1 equiv of HPOD (the hydroperoxide product of sLO) to ferrous N694C oxidized the iron to the catalytically competent ferric species. The ferric N694C at pH 9.2 (0.1 M borate, 0.1 M ionic strength) exhibited a complex multicomponent EPR signal, centered around g ~ 4.3, with a minor species at g ~ 6 (Figure 3A). The ferric species was also generated at pH 7.2 (0.1 M Tris, 0.1 M ionic strength) and only slight perturbations were observed.
Figure 3.
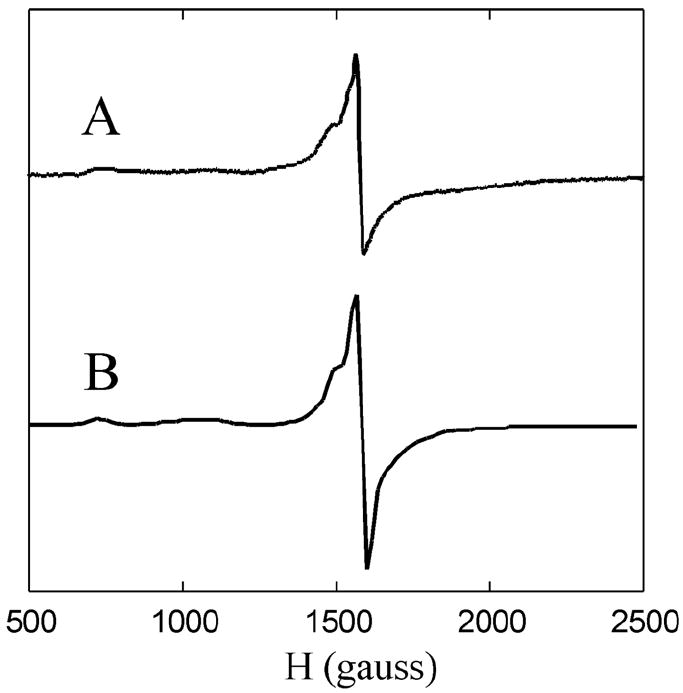
EPR spectra of the N694C: A) Ferric form, 0.1 M Borate, pH 9.2; B) Simulation with 80 ± 8% rhombic and 20 ± 2% axial; Experimental parameters for all spectra: Concentration = 50 μM FeIII, Temperature = 4 K, field set = 4000 G, scan range = 8 kG, time const. = 82 ms, mod. amplitude = 8 G, microwave power = 615 μW, freq. = 9.44 GHz, gain = 60.
The quantification of the amount of FeIII contributing to the multicomponent signal was complicated by the fact that the signal is spread over a large field range (~2300 gauss). This problem was addressed by the EPR spectral simulations (see S.I.). The simulated spectra were integrated over the entire field range to obtain the total EPR intensity from transitions within the middle Kramers doublet for the rhombic signal and the MS = ± ½ for the axial signal (see Figure 3B and S.I.). Utilizing the simulated (gave corrected) intensity, it was determined that greater than 85% of the expected ferric signal was observed and that the relative concentrations of the EPR species for ferric N694C (pH 9.2) were 80 ± 8 % for the rhombic signal and 20 ± 2 % for the axial signal.
An as-isolated, reduced N694C sample was oxidized with one equivalent of HPOD, anaerobically. An increase at 762 nm and an increase of the rhombic EPR signal were observed. One equivalent of anaerobic LA substrate was subsequently added and the rate of the disappearance of the 762 nm band was observed to be 0.017 +/- 0.003 sec-1 (Figure S2). This value is consistent with the rate of enzymatic turnover, considering that substrate is not saturating. The EPR signal of the ferric N694C disappeared upon substrate addition (paralleling the Cys→FeIII charge transfer band in absorption), indicating reduction of the ferric species to be associated with the anaerobic reaction of the enzyme with substrate.
(2) FeII site
Near-infrared CD and MCD spectroscopies were employed to study the high-spin ferrous active site in N694C in solution as analysis of the energies and splitting pattern of CD/MCD bands provides information about the geometric and electronic structure of the ferrous active site.28, 35 The 278 K CD spectrum of N694C/FeII contains a single negative ligand field band at ~11300 cm-1 (Figure 4A). CD spectra measured both without and with the glassing agent glycerol present were unchanged. The 1.6 K, 7T MCD spectrum of N694C/FeII (Figure 4B) contains a large positive ligand field feature at ~6050 cm-1 and a weak negative feature at ~11450 cm-1. The saturation magnetization behavior for N694C/FeII collected at 6250 cm-1 (Figure 4B, inset) is well-fit by a positive ZFS non-Kramers doublet model (i.e. large nesting) with ground-state spin Hamiltonian parameters of D = 12.5 ± 0.3 cm-1 and |E| = 2.6 ± 0.2 cm-1, corresponding to Δ = +500 ± 150 cm-1 and |V/2Δ| = 0.29 ± 0.02 which reflect the axial (Exz,yz-Exy = Δ) and rhombic (Exz-Eyz = V) dπ splitting of the t2g set of d orbitals on the FeII. The observed CD and MCD excited state splittings and energies of the two transitions of N694C/FeII are consistent with a square pyramidal 5C ferrous site. It should be noted that Fe K-near-edge X-ray absorption analysis performed on N694C/FeII indicates that the cysteine sulfur is not bound to the FeII site and, thus, is the ligand lost to generate the 5C site.36
Figure 4.
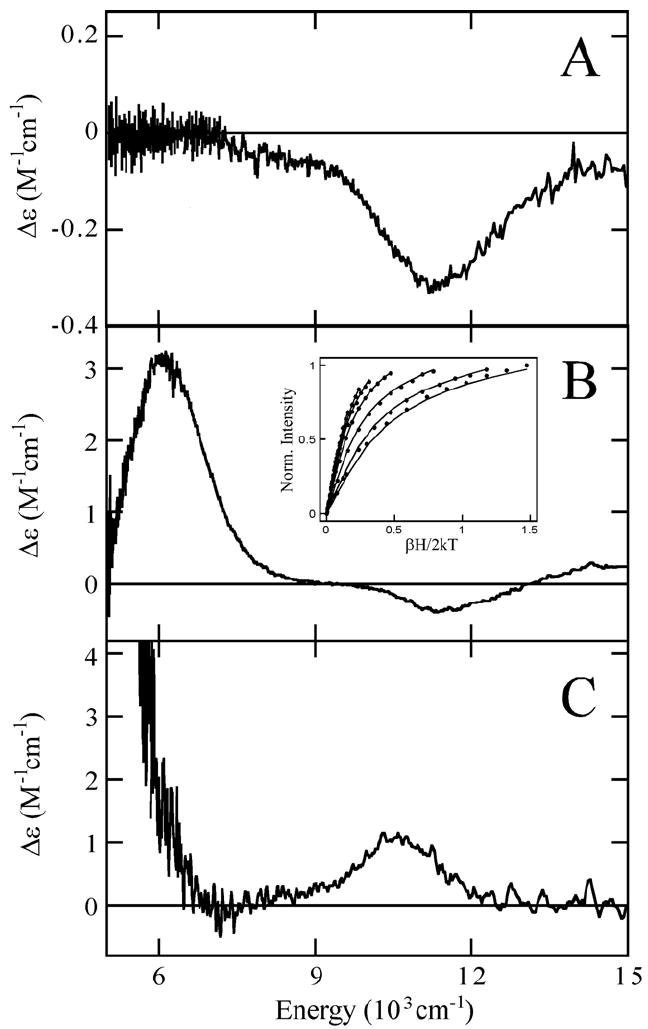
The 278K CD (A) of N694C/FeII and the low-temperature MCD of (B) N694C/FeII and (C) the 5C component of WT sLO-1/FeII. VTVH-MCD (B, inset) data (symbols) and their best fit (lines) for N694C/FeII collected at 6250 cm-1.
Discussion
A first sphere mutant of sLO-1 (N694C) has been prepared in which the Asn694 ligand responsible for the coordination flexibility observed in WT sLO-1 has been selectively mutated to a Cys. A combination of CD and VTVH MCD spectroscopies indicates that the FeII site of N694C is a 5C square pyramidal species in contrast to the 40/60 % mixture of 5C and 6C ferrous species in WT sLO-1/FeII. The Cys residue is not coordinated to the FeII site of the mutant, but oxidation of the site leads to ligation of the cysteine sulfur to the FeIII based upon the presence of a Cys-FeIII charge transfer band at 762 nm (ε = 760 M-1 cm-1).
The observed Cys-FeIII charge transfer band in N694C is at lower energy with decreased intensity compared to the Cys-FeIII charge transfer transition in ferric high-spin sites such as superoxide reductase.33, 34 One possible contribution to the decreased intensity of the transition could be partial coordination of the ferric site by Cys. However, from EPR of ferric N694C the rhombic signal, indicative of a 6C site in sLO where the Cys S occupies the sixth coordination site, is present at 80%. Correcting for this still leads to a factor of ~2.5 lower extinction coefficient relative to superoxide reductase (ε(SOR at 660 nm) = 2500 M-1cm-1).34 However, a weakened FeIII-S bond in N694C compared to superoxide reductase would both decrease the intensity of the transition and shift it to lower energy as observed. A weaker Cys-FeIII bond could reflect the fact that the cysteine side chain is one carbon shorter than the Asn ligand it replaced. This is also consistent with the loss of the Cys ligand upon reduction of the site.
Previous studies of WT sLO-1 have shown that it is converted from a 6C FeII site upon substrate analog binding to a 5C FeIII site.12, 14 In contrast, the present study indicates that N694C sLO-1 is converted from a 5C FeII site with a substrate analog bound (oleic acid, see S.I.) to a 6C FeIII site as determined from EPR data which show a predominately rhombic site indicative of 6C.37 The FeIII form of N694C is the catalytically active species analogous to WT sLO-1 as determined by UV/Vis absorption and EPR studies of the decay of this species at a catalytically relevant rate in the anaerobic reaction with substrate. Importantly, the kcat for N694C is significantly reduced (~ 3000-fold) relative to WT sLO-1 and the iron active site in the mutant is anaerobically reduced upon substrate addition.
Our spectroscopic studies of N694C support the presence of an iron active site for N694C with a lowered Eo of the FeII/FeIII couple compared to WT sLO-1 due to the presence of a 6C ferric site, with the cysteine sulfur coordinated in the ferric but not ferrous state. This stabilizes the FeIII site relative to the FeII site. The pKa of the coordinated water in the 6C ferric site of the mutant should be increased as the site is 6C and the cysteine donor ligand would tend to decrease the donation of coordinated H2O to FeIII. Thus, the Asn694→Cys694 mutation results in an iron active site with an Eo of the FeII/FeIII couple and a pKa of water bound to the ferric site that would, in principle, be more effective than WT sLO-1 in a σ-organoiron mechanism. Alternatively, with respect to H-atom abstraction, the coordination change relative to WT sLO-1 (loss of the Cys ligand to produce a 5C FeII site) would both lower Eo of the FeII/FeIII couple and lower the pKa of water bound to the ferrous site which would disfavor this reaction. In fact, the enzymatic turnover data are consistent with an H-atom abstraction mechanism for N694C due to the observation of a significant deuterium isotope effect on rate, a substantially reduced kcat (~ 3000-fold) of N694C relative to WT sLO-1 and the observed anaerobic reduction of iron by substrate. The iron active site would remain in the ferric state throughout the catalytic cycle in a σ-organoiron mechanism.
In addition to Eo and pKa, possible structural changes due to the Asn694→Cys694 mutation could contribute to the decreased reactivity. Since the cysteine sulfur is not coordinated in N694C/FeII, the ligands likely remain unchanged relative to the WT sLO-1/FeII 5C component (3His, 1 C-terminal carboxylate from Ile839 and H2O). However, a perturbation of the 5C ferrous site of N694C is observed compared to the 5C site in WT sLO-1/FeII (Figure 4B,C) in that both excited state transitions have shifted to higher energy and the sign of the higher energy transition has become negative.12 In addition, the splitting of the 5T2g ground-state obtained from the VTVH MCD data in Figure 4B (inset) has decreased in the mutant. Previously it has been shown that the N694 ligand is involved in a H-bonding network with residues Q697 and Q495 in the second sphere, linking the substrate binding pocket to the flexible N694 ligand and, thus, playing a role in the coordination flexibility of this residue (Figure 5).11 Binding of substrate, glycerol or inhibitor (oleic acid) affects the H-bonding, resulting in conversion to a purely 6C site.20 However, the N694C mutation eliminates the possibility of a H-bond from residue 694 to Q697, supported by CD studies showing no perturbations of the FeII due to glycerol or substrate analog oleic acid binding (see Figure S3). Crystallographic studies of other first sphere mutants (N694H and N694G) 11, 24 which also lack H-bonding from residue 694 to Q697 indicate that perturbing this network also can affect the interactions of these second sphere residues with the coordinating H499 ligand and, thus, its interaction with the FeII. Therefore, the changes in the MCD spectrum of N694C/FeII relative to WT sLO-1/FeII are consistent with the perturbation of the H-bonding network and the effects of this on the H499 ligand. However, since both N694C/FeII and WT sLO-1/FeII are 5C with the same ligation, this structural perturbation should not have a significant effect on Eo. For the catalytically relevant ferric site of N694C sLO-1, the structural difference from WT sLO-1/FeIII is the ligation of Cys which could sterically affect substrate binding. However, the fact that the KM value for the N694C mutant is similar to that for WT sLO-1 and to that previously reported for another first sphere mutant, N694H (KM ~ 4 μM),23 indicates that the mutation results in only minor changes in the substrate binding pocket.
Figure 5.
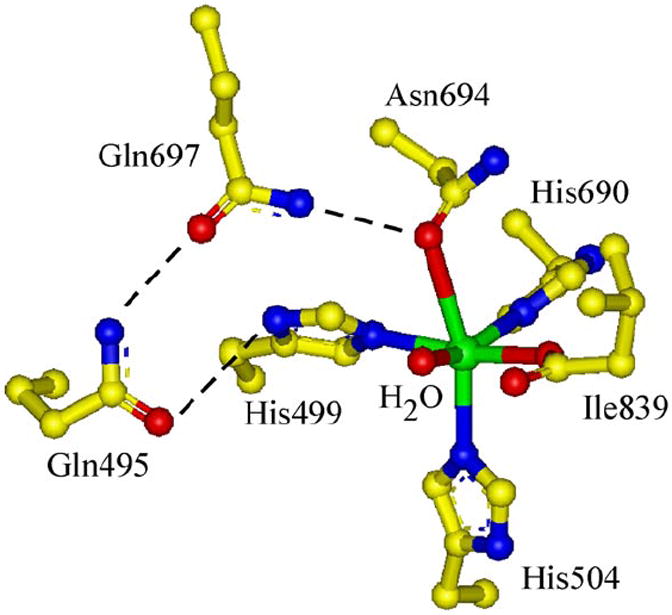
Active Site of WT sLO-1 including the second sphere H-bonding network.
Finally, based on crystallographic studies of WT sLO-1 in the absence of substrate, it was proposed that the presence of certain active site residues such as Gln495 and Gln697 restrict access to the Fe site to disfavor formation of the σ-organoiron intermediate.10 However, the possibility of structural rearrangement of the protein active site to accommodate this species could not be eliminated. Subsequent crystallographic studies do indicate the possibility of structural changes in some of these residues upon substrate binding.11 However, the spectroscopic and kinetic data presented here argue against a σ-organoiron intermediate.
In summary, a first sphere mutant of sLO-1, N694C, has been prepared and characterized which allows evaluation of the σ-organoiron mechanism previously proposed for the reaction of lipoxygenases. While the mutant has a lowered Eo of the FeII/FeIII couple and a raised pKa of water bound to the ferric site which would favor a σ-organoiron reaction, the kinetic data of N694C show a significant deuterium isotope effect on rate, an observed anaerobic reduction of iron by substrate and a substantially reduced kcat (~ 3000-fold) of N694C relative to WT sLO-1. These results experimentally evaluate the organoiron mechanism and provide further support for H-atom abstraction as the catalytically relevant mechanism in lipoxygenase.
Supplementary Material
Acknowledgments
This research has been supported by NIH grant GM56062-06 (T.R.H.), GM40392 (E.I.S.), and the NIH instrument grant DBI-0217922 (UCSC EPR).
ABREVIATIONS
- LO
lipoxygenase
- sLO-1
soybean lipoxygenase-1
- WT sLO-1
wild type soybean lipoxygenase-1
- PCET
proton coupled electron transfer
- CD
circular dichroism
- MCD
magnetic circular dichroism
- VTVH MCD
variable-temperature, variable-field magnetic circular dichroism
- EPR
electron paramagnetic resonance
- LA
linoleic acid
- HPOD
13-(S)-hydroperoxy-9,11-(Z,E)-octadecadienoic acid (the oxidation product of LA)
- kcat
the rate constant for product release
- kcat/Km
the rate constant for substrate capture
- Dkcat/Km
the competitive primary kinetic isotope effect
- ZFS
zero-field splitting
Footnotes
EPR simulation and integration for ferric N694C, LA oxidation by N694C/FeIII monitored by absorption and the 278K CD spectra of N694C/FeII + 60% (v/v) glycerol and N694C/FeII + oleic acid (saturating) are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee S-K, Lehnert N, Neese F, Skulan AJ, Yang Y-S, Zhou J. Chem Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 2.Neidig ML, Solomon EI. Chem Commun. 2005;47:5843–5863. doi: 10.1039/b510233m. [DOI] [PubMed] [Google Scholar]
- 3.Siedow JN. Annu Rev Plant Physiol Plant Mol Biol. 1991;42:145–188. [Google Scholar]
- 4.Yamamoto S. Biochim Biophys Acta. 1992;1128:117–131. doi: 10.1016/0005-2760(92)90297-9. [DOI] [PubMed] [Google Scholar]
- 5.Ford-Hutchinson AW, Gresser M, Young RN. Annu Rev Biochem. 1994;63:383–417. doi: 10.1146/annurev.bi.63.070194.002123. [DOI] [PubMed] [Google Scholar]
- 6.Solomon EI, Zhou J, Neese F, Pavel EG. Chem Biol. 1997;4:795–808. doi: 10.1016/s1074-5521(97)90113-7. [DOI] [PubMed] [Google Scholar]
- 7.Sigal E. Am J Physiol. 1991;260:L13–L28. doi: 10.1152/ajplung.1991.260.2.L13. [DOI] [PubMed] [Google Scholar]
- 8.Samuelsson B, Dahlen SE, Lindgren JA, Rouzer CA, Serhan CN. Science. 1987;237(4819):1171–1176. doi: 10.1126/science.2820055. [DOI] [PubMed] [Google Scholar]
- 9.Boyington JC, Gaffney BJ, Amzel LM. Science. 1993;260:1482–1486. doi: 10.1126/science.8502991. [DOI] [PubMed] [Google Scholar]
- 10.Minor W, Steczko J, Stec B, Otwinowski Z, Bolin JT, Walter R, Axelrod B. Biochemistry. 1996;35:10687–10701. doi: 10.1021/bi960576u. [DOI] [PubMed] [Google Scholar]
- 11.Tomchick DR, Phan P, Cymborowski M, Minor W, Holman TR. Biochemistry. 2001;40:7509–7517. doi: 10.1021/bi002893d. [DOI] [PubMed] [Google Scholar]
- 12.Pavlosky MA, Zhang Y, Westre TE, Gan Q-F, Pavel EG, Campochiaro C, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1995;117:4316–4327. [Google Scholar]
- 13.Scarrow RC, Trimitsis MG, Buck CP, Grove GN, Cowling RA, Nelson MJ. Biochemistry. 1994;33:15023–15035. doi: 10.1021/bi00254a011. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Gebhard MS, Solomon EI. J Am Chem Soc. 1991;113:5162–5175. [Google Scholar]
- 15.Gardner HW. Biochim Biophys Acta. 1989;1001:274–281. doi: 10.1016/0005-2760(89)90111-2. [DOI] [PubMed] [Google Scholar]
- 16.Glickman MH, Klinman JP. Biochemistry. 1996;35:12882–12892. doi: 10.1021/bi960985q. [DOI] [PubMed] [Google Scholar]
- 17.Rickert KW, Klinman JP. Biochemistry. 1999;38:12218–12228. doi: 10.1021/bi990834y. [DOI] [PubMed] [Google Scholar]
- 18.Knapp MJ, Seebeck FP, Klinman JP. J Am Chem Soc. 2001;123:2931–2932. doi: 10.1021/ja003855k. [DOI] [PubMed] [Google Scholar]
- 19.Knapp MJ, Klinman JP. Biochemistry. 2003;42:11466–11475. doi: 10.1021/bi0300884. [DOI] [PubMed] [Google Scholar]
- 20.Schenk G, Neidig ML, Zhou J, Holman TR, Solomon EI. Biochemistry. 2003;42:7294–7302. doi: 10.1021/bi027380g. [DOI] [PubMed] [Google Scholar]
- 21.Lehnert N, Solomon EI. J Biol Inorg Chem. 2003;8:294–305. doi: 10.1007/s00775-002-0415-6. [DOI] [PubMed] [Google Scholar]
- 22.Gardner KA, Mayer JM. Science. 1995;269:1849–1851. doi: 10.1126/science.7569922. [DOI] [PubMed] [Google Scholar]
- 23.Holman TR, Zhou J, Solomon EI. J Am Chem Soc. 1998;120:12564–12572. [Google Scholar]
- 24.Segraves EN, Chruszcz M, Neidig ML, Ruddat V, Zhou J, Wecksler AT, Minor W, Solomon EI, Holman TR. Biochemistry. 2006;45:10233–10242. doi: 10.1021/bi060577e. [DOI] [PubMed] [Google Scholar]
- 25.Corey EJ, Nagata R. J Am Chem Soc. 1987;109:8107–8108. [Google Scholar]
- 26.Corey EJ, Walker JC. J Am Chem Soc. 1987;109:8108–8109. [Google Scholar]
- 27.Lewis ER, Johansen E, Holman TR. J Am Chem Soc. 1999;121:1395–1396. [Google Scholar]
- 28.Pavel EG, Kitajima N, Solomon EI. J Am Chem Soc. 1998;120:3949–3962. [Google Scholar]
- 29.Campochiaro C, Pavel EG, Solomon EI. Inorg Chem. 1995;34:4669–4675. [Google Scholar]
- 30.Glickman MH, Klinman JP. Biochemistry. 1995;34:14077–14092. doi: 10.1021/bi00043a013. [DOI] [PubMed] [Google Scholar]
- 31.Klinman JP. Philos Trans R Soc B. 2006;361:1323–1331. doi: 10.1098/rstb.2006.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jonsson T, Glickman MH, Sun S, Klinman JP. J Am Chem Soc. 1996;118:10319–10320. [Google Scholar]
- 33.Tavares P, Ravi N, Moura JJG, LeGall J, Huang Y-H, Crouse BR, Johnson MK, Huynh BH, Moura I. J Biol Chem. 1994;269:10504–10510. [PubMed] [Google Scholar]
- 34.Clay MD, Jenney FE, Jr, Hagedoorn PL, George GN, Adams MWW, Johnson MK. J Am Chem Soc. 2002;124:788–805. doi: 10.1021/ja016889g. [DOI] [PubMed] [Google Scholar]
- 35.Solomon EI, Pavel EG, Loeb KE, Campochiaro C. Coord Chem Rev. 1995;144:369–460. [Google Scholar]
- 36.Sarangi R, Hocking RH, Neidig ML, Benfatto M, Holman TR, Solomon EI, Hodgson KO, Hedman B. doi: 10.1021/ic800580f. Manuscript submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Gebhard MS, Solomon EI. J Am Chem Soc. 1995;113:5162–5175. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.