

Abstract
A series of diaroyl phosphates was employed to assess the general reactivity of this class of molecule against classical class A and class C β-lactamases. The compounds were found, in general, to be inhibitory substrates of both classes of enzyme. In each case, they reacted rapidly with the enzyme (104 – 106 s−1M−1) to yield transiently stable intermediates, most likely acyl-enzymes, which slowly (10−3 – 10−1 s−1) regenerated free enzyme. In certain cases, side branches from direct turnover produced EII complexes (“substrate” inhibition), more inert EI′ complexes, and, in one case, a completely inactive EI′ complex. Deacylation, but not acylation, was enhanced by electron-withdrawing substituents. Acylation rates were enhanced by hydrophobic substitution, both in the diaroyl phosphate and at the enzyme active site. The latter factor led to the general order of β-lactamase acylation rates: class D (previous results) > class C > class A. It is likely that nanomolar inhibitors of all serine β-lactamases could be achieved by rational exploitation of diacyl phosphates.
The threat of pathogenic bacteria to human health continues to rise. Such bacteria are now often resistant to many, if not all, of the antibiotics that we routinely use to defend ourselves from them (1). Among those antibiotics whose effectiveness is threatened by the spread of bacterial resistance are the β-lactams, which have been in constant clinical use since 1945, and which are probably the generally most effective and safe class of such compounds. Although there are many sources of bacterial resistance to β-lactams, the most frequently encountered derives from the β-lactamases, bacterial enzymes that catalyze the hydrolysis of β-lactams and thus destruction of their antibiotic activity (2).
Resistance to β-lactam antibiotics arising from β-lactamases can, both in principle and in practice, be reduced by the administration of effective β-lactamase inhibitors together with β-lactams (3). Therefore, just as the search for new antibiotics continues, so too does the search for new β-lactamase inhibitors (4, 5). It is now very well established that there are a large number of β-lactamases, differing in active site structure and substrate specificity, and spanning four molecular homology classes A-D (6). It is unlikely that a common and yet potent non-covalent inhibitor can be found to span all of these enzymes. Experience with class A, C and D β-lactamases, the serine enzymes, has shown that covalent inhibitors of the mechanism-based variety and transition state analogue inhibitors (also most likely acting covalently since the chemical transition states are covalently bound to the enzymes), aimed at the reaction center, are most likely to be broadly successful.
Our studies of phosphorus-based inhibitors of serine β-lactamases have demonstrated two important points. First, that anionic phosphylating agents (1) can be effective inhibitors (7), and second, that negatively charged phosphate leaving groups

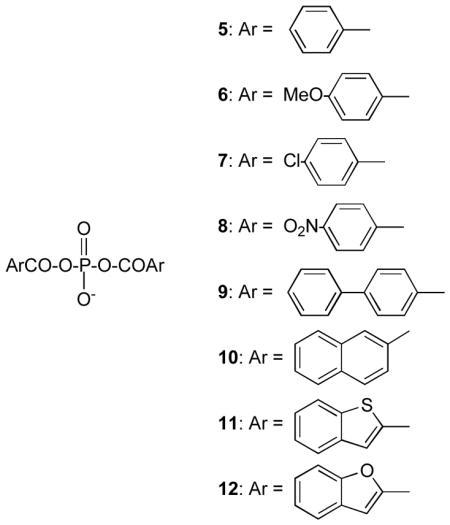
enhance acylation rates by interaction with the active site (2) (8). With respect to the second of these points, the monoacyl (3) and diacyl (4) phosphates have been identified as structures having considerable potential as inhibitory substrates of serine β-lactamases (8,9). The diacyl phosphates are particularly effective against typical class D enzymes (10) and also show activity against classes A and C (9). In this paper, we examine in more detail the potential of diacyl phosphates against the latter classes, and explore the structural basis of their activity. In order to facilitate these studies, the diacyl phosphates 5–12 have been employed and the kinetics of their interactions with typical class A and class C β-lactamases determined.

EXPERIMENTAL
The purified Enterobacter cloacae P99 and Escherichia coli W3310 TEM-2 β-lactamases were purchased from the Centre for Applied Microbiology and Research (Porton, Down, Wiltshire, UK) and used as received. The chromogenic substrate CENTA was prepared in this laboratory by Dr. Rajesh Nagarajan, and cephalothin was a gift from Eli Lilly and Co. Benzylpenicillin was purchased from Sigma-Aldrich. The preparation of the diacyl phosphates 5–10 has been previously described (11); compounds 11 and 12 were prepared as described below.
Synthesis of Diaroyl Phosphates 11 and 12
The relevant aryl carboxylic acid (2.2 mmol) and monobasic tetrabutylammonium phosphate (747 mg, 2.2 mmol) were dissolved in dry acetonitrile (20 ml). N,N-diisopropylethylamine (380 μL, 2.2 mmol) and O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU) (1.0 g, 2.6 mmol) were then added to the reaction mixture at ice temperature with continuous gentle stirring. After 15 min, the reaction mixture was allowed to come to room temperature, and stirred for a further 22 hr. At that time, N,N-diisopropylethylamine (760 μL, 4.4 mmol) and benzoyl chloride (503 μL, 4.4 mmol) were added to the reaction mixture and stirring continued for another 22 h at room temperature. After removal of the acetonitrile by rotary evaporation, saturated NaCl (15 mL) was added and the mixture stirred at 0 °C for 30 min. The sodium salts of the products formed a sticky layer on the wall of the flask. The NaCl solution was decanted off and the sticky product was washed with water to remove excess NaCl. Acetonitrile (15 mL) was added to the flask and the mixture gently stirred for 10 min. The suspended colorless solid was removed by filtration, washed very well with acetonitrile and dried under vacuum overnight. Approximately 350 mg of crude product was collected.
A 31P NMR spectrum in DMSO showed that the crude product was a mixture of all three possible aroyl phosphates 5, 11 (or 12), and the corresponding asymmetric compounds. The sodium salts of 11 and 12 were purified by HPLC (elution by a 0–100% MeOH/H2O gradient over 30 min from a Macherey-Nagel SS 250/0.5in./10-nucleosil 300-7 C18 reverse phase column; retention times 27.1 min (11) and 19.2 min (12) at a flow rate of 3 ml/min). The products were characterized by NMR, IR and mass spectra.
Sodium bis[benzo(b)thiophenecarbonyl] phosphate, 11
1H NMR [(DMSO)-d6] δ 7.37 (t, J = 7.5 Hz, 1H), 7.45 (t, J = 7.8 Hz, 1H), 7.86 (d, J = 6.6 Hz, 1H), 7.89 (d, J = 7.5 Hz, 1H), 8.17 (s, 1H). 31P NMR (DMSO-d6) δ −19.13. FTIR (KBr, cm−1) 1719.6. ES(−) MS m/z 417.03.
Sodium bis[2-benzo(b)furancarbonyl] phosphate, 12
1H NMR (DMSO)-d6) δ 7.25 (t, J = 7.4 Hz, 1H), 7.43 (t, J = 7.4 Hz, 1H), 7.48 (d, J = 7.2 Hz, 1H), 7.68 (d, J = 7.5 Hz, 1H), 7.72 (s, 1H). 31P NMR (DMSO-d6) δ −19.26. FTIR (KBr, cm−1) 1731. ES(−) MS m/z 385.08.
Inhibition Kinetics
All kinetics experiments were performed at 25 °C in a buffer at pH 7.5 containing 20 mM MOPS. A combination of several different experiments, listed below, was employed for each inhibitor to obtain values for the inhibition parameters of
Direct turnover of I could generally be followed spectrophotometrically under steady state conditions. The wavelengths employed for 5 – 10 were those used previously (11), while 11 and 12 were studied at 300 nm, where large changes in absorption were observed during hydrolysis. In cases where Ki was very low, k2 values could be obtained directly from initial rates. Otherwise, the measured initial rates were fitted to the Henri-Michaelis-Menten equation by a non-linear least squares procedure to obtain k2 and Ki. With the P99 β-lactamase, concentrations of enzyme and inhibitor were 1.02 μM and 10–20 μM, respectively, for 5, 0.20 μM and 4.0–12 μM, respectively, for 6, 0.20 μM and 2.0–30 μM, respectively, for 7, 0.31 μM and 2.0–70 μM, respectively, for 8, 0.48 μM and 2.0–20 μM, respectively, for 9, 0.46 μM and 4.0–12 μM, respectively, for 10, 0.10 μM and 2.0–6.0 μM, respectively, for 11, and 0.10 μM and 1.0–10 μM, respectively, for 12. With the TEM β-lactamase, the concentrations of enzyme and inhibitor were 0.98 μM and 2.0–24 μM, respectively, for 6, 0.20 μM and 1.0–12 μM, respectively, for 7, 0.98 μM and 10–500 μM, respectively, for 8, 0.20 μM and 1.0–12 μM, respectively, for 9, 0.95 μM and 4.0–10 μM, respectively for 10, 0.14 μM and 2.0–6.0 μM, respectively, for 11, and 0.35 μM and 2.0–32 μM, respectively, for 12.
-
In a number of cases, Ki values were obtained directly from steady state inhibition experiments, from initial rates of hydrolysis of a good substrate in the presence of several concentrations of the inhibitor. For example, cephalothin was used as substrate for Ki determinations of 9 and 10 with the TEM β-lactamase. Initial rates of cephalothin (1 mM, Km = 147 μM) hydrolysis were measured spectrophotometrically at 290 nm. The enzyme concentration was 0.12 μM and concentrations of 9 and 10 were 0–20 μM. For compound 8, CENTA (50 μM, Km = 42.5 μM) was used as substrate and its hydrolysis monitored at 410 nm. Enzyme and inhibitor concentrations were 0.02 μM and 0–2.0 μM, respectively. For compounds 11 and 12, this method of Ki determination was employed for both the P99 and TEM enzymes. With compound 11 and the P99 enzyme, nitrocefin was used as a substrate (100 μM, Km = 50 μM) and its hydrolysis at 482 nm monitored. Enzyme and inhibitor concentrations were 5.0 nM and 0–1.0 μM, respectively. In all cases, competitive inhibition was assumed and the data were fitted to equation 1 to obtain Ki values.
(1) -
Values of ki were obtained by one of three methods. In some cases, direct fitting of total progress curves to Scheme 1 by means of the Dynafit program (12) was successfully employed. This was done, for example, with data for turnover of 5, 7 and 10 by the P99 β-lactamase, where concentrations of enzyme and inhibitor were 1.0 μM and 10 – 20 μM (5), 0.2 μM and 10–20 μM (7), and 0.46 μM and 4.0–12 μM (10), respectively. In other cases, competitive experiments where spectrophotometric total progress curves for hydrolysis of good substrates in the presence of the diacyl phosphate inhibitors were recorded and the data were fitted to Scheme 2 by the Dynafit program (12). This method was used, for example, to obtain ki values for inhibition of the TEM β-lactamase by 6, 7 and 10.
Cephalothin, enzyme and inhibitor concentrations were 1.0 mM, 0.12 μM and 10–20 μM (6), 20–40 μM (7), and 4.0–12 μM (10), respectively. Two or three separate curves were obtained in the inhibitor concentration range specified and the data fitted simultaneously. This method was also used to obtain ki values for inhibition of the P99 β-lactamase by 6, 9 and 10. The substrate was cephalothin (200 μM). Enzyme and inhibitor concentrations were 1 nM and 3.0–10 μM, respectively, for 6 and, for both 9 and 10, 2.0–4.0 nM and 0.1–0.6 μM, respectively. In all other cases, ki values were determined from k2 and Ki values (ki = k2/Ki), obtained as described above.
On some occasions, the value of the rate constant k2 was checked by means of a return of activity experiment, where an aliquot of a reaction mixture was diluted out into an assay solution containing a good substrate at high concentration, assuring saturation of free enzyme by the latter, and the time dependent restoration of enzyme activity monitored. This was done, for example, in studies of the reaction between the TEM β-lactamase and 9. The enzyme (3 μM) and 9 (100 μM) were incubated together and after 4 min and 60 min, aliquots were diluted 300-fold into benzylpenicillin solutions (500 μM, Km = 16 μM) and the spectrophotometric progress curve at 232 nm monitored. These data were also fitted to Scheme 2.
-
In certain individual cases, more complex schemes were required to fit the data. For example, the reaction between the P99 β-lactamase and 9 and 10, studied directly by measurement of initial rates [see (i) above], showed indications of substrate inhibition. Accordingly, Scheme 3 was used to fit the data. Compound 8 was unusual in also exhibiting a degree of irreversible inhibition of the P99 enzyme. This was directly detected by assay of the enzyme activity after complete turnover of 8. The total progress curves used in this case to obtain ki were therefore fitted to Scheme 4, where the relatively rapid spontaneous hydrolysis of 8 was also included.
At high inhibitor/enzyme concentration ratios (>100), the reaction between the TEM β-lactamase and 5 also displayed branched kinetics, and, as found previously (9), Scheme 5 was required to fit the data.
Routinely, however, in the experiments reported here, the inhibitors were studied at low concentrations where this complexity was not observed because fewer turnovers occurred and k2 ≫ k3.
Access of the alternative nucleophile methanol to the acyl-enzyme intermediate EI was also determined for turnover of 9, as previously described for 5 (11, 13). The enzyme and inhibitor (9) concentrations were 0.24 μM and 2.0 μM, respectively, and methanol concentrations were 0–2.5 M.
Scheme 1.

Under steady state conditions, the effective thermodynamic inhibition constant, Ki, is given by k2/ki. No evidence of non-covalent binding of the inhibitors to these enzymes was observed at the concentrations employed.
Scheme 2.

Scheme 3.

Scheme 4.

Scheme 5.

Computational Methods
Simulations were performed on a SGI workstation running the program Insight II, essentially as previously described (8,11). The crystal structure of the P99 β-lactamase [PDB entry 1XX2 (14)], including crystallographic water molecules, was modified to construct the anionic acylation tetrahedral intermediate 13. The pH was set to 7.5 and the total charge on the complexes was −2.0. The side chain of Tyr 150 was neutral and those of Lys 67 and 315 were cationic. The partial charges of the enzyme were assigned by Insight II. Partial charges (MNDO) on atoms of the inhibitor in the tetrahedral intermediate 13 were calculated from a model adduct with serine. The active site was further hydrated with a 15 Å sphere of water centered on Oγ of the nucleophilic serine 64. The model was then subjected to molecular dynamics runs of 100 ps. Several typical snapshots from the molecular dynamics runs were selected. These structures were then each subjected to 1000 steepest descent energy minimization, followed by 2000 steps of conjugate gradients. Interaction energies between enzyme and ligand (Eint) were calculated for each of the minimized structures, as previously described (15).
The TEM-2 model was constructed in exactly the same way from the crystal structure of the enzyme [PDB entry 1BTL (16)]. In this model, the Lys 73 and Lys 234 side chains were cationic and that of Glu 166 neutral (in the tetrahedral intermediate, Glu 166, after having taken a proton, directly or indirectly, from the Ser 70 hydroxyl group, will be neutral).
RESULTS AND DISCUSSION
Diaroyl phosphates are inhibitory substrates of representative class A and class C serine β-lactamases, the TEM-2 and Enterobacter cloacae P99 enzymes, respectively. This means that they react with these enzymes to form relatively inert complexes that break down slowly to regenerate free enzyme. The compounds themselves, 5 – 12, are intrinsically quite stable in solution although electron-withdrawing substrates lead to greater lability. The spontaneous hydrolysis rates constants, k0, for 5–12 in 25 mM MOPS buffer, pH 7.5 at 25 °C are listed in Table 1.
Table 1.
Rate and Equilibrium Constants For Inhibition and Turnover of Diaroyl Phosphates by β-Lactamases
| ko × 106 (s−1) | P99 | TEM | |||||
|---|---|---|---|---|---|---|---|
| ki (s−1mM−1) | k2 × 103 (s−1) | K1 (μM) | ki (s−1mM−1) | k2 × 103 (s−1) | Ki (μM) | ||
| 5 | 0.78 | 7 ± 1 | 9.3 ± 0.2 | 1.33a | 0.5 ± 0.1 | 38 ± 3 | 76a |
| 6 | 0.31 | 9 ± 1 | 1.28 ± 0.01 | 0.147a | 6.1 ± 0.1 | 1.98 ± 0.04 | 0.32a |
| 7 | 4.2 | 14 ± 1 | 58 ± 5 | 4.21a | 19.5 ± 3.0 | 39.5 ± 3.0 | 2.0a |
| 8 | 52 | 6 ± 1 | 65 ± 8 | 12.1a | 0.65b | 124 ± 5 | 190 ± 20 |
| 9 | 1.31 | 870 ± 20 | 27 ± 1 | 0.032a | 296b | 23.7 ± 1.0 | 0.08 ± 0.01 |
| 10 | 1.47 | 1810 ± 360 | 28 ± 1 | 0.016a | 40 ± 5 | 12.6 ± 0.1 | 0.31 |
| 11 | 54.1 | 69600b | 211 ± 6 | (3.0 ± 0.2) × 10−3 | 53b | 79 ± 2 | 1.47 ± 0.06 |
| 12 | 285 | 265b | 354 ± 26 | 1.3 ± 0.3 | 1.8b | 74 ± 1 | 41 ± 9 |
Calculated from Ki = k2/ki
Calculated from ki = k2/Ki
Figures 1–6 illustrate the data obtained to reach the above conclusions for various combinations of the TEM-2 and P99 β-lactamases and the diacyl phosphates 5–12. Figure 1, for example, shows total progress curves for slow turnover of 10 by the P99 β-lactamase. The linear initial rates and sharp curvature close to the completion of the reaction indicate Km (Ki) values much below the initial concentration of 10. Figure 2 shows a biphasic total progress curve, characteristic of reactions of 8 with the P99 enzyme. The first phase here reflects turnover of 8 by the enzyme, and the second, background hydrolysis of 8. As noted in the Experimental section, 8 is unique in also leading to irreversible inhibition of the enzyme at the inhibitor concentrations routinely employed in these experiments, and hence it is possible, at high inhibitor/enzyme ratios, to fully inactivate the enzyme before turnover of 8 is complete, as seen in the two phases of Figure 2. Figure 3 shows steady state initial rate inhibition of nitrocefin turnover by 7, and indicates a very low Ki value. Figure 4 illustrates the inhibition of cephalothin hydrolysis by 5 in a total progress curve. Analysis of such curves allowed determination of ki values (see Experimental section). Figures 5 and 6 illustrate typical results with the TEM-2 enzyme. For example, Figure 5 shows initial rates of hydrolysis of 8 by this enzyme. The data is fitted to a Henri-Michaelis-Menten equation (see Experimental section) to obtain Km (i.e. Ki). Finally, Figure 6 shows the results of a return of activity experiment where a pre-incubated mixture of the TEM-2 enzyme and 9 is added to a solution containing a saturating concentration of the good substrate benzylpenicillin The rate constant for return of activity derived from this curve, 0.024 s−1, was identical to that derived from a direct turnover experiment (see below).
Figure 1.

Total progress curves for turnover of 10 (○, 4 μM; □, 8 μM) by the P99 – β-lactamase (0.46 μM). Absorbance changes at 254 nm are shown.
Figure 6.

Return of activity against benzylpenicillin (0.5 mM) of the TEM β-lactamase (10 μM) after incubation (4 min) with 9. Concentrations of the enzyme and 9 in the incubation mixture were 3 μM and 100 μM, respectively. The points are experimental and the solid line calculated from the fit of the data to Scheme 2.
Figure 2.

Total progress curve for turnover of 8 (20 μM) by the P99 β-lactamase (0.46 μM). The absorbance change at 260 nm is shown. The circles represent experimental points and the solid line was calculated from the fit of the data to Scheme 4.
Figure 3.

Inhibition by 11 of nitrocefin (100 μM) hydrolysis catalyzed by the P99 β-lactamase. The circles represent experimental points and the solid line was calculated as described in the text.
Figure 4.

Total progress curves for turnover of cephalothin (200 μM) by the P99 β-lactamase (2.0 nM) in the absence (□) and presence (○) of 9 (0.1 μM). Absorbance changes at 278 nm are shown. The points are experimental and the lines calculated from the fit of the data to Scheme 2.
Figure 5.

Initial rates of turnover of 8 by the TEM β-lactamase (0.98 μM) as a function of concentration of 8. The circles represent experimental points and the solid line was calculated as described in the text.
Analysis of data of the kind shown in Figures 1–6 led to the derived rate and equilibrium constants of Table 1, which gives an overview of the effectiveness of 5–12 as inhibitors of the two β-lactamases. At concentrations of these compounds ≥100 μM, and at lower concentrations than this for many of them, the enzyme would be held in the steady state essentially completely as EI (Scheme 1) and thus would be effectively inactive against other substrates. The half-lives of these complexes with respect to breakdown to free enzyme are in the 2 s to 9 min range, depending on the enzyme and diacyl phosphate. All precedent indicates that these slowly dissociating complexes are aroyl-enzymes (8,9) so that the ki values of Table 1 represent rate constants of acylation of the active site, and k2 of deacylation. This interpretation was supported by observations of the effect of methanol on the steady state rates of turnover of 9 by the P99 β-lactamase. The observed rate increased linearly with methanol concentration (not shown), as usually observed in substrate [including 5 (9)] turnover by this enzyme where deacylation is usually rate-determining (13,17), and, after quantitative analysis of the data in terms of Scheme 6, a k3/k2 value of 28.5 ± 0.8 was obtained.
Scheme 6.

This is similar to a previously determined ratio of 23.6 for 5 and indicates, as might be expected, that methanol has similar access to the acyl-enzyme derived from 9 as from 5. It will be assumed for further discussion that 5–12 form covalent acyl-enzyme intermediates with all serine β-lactamases; note that this has previously been established for a typical class D enzyme (11),
Complications beyond Scheme 1 were observed in certain cases. Uniquely, compound 8 irreversibly inactivated the enzyme. In terms of a branched reaction path (Scheme 4), the value of k3 was (4.3 ± 0.1) × 10−3 s−1; note, however, that a parallel reaction scheme would have fitted the data equally well. It seems possible that 8, the most chemically reactive of the substituted benzoyl derivatives, may also be able to acylate an amino acid residue other than the active site serine and thereby inactivate the enzyme. Direct acylation of or intramolecular acyl transfer to an active site lysine residue is one possibility (18). The other compounds may also be able to achieve this at concentrations higher than those used in the described experiments (i.e. after more turnovers per enzyme).
A branched reaction scheme (Scheme 5) was also needed to fully explain the kinetics of reaction between 5, at high concentrations (>100 μM), and the TEM enzyme. As described previously, it appears that the acyl enzyme, EI, in this case can also rearrange to a more inert complex EI′ [k3 = (3.3 ± 0.7) × 10−4 s−1] but which, unlike that from 8 (see above), can slowly break down to free enzyme (k4 = 1.5 × 10−4 s−1). Again, the other compounds may also display this mode of reaction if studied at higher concentrations.
Finally, the very hydrophobic compounds 9 and 10 were found to exhibit “substrate” inhibition of the P99 enzyme (Scheme 3) at higher concentrations. Kii values were (20 ± 3) μM and (115 ± 20) μM for 9 and 10, respectively. The P99 β-lactamase is well known to have an extended binding site where a variety of ligands can bind in addition to substrate at the active site (19, 20). One would expect that 11 and 12 would also exhibit this behavior at concentrations above those employed (i.e. > 10 μM).
Inspection of Table 1 shows that the acylation and deacylation rate constants vary significantly among the various compounds. The effect of electronic effects can be assessed by considering compounds 5 – 8. It seems that although the acylation rates do not vary systematically, the deacylation rates generally increase as electron withdrawing substituents are added (the Hammett ρ value is ca 1.5 for both enzymes). This suggests that direct nucleophilic attack of water on the acyl-enzyme is a major contributor to the rate-determining process in deacylation. That the rate of acylation does not also directly reflect these electronic effects demonstrates that the observed rates are modulated by strong interaction of the leaving group with the enzyme (see below), with the latter perhaps acting as an electrophile.
The kinetics data for compounds 9–12 with both enzymes demonstrate the dramatic effect of hydrophobic substitution. Although the deacylation rates of 9 and 10 are not significantly different from those of 5–8, the acylation rates are strikingly larger. The latter applies to 11 and 12 also, although less so to the more polar 12. The deacylation rates of 11 and 12 are also much larger than those of 5–10, reflecting the electronic effect of the heteroatom, which is seen also in spontaneous hydrolysis rates (see above).
Another measure of the effectiveness of these compounds as β-lactamase inhibitors, taking into account both acylation and deacylation rates, is the ratio of k2 and ki, the effective thermodynamic inhibition constant Ki. By this measure, as indicated by the data in Table 1, 11 stands out as the most effective inhibitor of the P99 β-lactamase, followed by 10 and 9, while 9, followed by 10 and 6, is most effective against the TEM-2 enzyme. The effectiveness of 11 against the P99 enzyme is particularly striking and reflects the already noticed affinity of the benzothiophene moiety for the class C β-lactamase active site (8,21,22). The question of whether this effectiveness is dictated by the acyl group, the leaving group, or both, is addressed in the following paper.
The data of Table 1 also demonstrate that the diaroyl phosphates selected for study here are generally more effective against the P99 β-lactamase than against TEM-2. Comparison of these results with those previously published for a class D β-lactamase (OXA-1) suggests that the latter enzyme is generally more strongly inhibited (Ki) than the class C P99 enzyme (although 11 and 12, with Ki values for OXA-1 determined to be 0.06 μM and 14.2 μM, respectively, are somewhat more effective against the class C enzyme than the class D). The general result, however, and particularly with respect to the enhanced activity of the more hydrophobic molecules 9 and 10, probably reflects the relative general hydrophobicity of the active sites involved, viz. class D > class C > class A. These structural differences are seen in the molecular models described below.
In order to understand how the diacyl phosphates might engage the active site functionality of the class A and class C β-lactamases, molecular models of acylation tetrahedral intermediates (13) were constructed. These were based on the crystal structures of these enzymes (14,16) and on those of various covalent complexes with poor substrates/inhibitors (23–26). The constructed models involved deliberate placement of the acyl donor phenyl group in the side chain site of the enzymes and the phosphate leaving group in the leaving group site. We felt that there was a better match of polarities with the inhibitor oriented in this fashion rather than in the reverse, particularly with respect to that of the very polar leaving group with the polar side chains (Lys 73, Ser 130, Lys 234, Ser 235, and Arg 244 in the TEM β-lactamase and Lys 67, Tyr 150, Lys 315, and Thr 316 in the P99 enzyme) of the leaving group site. After construction of the models, a short (100 psec) molecular dynamics equilibration of the structure was run, followed by energy-minimization of dominant conformers. From these studies, the structures shown in Figure 7 emerged as those containing ligands that most strongly (by the criterion of Eint values- see Experimental section) interacted with each enzyme active site.
Figure 7.

A. Stereoview of an energy minimized tetrahedral intermediate structure, 13, formed on reaction of the TEM β-lactamase with dibenzoyl phosphate. Only heavy atoms are shown. B. As for A, but with the P99 β-lactamase.
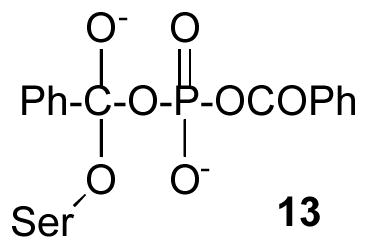
Figure 7A shows the complex 13 with the class A TEM β-lactamase. The active site constituents appear in their normal interactive mode. The side chains of Glu 166 and Asn 170 are hydrogen-bonded to each other (N-O distance ≤ 3.0 Å) and each to a common water molecule (“the deacylating water molecule”), and the side chain of Asn 132 to that of Lys 73. The latter is hydrogen-bonded to Ser 70 Oγ as it might be in an acylation transition state (24,25). Ser 130 Oγ is most directly interacting, presumably by means of a hydrogen bond, with one of the phosphoryl oxygens. This phosphoryl oxygen is also hydrogen bonded to the cationic ammonium ion of Lys 234. The second phosphoryl oxygen appears to be hydrogen-bonded to the side chains of Ser 235 and Arg 244. A nitrogen atom of the latter side chain is also only 3.2 A from the leaving group carbonyl oxygen. The tetrahedral oxyanion finds itself in an asymmetric position in the usual oxyanion hole – within strong hydrogen bonding distance of Ser 70 N but much further (3.5 Å) from Ala 237 N. The tetrahedral adduct seems to have tilted somewhat, such that the leaving group oxygen is hydrogen bonded to Ala 237 N. This may reflect the effort of one phosphoryl oxygen to come into direct contact with Arg 244. In several crystal structures of complexes between penicillins and TEM β-lactamases, this arginine is found to be hydrogen-bonded to the penam carboxylate (25,27,28). In class C enzymes, however, a water molecule is often found interceding between this carboxylate and a comparably placed arginine residue (26,29). If such a water molecule were inserted between Arg 244 and the phosphoryl oxygen in Figure 7A, the leaving group would tilt back, putting the tetrahedral oxyanion oxygen into hydrogen bonding contact with Ala 237 N. Despite the uncertainty of the meaning of this last described issue, the structure does clearly show that the acyl phosphate leaving group is able to interact very strongly with the polar residues of the active site, in a way that would enhance the leaving group ability of the former.
A similar picture for the class C P99 β-lactamase emerges on inspection of Figure 7B. The Lys 67 ammonium ion is hydrogen-bonded to Ser 64 Oγ. The Tyr 150 hydroxyl oxygen is within hydrogen-bonding distance of both Ser 64 Oγ and the leaving group oxygen. The leaving group carbonyl oxygen is apparently hydrogen-bonded to both the Lys 315 ammonium ion and the Thr 316 side chain hydroxyl oxygen. The latter is also close to one phosphoryl oxygen. The tetrahedral oxyanion lies asymmetrically in the oxyanion hole, closer to Ser 318 N. The second phosphoryl oxygen in this case, unlike with the TEM enzyme, does not appear in direct contact with the enzyme. Nonetheless, the interactions between the enzyme and leaving group that are present would also promote fission of the leaving group from the tetrahedral intermediate.
Diacyl phosphates, therefore, are inhibitory substrates of class A and class C β-lactamases that strongly interact with these enzymes. Their rapid acylation of the enzymes (104 – 10 6 s−1M−1) probably largely derives from interactions between the anionic phosphate leaving group and the enzyme active site. Such interactions probably enhance both the electrophilicity of the acylating carbonyl group and also the ease of departure of the leaving group. These interactions are further enhanced by hydrophobic acyl groups and, as demonstrated in the accompanying paper, by cooperative interactions involving the diacyl phosphates themselves. The ensuing acyl-enzymes hydrolyze much more slowly, 10−3 – 10−1 s−1, than those derived from good substrates (100–1000 s−1) (30), probably because of the absence of specific interactions between the substrate and the enzyme afforded, with good substrates, by amidoacyl groups. A study of the reactivity of diacyl phosphates with class D β-lactamases revealed similar results (11). It does seem possible, therefore, that a subnanomolar (Ki) inhibitor of all serine β-lactamases might be achievable based on the diacyl phosphate platform.
Abbreviations
- CENTA
7β-[(thien-2-yl)acetamido]-3-[(4-nitro-3-carboxyphenylthio)methyl]-3-cephem-4-carboxylic acid
- DMSO
dimethyl sulfoxide
- ESMS
electrospray mass spectrometry
- FTIR
Fourier transform infra-red
- MOPS
3-(N-morpholino)propanesulfonic acid
- NMR
nuclear magnetic resonance
Footnotes
This research was supported by National Institutes of Health, Grant R01 AI-17986
References
- 1.Hawkey PM. The growing burden of antimicrobial resistance. J Antimicrob Chemother. 2008;62(Suppl 1):1–9. doi: 10.1093/jac/dkn241. [DOI] [PubMed] [Google Scholar]
- 2.Helfand MS, Rice LB. Problematic β-lactamases: an update. Infect Disease Ther. 2008;48:169–182. [Google Scholar]
- 3.Buynak JD. Understand the longevity of the β-lactam antibiotics and of antibiotic/β-lactamase inhibitor combinations. Biochem Pharmacol. 2006;71:930–940. doi: 10.1016/j.bcp.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 4.Silver LL. Novel broad-spectrum β-lactamase inhibitors. Expert Opin Ther Pat. 2007;17:1175–1181. [Google Scholar]
- 5.Bassetti M, Righi E, Viscoli C. Novel β-lactam antibiotics and inhibitor combinations. Expert Opin Invest Drugs. 2008;17:285–296. doi: 10.1517/13543784.17.3.285. [DOI] [PubMed] [Google Scholar]
- 6.Matagne A, Dubus A, Galleni M, Frere JM. The β-lactamase cycle: a tale of selective pressure and bacterial ingenuity. Nat Prod Rep. 1999;16:1–19. doi: 10.1039/a705983c. [DOI] [PubMed] [Google Scholar]
- 7.Rahil J, Pratt RF. Mechanism of inhibition of the class C β-lactamase of Enterobacter cloacae by phosphonate monoesters. Biochemistry. 1992;31:5869–5878. doi: 10.1021/bi00140a024. [DOI] [PubMed] [Google Scholar]
- 8.Kaur K, Pratt RF. Mechanism of reaction of acyl phosph(on)ates with the β-lactamase of Enterobacter cloacae P99. Biochemistry. 2001;40:4610–4621. doi: 10.1021/bi002243+. [DOI] [PubMed] [Google Scholar]
- 9.Li N, Pratt RF. Inhibition of serine β-lactamases by acyl phosph(on)ates: a new source of inert acyl (and phosphyl) enzymes. J Amer Chem Sec. 1998;120:4264–4268. [Google Scholar]
- 10.Adediran SA, Nukaga M, Baurin S, Frere JM, Pratt RF. Inhibition of class D β-lactamases by acyl phosphates and phosphonates. Antimicrob Ag Chemother. 2005;49:4410–4412. doi: 10.1128/AAC.49.10.4410-4412.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Majumdar S, Adediran SA, Nukaga M, Pratt RF. Inhibition of class D β-lactamases by Diaroyl Phosphates. Biochemistry. 2005;44:16121–16129. doi: 10.1021/bi051719s. [DOI] [PubMed] [Google Scholar]
- 12.Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 13.Xu Y, Soto G, Hirsch KR, Pratt RF. Kinetics and mechanism of the hydrolysis of depsipeptides catalyzed by the β-lactamase of Enterobacter cloacae P99. Biochemistry. 1996;35:3595–3603. doi: 10.1021/bi952106q. [DOI] [PubMed] [Google Scholar]
- 14.Lobkovsky E, Moews PC, Liu H, Zhao H, Frere J-M, Knox JR. Evolution of an enzyme activity: crystallographic structure at 2 A resolution of cephalo-sporinase from the amp C gene of Enterobacter cloacae P99 and comparison with a class A penicillinase. Proc Natl Acad Sci. 1993;90:11257–11261. doi: 10.1073/pnas.90.23.11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Curley K, Pratt RF. Effectiveness of tetrahedral adducts as transition-state analogs and inhibitors of the class C β-lactamase of Enterobacter cloacae P99. J Am Chem Soc. 1997;119:1529–1538. [Google Scholar]
- 16.Jelsch C, Mourey L, Masson JM, Samama JP. Crystal structure of Escherichia coli TEM1 β-lactamase at 1·8 A resolution. Proteins: Struct Funct Genet. 1993;16:364–383. doi: 10.1002/prot.340160406. [DOI] [PubMed] [Google Scholar]
- 17.Govardhan CP, Pratt RF. Kinetics and mechanism of the serine β-lactamase catalyzed hydrolysis of depsipeptides. Biochemistry. 1987;26:3385–3395. doi: 10.1021/bi00386a021. [DOI] [PubMed] [Google Scholar]
- 18.Wyrembak PN, Babaoglu K, Pelto RB, Shoichet BK, Pratt RF. O-Aryloxycarbonyl hydroxamates: new β-lactamase inhibitors that crosslink the active site. J Amer Chem Soc. 2007;129:9548–9549. doi: 10.1021/ja072370u. [DOI] [PubMed] [Google Scholar]
- 19.Pazhanisamy S, Pratt RF. β-Lactamase-catalyzed aminolysis of depsipeptides: peptide inhibitors and a new kinetic mechanism. Biochemistry. 1989;28:6875–6882. doi: 10.1021/bi00443a015. [DOI] [PubMed] [Google Scholar]
- 20.Dryjanski M, Pratt RF. Inactivation of the Enterobacter cloacae P99 β-lactamase by a fluorescent phosphonate: direct detection of ligard binding at the second site. Biochemistry. 1995;34:3569–3575. doi: 10.1021/bi00011a011. [DOI] [PubMed] [Google Scholar]
- 21.Weston GS, Blazquez J, Baquero F, Shoichet DK. Structure-based enhancement of boronic acid-based inhibitors of Amp C β-lactamase. J Med Chem. 1998;41:4577–4586. doi: 10.1021/jm980343w. [DOI] [PubMed] [Google Scholar]
- 22.Powers RA, Blazquez J, Weston GS, Morosini MI, Baquero F, Shoichet BK. The complexed structure and antimicrobial activity of a non-β-lactam inhibitor of Amp C β-lactamase. Protein Sci. 1999;8:2330–2337. doi: 10.1110/ps.8.11.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lobkovsky E, Billings E, Moews PC, Rahil J, Pratt RF, Knox JR. Crystallographic structure of a phosphonate derivative of the Enterobacter cloacae P99 cephalosporinase: mechanistic interpretation of a β-lactamase transition-state analog. Biochemistry. 1994;33:6762–6772. doi: 10.1021/bi00188a004. [DOI] [PubMed] [Google Scholar]
- 24.Maveyraud L, Pratt RF, Samama JP. Crystal structure of an acylation transition-state analog of the TEM-1 β-lactamase. Mechanistic implications for class A β-lactamases. Biochemistry. 1998;37:2622–2628. doi: 10.1021/bi972501b. [DOI] [PubMed] [Google Scholar]
- 25.Strynadka NCJ, Adashi H, Jensen SE, Johns K, Sielecki A, Betzel C, Sutoh K, James MNG. Molecular structure of the acyl-enzyme intermediate in β-lactam hydrolysis at 1·7 A resolution. Nature. 1992;359:700–705. doi: 10.1038/359700a0. [DOI] [PubMed] [Google Scholar]
- 26.Patera A, Blaszczak LC, Shoichet BK. Crystal structures of substrate and inhibitor complexes with amp C β-lactamase: possible implications for substrate assisted catalysis. J Am Chem Soc. 2000;122:10504–10512. [Google Scholar]
- 27.Maveyraud L, Mourey L, Pedelacq JD, Guillet U, Kotra LK, Mobashery S, Samama JP. Structural basis for clinical longevity of carbapenem antibiotics in the face of the challenges by the common class A beta-lactamases from antibiotic-resistant bacteria. J Am Chem Soc. 1998;120:9748–9752. [Google Scholar]
- 28.Maveyraud L, Massova I, Samama JP, Mobashery S. Crystal structure of 6-alpha-hydroxymethylpenicillanate complexed to the TEM-1 beta-lactamase from Escherichia coli; Evidence on the mechanism of action of a novel inhibitor designed by a computer-aided process. J Am Chem Soc. 1996;118:7435. [Google Scholar]
- 29.Trehan I, Morandi F, Blaszczak LC, Shoichet BK. Crystal structure of Amp C beta-lactamase from E. coli in complex with amoxicillin. Chem Biol. 2002;9:971–980. doi: 10.1016/s1074-5521(02)00211-9. [DOI] [PubMed] [Google Scholar]
- 30.Matagne A, Misselyn-Bauduin AM, Joris B, Erpicum T, Granier B, Frere JM. The diversity and the catalytic properties of class A β-lactamases. Biochem J. 1990;265:131–146. doi: 10.1042/bj2650131. [DOI] [PMC free article] [PubMed] [Google Scholar]