

Abstract
Structural information such as orientations of interfacial proteins and peptides is important for understanding properties and functions of such biological molecules, which play crucial roles in biological applications and processes such as antimicrobial selectivity, membrane protein activity, biocompatibility, and biosensing performance. The α-helical and β-sheet structures are the most widely encountered secondary structures in peptides and proteins. In this paper, for the first time, a method to quantify the orientation of the interfacial β-sheet structure using a combined Attenuated Total Reflectance Fourier Transformation Infrared Spectroscopic (ATR-FTIR) and Sum Frequency Generation (SFG) vibrational spectroscopic study was developed. As an illustration of the methodology, the orientation of tachyplesin I, a 17-amino acid peptide with an anti-parallel β-sheet, adsorbed to polymer surfaces as well as associated with a lipid bilayer was determined using the regular and chiral SFG spectra, together with polarized ATR-FTIR amide I signals. Both the tilt angle (θ) and the twist angle (ψ) of the β-sheet at interfaces are determined. The developed method in this paper can be used to obtain in situ structural information of β-sheet components in complex molecules. The combination of this method and the existing methodology that is currently used to investigate α-helical structures will greatly broaden the application of optical spectroscopy in physical chemistry, biochemistry, biophysics, and structural biology.
1. Introduction
Structural information such as orientations of interfacial proteins and peptides are important for understanding properties and functions of such biological molecules, which play crucial roles in a variety of biological applications and processes. For example, the adsorbed proteins on a biomedical material surface determine the biocompatibility of the material. Surface immobilized proteins serve as sensing units for biosensors; their interfacial structures play crucial roles in biosensing performance. Membrane associated proteins are important for trans-membrane transport and cellular communication. The structure and orientation of antimicrobial peptides in cell membranes control their activity and selectivity. In order to study the interfacial orientation of complex proteins, it is necessary to examine orientations of common secondary structures that are components of interfacial peptides and proteins. The most commonly seen secondary structures are α-helices and β-sheets. We have developed systematic means to determine interfacial orientation of α-helices.1–3 In this research, we will present a systematic way to study orientation of β-sheet structure on surfaces/at interfaces.
The β-sheet structure was first proposed by Astbury in 1931; that model was not accurate until the refinements made by Pauling and Corey in 1953.4 It is a widely distributed secondary structure found in a diverse range of proteins. Many membrane-associated proteins and peptides have β-sheet or β-barrel components. Determining the orientation of this structure, is therefore crucial in having a detailed understanding of the protein’s functional mechanisms.5 Transformational changes of protein components to β-sheets have also been observed and are believed to be crucial malfunctions that lead to diseases such as mad cow disease and Creutzfeldt-Jakob disease.6,7 The formation of β-sheets at the interface can lead to the accumulation of fibrous structures that can disrupt cellular processes.7 These amyloid β-peptide (Aβ-) fibrous plaques can accumulate on the brain, leading to Alzheimer’s disease.8 The occurrence of β-sheet structures in membrane-associated proteins as well as in Aβ-plaques raises much interest about β-sheet orientation. The ability to determine the structural information of the β-sheets, including orientation of β-sheets at interfaces and on surfaces, could aid in the understanding of such important protein behaviors.
A wide range of spectroscopic techniques have been applied to the study of interfacial proteins and peptides. For instance, Attenuated Total Reflectance Fourier Transform Infrared spectroscopy (ATR-FTIR) and Raman spectroscopic techniques have been extensively used to study surface proteins/peptides, providing orientation information of these molecules while they are adsorbed on surfaces.9–32 Nuclear Magnetic Resonance (NMR) techniques have also been successfully applied to determine detailed structural information regarding membrane-associated proteins/peptides, including interaction mechanisms of adsorbed proteins/peptides.33–43
Recently, a nonlinear optical spectroscopy, sum frequency generation (SFG) vibrational spectroscopy, has been developed into a powerful technique to investigate surfaces and interfaces.44–64 SFG measures the second order nonlinear response of a system, χ(2), which is intrinsically surface sensitive, allowing peptides and proteins to be selectively studied while they are in interfacial environments such as in lipid bilayers or on polymer surfaces. In addition, SFG has been demonstrated to be an extremely sensitive technique, allowing interfacial proteins and peptides to be studied in situ and at physiological concentration.58,65–87 Protein (or peptide) - membrane interactions can also be studied in real time, revealing adsorption kinetics as well as orientation and structural changes as the proteins interact with surfaces or cell membranes. Therefore, in comparison with ATR-FTIR, Raman spectroscopy and NMR, SFG has many significant advantages such as superb detection limit, near background free detection and real time in situ study capability. SFG has been successfully applied to the orientation determination of interfacial α-helices.1–3, 85 Combination of SFG measurements with linear optical spectroscopic studies (e.g., ATR-FTIR studies) has been used to determine complicated orientation distribution of α-helices.
In this work, we successfully applied the bond additivity model to calculate the IR transition dipole moment, the Raman polarizability and eventually the SFG hyperpolarizability of the anti-parallel β-sheet. The hyperpolarizability tensor components are related to the SFG chiral and achiral signal strength, which allows for the orientation analysis of this particular structure at interfaces. Using combined ATR-FTIR and SFG studies, both the tilt angle and the twist angle of a β-sheet peptide, tachyplesin I, on polymer surfaces and in a lipid bilayer were deduced using the vibrational amide I signals. The possibility of determining the orientation of β-sheet structure is of great importance in the structural analysis of large proteins which consist of both α-helical and β-sheet structures. The orientation information of these proteins now can be investigated by two independent yet complementary SFG approaches (by studies of α-helical and β-sheet components) in situ in real time.
2. Experimental
2.1 Materials
C-terminal amidated tachyplesin I (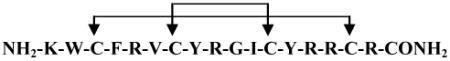 ) was purchased from GenScript USA Inc. (Piscataway, NJ) with >95% purity. Polystyrene (PS) used in this research was PS standard, purchased from Scientific Polymer Products Inc, with a molecular weight of 393,400. The sulfonated PS (s-PS) was prepared from such PS standard. The sulfonation reaction has been described previously.86 PS and PS with 15% sulfonation were used for the SFG experiments. Both the lipids, hydrogenated and deuterated 1,2-dipalmitoyl (D26)-sn-glycero-3-phosphoglycerol (DPPG and dDPPG), used in this research were purchased from Avanti Polar Lipids Inc (Alabaster, AL).
) was purchased from GenScript USA Inc. (Piscataway, NJ) with >95% purity. Polystyrene (PS) used in this research was PS standard, purchased from Scientific Polymer Products Inc, with a molecular weight of 393,400. The sulfonated PS (s-PS) was prepared from such PS standard. The sulfonation reaction has been described previously.86 PS and PS with 15% sulfonation were used for the SFG experiments. Both the lipids, hydrogenated and deuterated 1,2-dipalmitoyl (D26)-sn-glycero-3-phosphoglycerol (DPPG and dDPPG), used in this research were purchased from Avanti Polar Lipids Inc (Alabaster, AL).
2.2 Lipid bilayer deposition
Single lipid bilayers, which can have two different leaflets, were prepared on CaF2 right angle prisms (Altos Photonics, Bozeman, MT). Langmuir-Blodgett and Langmuir-Schaefer (LB/LS) methods were used to deposit the proximal and then the distal leaflets, respectively. A KSV2000 LB system and ultrapure water from a Millipore system (Millipore, Bedford, MA) were used throughout the experiments for bilayer preparation. The bilayer was immersed in water inside a 2-mL reservoir during the experiment and a small amount of water could be added to the reservoir to compensate for evaporation when needed for long timescale experiments. For tachyplesin I-bilayer interaction experiments, 10 μL of 0.25 mg/ml tachyplesin I solution was injected into the reservoir. A magnetic micro-stirrer was used at a rate of 100 rpm to ensure a homogeneous concentration distribution of peptide molecules in the subphase below the bilayer.
2.3 SFG
The details of our SFG setup and experimental design have been previously described.2, 66 Polymer films were prepared by spin coating 1 wt% polymer solutions onto CaF2 prisms at a speed of 2400 rpm. Spectra were collected in ssp (s-SFG, s-visible, p-IR), ppp and spp polarizations using our previously reported near total reflection geometry.78,81
2.4 ATR-FTIR
The ATR-FTIR spectra were collected on a Nicolet 550 spectrometer (Thermo Fisher Scientific. Inc., MA, USA). A thin polymer film was solution cast onto a germanium substrate from a 0.1 wt % polymer solution. 50 μL of 0.05 mg/ml tachyplesin I solution in D2O (Sigma Aldrich, St. Louis, MO) was injected into the ATR trough, and S and P polarized spectra were collected approximately one hour after the injection of peptide. The chamber was purged with nitrogen before and during the data collection, and the spectra were collected using 256 scans/spectrum.
3. Orientation determination of β-sheet
The SFG hyperpolarizability tensor, β, can be described as a tensor product of the IR transition dipole moment and the Raman polarizability tensor,3,44,88,89
| (1) |
where l, m, and n are the molecular coordinates, and are the Raman polarizability and IR dipole moment derivatives with respect to the normal mode coordinate of the qth vibrational mode, respectively. Throughout this paper these derivatives will be referred to as the components of the Raman polarizability tensor and IR transition dipole moment. As equation 1 indicates, if both the IR transition dipole moment and the Raman polarizability tensor are known, the SFG hyperpolarizability tensor for the vibrational mode of interest can be deduced. It is widely accepted that the anti-parallel β-sheet adopts D2 symmetry.90–92 Applying this symmetry point group, the Raman polarizability tensor and the IR transition dipole of the four peptide units that comprise the β-sheet repeating unit can be projected onto the molecular coordinate system using the bond additivity model. This treatment is similar to that used by Higgs for helical molecules.13
3.1 Anti-parallel β-sheet structure and D2 point group symmetry
The anti-parallel β-sheet belongs to D2 symmetry. For a structure with D2 point group symmetry, there are three IR active (B1, B2 and B3) and four Raman active amide I vibrational modes (A, B1, B2 and B3). The four modes are:92–96
| (2) |
where ν0 is the unperturbed peptide unit frequency and D10 and D01 account for the intrachain and interchain couplings, respectively. The term D11 represents the coupling of the transition dipoles between adjacent strands. A graphical illustration of these four vibrational modes is presented in Figure 1.
Figure 1.

Four amide I vibrational modes of a repeating unit of an anti-parallel β-sheet. For each mode, the peptide units 1, 2, 3, and 4 refer to the lower right unit, the lower left unit, the upper left unit, and the upper right unit, respectively.
3.2 The Raman polarizability tensor of an anti-parallel β-sheet
The Raman tensor for the amide I mode (Figure 2) has been proposed by Tsuboi and colleagues by investigating Raman spectra of a uniaxial tetragonal aspartame.30 For β-sheet structures, the principle x-axis of Tsuboi’s polarizability tensor is found to be significantly more in line with the C=O bond; this tilt angle is only 22 degrees (instead of 34.5 degrees in α-helical structures).95 This refinement was made by adjusting this tilt angle so that the Raman intensity ratio matches the experimental observations on Bombyx mori silk ( ), Nephila edulis spidroin ( ), S. c. ricini fibroin silk ( ), and fowl feather barb ( ).94,95,97 For the β-sheet, the polarizability tensor can be written in the given molecular frame as defined in Figure 2.
Figure 2.
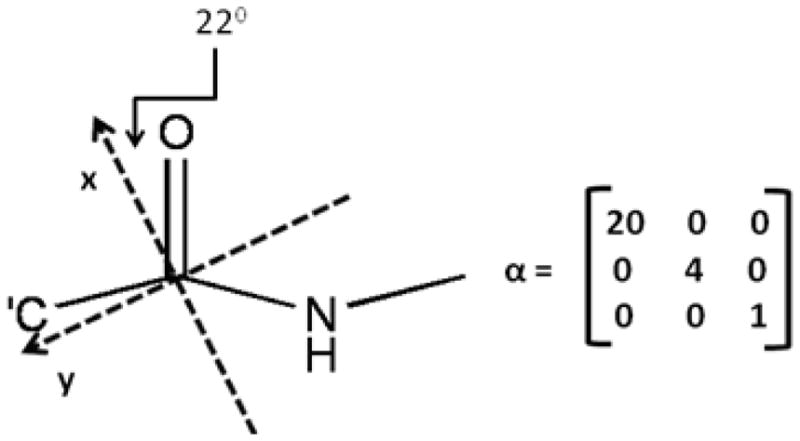
A peptide unit and its transition Raman polarizability tensor in the given molecular frame
Given this polarizability tensor and its corresponding molecular frame, an Euler transformation can be used to impose this tensor in the molecular coordinate system onto the four peptide units of the Pauling-Corey β-sheet. The Euler transformation used here follows the z-y-z convention, which has a matrix in the form of
| (3) |
and the rotations are carried out using equation (4).
| (4) |
We define a positive rotation as a rotation in the counter-clockwise direction. The four Euler angle sets used to rotate the peptide units comprising the β-sheet repeating unit from Tsuboi’s frame into Pauling-Corey’s frame are: (ψ1=292°, θ1= 120°, ϕ1= 0°), (ψ2=112°, θ2= 240°, ϕ2= 0°), (ψ3=112°, θ3= 60°, ϕ3= 0°) and (ψ4=292°, θ4= 300°, ϕ4= 0°). The first Raman polarization tensor of the anti-parallel β-sheet structure in the Pauling-Corey coordinate system can be calculated as:
| (5) |
The four resulting Raman polarization tensors after the rotation are
| (6) |
The transition Raman polarizability tensor of the A, B1, B2 and B3 modes of the repeating unit of the β-sheet structure can be calculated from these Raman polarizability tensors, with the consideration of the phase of the amide I mode for each peptide unit (shown in Figure 1) in the A, B1, B2 and B3 modes.
A mode:
| (7) |
B1 mode:
| (8) |
B2 mode:
| (9) |
B3 mode:
| (10) |
As seen from these modes, the A mode dominates the Raman spectra of the anti-parallel β-sheet structures, which was also experimentally observed.94
3.3 IR transition dipole moment of an anti-parallel β-sheet
The calculation of the IR transition dipole moment of the individual peptide unit comprising one repeating unit of the anti-parallel β-sheet structure (shown in Figure 3)98 has been reported by Marsh.92 We slightly modified Marsh’s projection by transforming the dipole moment of the peptide unit into Tsuboi’s frame, followed by a transformation into the Pauling-Corey frame using the Euler transformation that was introduced previously in section 3.2. We have verified the outcome of this approach (projection followed by rotation) with the results obtained from the conventional projection to ensure that the proper rotational operations were performed. The final calculated dipole moments of the four peptide units are
Figure 3.

Orientation of the IR transition dipole moment of the individual peptide unit comprising the anti-parallel β-sheet structure. The dipole moment lies in the plane that is inclined at an angle of 30° to the strand axis, and makes an angle of 19° to the axis that connects the two successive α-carbons of the two strands.
| (11) |
From these transition dipole moments of the four individual peptide units in a β-sheet repeating unit, one can see that the symmetric A mode is inactive in IR spectroscopy. The overall transition dipole moment of the B1, B2 and B3 vibrational modes can be calculated: 93
B1 mode:
| (12) |
B2 mode:
| (13) |
B3 mode:
| (14) |
The calculated overall dipole moment is indeed supported by the experimental observations with a strong peak at 1635 cm−1. This low frequency mode was assigned to the B2 representation, while the high frequency absorption peak at 1685 cm−1 was assigned to be the B1 representation. If we define the molecular (a, b, c) frame to be superimposed with the lab (x, y, z) coordinate system, the B2 mode of the β-sheet structure is y polarized, and the B1 mode is z polarized. As one can see from the calculated dipole moments, the strong mode is along the y axis, which belongs to the B2 representation. Additionally, the calculated intensity ratio between the B1/B2 modes agrees fairly well with the value reported by Choi et al. Applying normal mode analysis of the amide I vibrations, Choi reported the B1/B2 intensity to be around 0.11 versus our calculated value of 0.09. However, Choi’s calculation predicts quite different peak centers for the B1 and B2 modes as previously done by the work of Miyazawa, Tsuboi, Krimm and Marsh.99
3.4 SFG data analysis for anti-parallel β-sheet structures based on the calculated IR transition dipole moment and Raman polarizability tensor
The SFG hyperpolarizability tensor, β, is a third-rank tensor with 27 elements. It is a tensor product of the Raman polarizability tensor and the IR transition dipole moment. Hence, a vibrational mode is only SFG active when it is both IR and Raman active. Therefore, B1 (components μc and αab), B2 (μb and αac) and B3 (μa and αbc) vibrational modes of the anti-parallel β-sheet are SFG active. Conventionally, chiral signal was believed to arise primarily from the double resonance SFG process, in which the Raman tensor is asymmetric.100,101 However, Shen and colleagues used perturbation theory of vibrational SFG to predict that the generation of chiral signal is plausible in vibrational SFG without the requirement of an asymmetric Raman tensor.100,102 Evidentially, our laboratory has also experimentally observed strong chiral vibrational SFG signal with the anti-parallel β-sheet, which was also predicted by Simpson and coworkers.81,103
In this paper we report the SFG achiral signal observed in ssp polarization combination and the SFG chiral signal observed in spp polarization.81,104
| (15) |
| (16) |
The normalized Fresnel factors Lyyz, Lyzx and Lyxz are calculated to be 11.0, 5.2 and 1.0, respectively for our experimental geometry which has been described in our previous publications.2,65 These Fresnel coefficients were normalized relative to Lyxz so that the refractive index of the interfacial medium (PS/peptide solution interface and lipid bilayer with the peptides) does not need to be determined explicitly.
To relate the molecular SFG hyperpolarizability, βabc, to the macroscopic SFG susceptibility, , we use a set of three Euler angles ϕ, θ, and ψ representing the in-plane rotation, the tilt angle and the twist angle, respectively. The transformation matrix therefore can be written as
| (17) |
Please notice that eqn (17) is the same as eqn (3). Using this transformation, the components can be related to hyperpolarizability components of anti-parallel β-sheet.
The macroscopic SFG susceptibility quantity, , can be described by the molecular hyperpolarizability tensor components: 44,89,105–108
| (18) |
The three vibrational modes of the anti-parallel β-sheet can be observed in the laboratory coordinate system by the following relations:81 B1 mode:
| (19) |
B2 mode:
| (20) |
B3 mode:
| (21) |
In the above equations, can be measured using different polarization combinations of the input and output beams in SFG experiments. The hyperpolarizability quantities βabc, βacb and βbca can be calculated from the Raman tensors and IR transition dipoles, details of which can be found in the supporting information:
| (22) |
Equations (18) to (21) show clearly that SFG vibrational spectroscopy is a technique in which signal strength is dependent on the orientation of the oscillators and is intrinsically sensitive to asymmetric systems (or systems with no inversion symmetry). It was questioned previously whether SFG can be used to study β-sheet structure due to its semi-symmetric structure. We successfully detected both chiral and achiral SFG amide I signals from interfacial β-sheet structures, even though such signals are weak.81,84 This demonstrates the feasibility of using SFG to study β-sheet structure. However, no quantitative orientation information on interfacial β-sheet structure has ever been obtained.
According to the above equations, measured values and calculated hyperpolarizability quantities, tile angle θ and twist angle ψ can be determined. For example, from the above equations, the relationship between the SFG susceptibility ratio and the two orientation angles θ and ψ is shown in Figure 4a, illustrating the ability to determine the two orientation angles based on observed SFG signal. Figure 4b shows the relationship between the SFG susceptibility component of the B1 mode and the tilt (θ) and twist angle (ψ) of the β-sheet. The same relationship for the B2 mode is displayed in Figure 4c.
Figure 4.
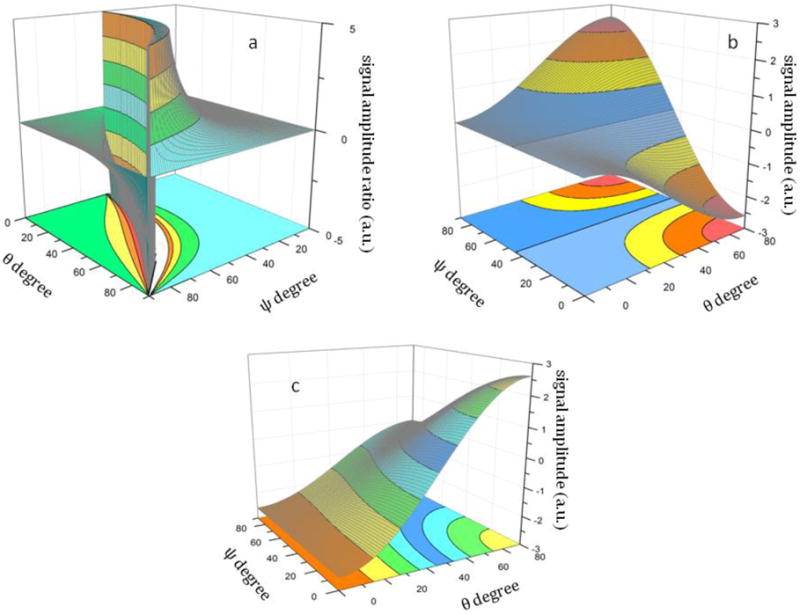
The relationship between the SFG susceptibility a). ratio; b.) c.) and the tilt (θ) and twist angle (ψ) of the β-sheet
In principle, all three SFG active modes, which are related to the molecular hyperpolarizability components βabc, βacb and βbca (equations 19, 20 and 21), can be observed using SFG. However, given that βacb is roughly 3 times higher than the next strongest vibrational mode (9 folds stronger in intensity), we will focus on the detection of achiral and chiral signals of the B2 vibrational mode for the orientation analysis of the anti-parallel β-sheet, if the B2 mode is observed experimentally. We will also demonstrate the feasibility of applying the interference enhancement method to enhance the chiral signal in situations in which none of the chiral signals is observed directly, as in the case of tachyplesin I adsorbed onto a lipid bilayer. The spp spectrum was deduced by using interference between ssp and spp, where the visible polarization was tuned ±20° away from the p polarization.81
| (23) |
Where K is a constant and is the phase difference between and .
4. Experimental Results and Discussion
4.1 The interaction between tachyplesin I and polystyrene (PS) polymer surface
SFG spectra were collected from the PS/tachyplesin I solution (~700 nM) using different polarization combinations such as ssp (s-SFG, s-visible, p-IR, Figure 5a) and spp (Figure 5b). As we discussed in our previous publication81, ssp polarization combination can be used to probe achiral signal, while spp polarization probes the chiral signal of the molecule. The ssp signal (Figure 5a) has several contributions which are centered at 1635 cm−1, 1642 cm−1, 1665 cm−1, 1685 cm−1 and 1730 cm−1, dominated by the 1635 cm−1 and 1730 cm−1 peaks. These signals correspond to the B2 mode, unordered structure, β-turns/unordered structures, B1/inter-β-strand and B3/side chains, respectively.20,81,92 The spp spectrum (Figure 5b) is dominated by a peak centered at 1635 cm−1, belonging to the B2 mode. A very weak peak centered at 1685 cm−1 from the B1 mode, along with two weak bands at 1615 cm−1 and 1710 cm−1, are also observed. As we discussed above, we can obtain some orientation information of the anti-parallel β-sheet structure of tachyplesin I using the signal strength measured in these two polarization combinations (ssp and spp). This is different from the α-helix orientation analysis in which the ppp and ssp signal strength ratio is used. Due to the D2 symmetry point group of the β-sheet structure, orientation information of this structure includes both the tilt angle (θ) and the twist angle (ψ). Because the SFG measurements in ppp and ssp polarization combination for the D2 symmetry point group are not independent to each other; their intensity ratio stays constant (about 2.0), regardless of the β-sheet orientation. Therefore the signal strength ratio measured in the ssp and ppp polarization combinations cannot be used for orientation determination here.
Figure 5.
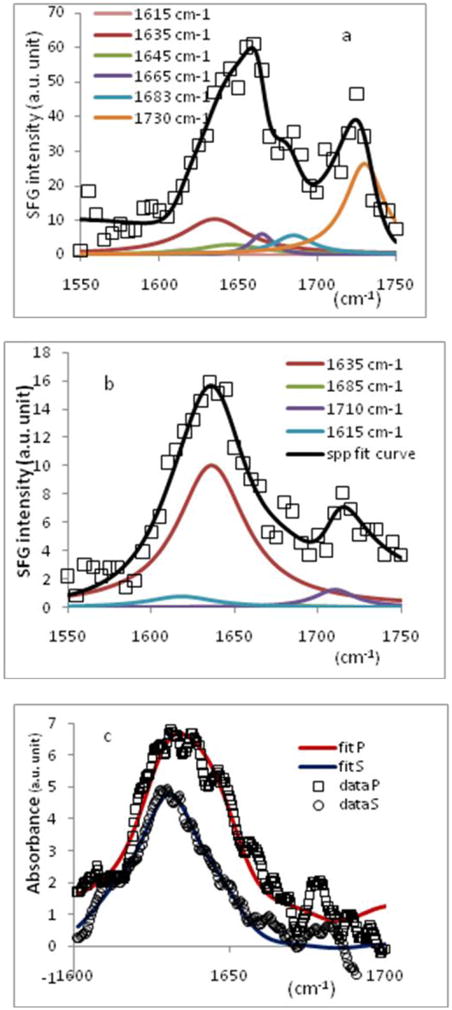
Amide I spectrum of tachyplesin I (~700 nM) adsorbed onto PS surface in a.) SFG ssp polarization combination; b.) SFG spp polarization combination and c.) ATR-FTIR amide I band obtained in s and p polarizations.
In this paper, the average orientation of tachyplesin I on a PS surface was determined by combining the results from ATR-FTIR and SFG measurements. Polarized ATR-FTIR spectra collected from the PS/tachyplesin I solution interface are presented in Figure 5c. The orientation determination methodology using ATR-FTIR of the anti-parallel β-sheet has been previously reported by Marsh.92 Assuming the staggering of the hydrogen bond between the adjacent strands is by one peptide unit, the strand tilt angle with the sheet was calculated to be 35° (or 60° if the staggering is caused by two peptide units).109 The tilt angle, θ, of the β-sheet structure can be written as
| (24) |
where Ex, Ey and Ez are the components of the electric field vector.
Combining ATR-FTIR with the SFG measurements, the following equations can be used to determine the complete orientation of the β-sheet.
| (25) |
From experimental measurements the ratio was found to be 1.04 (Figures 5a and 5b), and the RATR was determined to be 0.95 (Figure 5c). After inputting the appropriate radiation electric field vectors (Ex, Ey, Ez), we obtained two sets of solutions (θ= 76°, ψ= 86°) and (θ= 76°, ψ= 43°) for equations (25). However, according to the spectral fitting results of the chiral signal observed with spp polarization combination (Figure 5b), there was no discernable B1 mode, which excludes the solution set with ψ= 86° which would give relatively strong signal in B1 mode (Figure 4b). Therefore the tilt and twist angles for tachyplesin I at the PS/tachyplesin I solution are 76° and 43°, respectively. It is worth mentioning that around this particular orientation, the measured SFG signal ratio has a steep orientation angle dependent slope, therefore the orientation deduced should be relatively accurate.
In addition to the PS, we also applied a similar method to study molecular interactions between tachyplesin I and sulfonated polystyrene (s-PS). Similarly, both SFG chiral and achiral amide I signals were observed (Figure 6). To ensure that the observed SFG signal is indeed from the β-sheet structure, dithiothreitol (DTT) was added to the interaction medium before the additions of the peptide, which can denature the native structure of tachyplesin I by breaking the disulfide bonds. With the presence of DTT in the interaction medium, after the addition of the peptide, no chiral signal was observed from the s-PS/tachyplesin I solution interface in spp polarization combination and the ssp spectrum underwent a significant blue shift (these spectra can be found in the supporting material). The blue shift seen in the ssp spectrum corresponds to the loss of β-sheet signal centered at 1635 cm−1. The disappearance of the chiral signal and the spectral change in the achiral signal after the addition of DTT indicates the original chiral signal and the 1635 cm−1 peak in the ssp spectrum are contributed by the β-sheet structure. The detailed data analysis suggests a slightly different orientation of tachyplesin I at the s-PS/tachyplesin I solution interface.
Figure 6.

SFG amide I spectrum of tachyplesin I adsorbed to s-PS surface with and without addition of DTT in a). spp polarization combination and b.) ssp polarization combination
4.2 The interaction between tachyplesin I and lipid bilayer
Tachyplesin I is an effective antimicrobial peptide that interacts with bacterial and mammalian cell membranes differently.110,111 Using SFG, we successfully probed different interactions and orientations of tachyplesin I in both model mammalian and bacterial cell membranes. In brief, tachyplesin I does not bind effectively onto zwitterionic lipids such as 1-Palmitoyl-2-Oleoyl-sn-Glycero-3-Phosphocholine (POPC) or 1,2-Dimyristoyl-sn-Glycero-3-Phosphocholine (DMPC), which are similar to the major components of mammalian cell membranes. On the other hand, it binds quickly to the negatively charged lipid such as 1-Palmitoyl-2-Oleoyl-sn-Glycero-3-[Phospho-rac-(1-glycerol)] (POPG) and 1,2-Dipalmitoyl-sn-Glycero-3-Phosphoglycerol (DPPG) by the aid of the electrostatic attraction. PG lipids are the main components of bacterial cell membranes. We also obtained the SFG amide I achiral and chiral signal of membrane bound tachyplesin I (Figures 7a and 7b). The achiral signal from the peptide is relatively strong; however, the chiral signal could not be observed directly using the spp polarization combination. The interference enhancement method, which was discussed in our previous publication81, was implemented to deduce the weak chiral signal. A chiral spp spectrum was deduced, dominated by a clear peak centered at around 1685 cm−1, featuring the B1 vibrational mode (Figure 7b). By combining the signal strengths of the B1 and B2 modes, the orientation angles of tachyplesin I on the DPPG/deuterated DPPG (dDPPG) lipid bilayer can be estimated (Figure 4b and 4c): the tilt angle (θ) has a range of 75–90 degrees and the twist angle (ψ) has a range of 75–90 degrees. Tachyplesin I evidently adopts quite a different twist angle on DPPG/dDPPG lipid bilayer than it does on PS or sPS polymer surfaces. The different twist angles must be caused by the different molecular interactions between tachyplesin I and polymer surfaces and between tachyplesin I and negatively charged lipids. We believe that this is the first time the orientation information (θ, ψ) of antimicrobial peptides with β-sheet structure in cell membranes is determined using vibrational spectroscopies. Such information is important in understanding molecular mechanisms of interactions between β-sheet peptides and cell membranes, aiding in the design and development of improved anti-microbial peptides with β-sheet structures.
Figure 7.
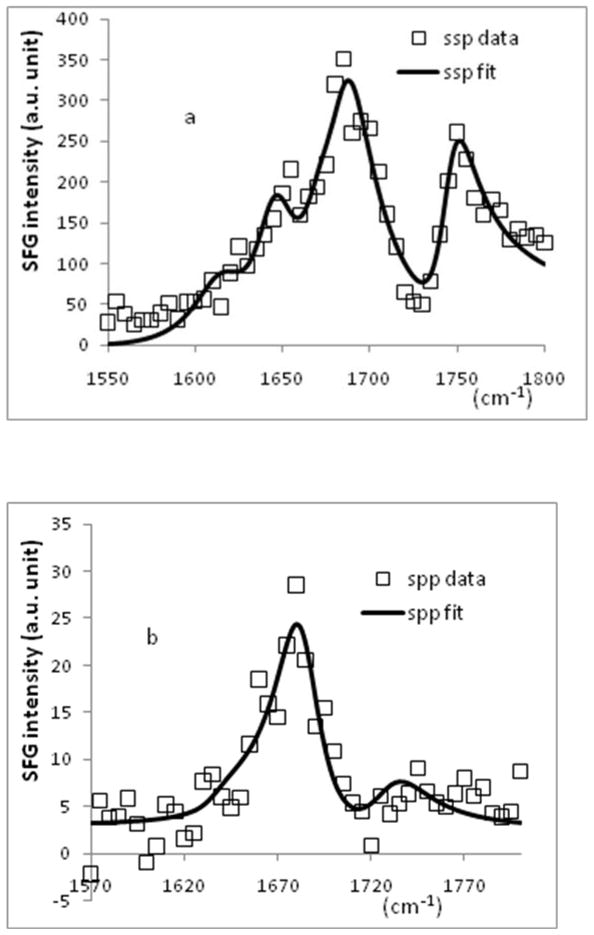
SFG spectrum of tachyplesin I (~700 nM) adsorbed onto DPPG/dDPPG lipid bilayer in a.) ssp polarization combination and b.) spp polarization combination.
Conclusion
In this paper, for the first time, a systematic method for determining β-sheet orientation on surfaces using SFG vibrational spectroscopy together with ATR-FTIR was presented. Using the bond additivity model, the IR transition dipole and the Raman polarizability tensor were calculated for the β-sheet structure. From the calculated IR dipole and Raman polarizability, the molecular hyperpolarizability was found, which was used to determine the tilt angle (θ) and the twist angle (ψ) of the β-sheet at different interfaces. Using SFG, the chiral and achiral components of the B2 vibrational mode were selectively probed and were ultimately used to deduce the orientation of tachyplesin I adsorbed to a PS surface. The relative SFG signal intensities of B1 and B2 modes were then used to determine the orientation angles of tachyplesin I in DPPG/dDPPG lipid bilayers. This methodology can be applied to the study of β-sheets and β-sheet containing proteins on surfaces such as polymer surfaces or cell membranes in situ, with the capability of performing real time studies on conformational changes that occur in many interfacial proteins. In the future, various signal enhanced methods will be adopted to directly probe modes other than B2 to provide additional measured parameters to determine more complicated orientations or orientation distributions. Coupled with previously reported orientation analysis of α-helices,1 this method lends itself to the study of larger and more complex interfacial proteins that have α-helix and/or β-sheet components.
Acknowledgments
This research is supported by National Institute of Health (1R01GM081655-01A2) and Office of Naval Research (N00014-08-1-1211). We thank Dr. Pei Yang for his fruitful discussion regarding the calculations.
Biographies

Khoi Nguyen received his first bachelor’s degree in electrical engineering from the Institute of Telecommunications of Saigon in 2002. He then received two bachelor’s degrees in mathematics and chemistry from the New Mexico Institute of Mining and Technology in 2006. He graduated with a PhD degree in chemistry from the University of Michigan in 2009. Currently he is an assistant professor in the School of Biotechnology, International University, National University of Ho Chi Minh City.

John King received his B.S. degree in chemistry from The State University of New York at Fredonia in 2009. He is currently a graduate student at the University of Michigan in Ann Arbor, working towards his Ph.D. in physical chemistry.

Zhan Chen received his B.S. degree in chemistry from Peking University in 1988. He then received his M.S. degree in physics from Institute of Physics, Chinese Academy of Sciences in 1991. After he worked in Institute of Physics for two years as a research scientist, he went to the Department of Chemistry at the University of California at Berkeley in 1993. He received his Ph.D. degree in 1998, advised by Prof. Herbert Strauss. He then worked as a postdoctoral research fellow in Prof. Gabor Somorjai’s group at Berkeley. He started his own research group in 2000 at the University of Michigan. Currently he is a professor of Chemistry there. His research is to understand molecular structures of polymers and biological molecules at interfaces.
References
- 1.Nguyen KT, Le Clair SV, Ye S, Chen Z. J Phys Chem B. 2009;113:12169–12180. doi: 10.1021/jp904153z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen X, Wang J, Boughton AP, Kristalyn CB, Chen Z. J Am Chem Soc. 2007;129:1420–1427. doi: 10.1021/ja067446l. [DOI] [PubMed] [Google Scholar]
- 3.Wang J, Lee SH, Chen Z. J Phys Chem B. 2008;112:2281–2290. doi: 10.1021/jp077556u. [DOI] [PubMed] [Google Scholar]
- 4.Pauling L, Corey RB. Proc Nat Acad Sci USA. 1953;39:253–256. doi: 10.1073/pnas.39.4.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nabedryk E, Garavito RM, Breton J. Biophys J. 1988;53:671–676. doi: 10.1016/S0006-3495(88)83148-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan K-M, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Melhorn I, Huang Z, Fletterick RJ, Cohen FE, Prusiner SB. Proc Natl Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sugiyama Y, Inoue Y, Muneyuki E, Haneda H, Fujimoto M. J Electron Micros. 2006;55:143–149. doi: 10.1093/jmicro/dfl024. [DOI] [PubMed] [Google Scholar]
- 8.Hardy J, Selkoe DJ. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 9.Axelsen PH, Citra MJ. Prog Biophys Molec Biol. 1996;66:227–253. doi: 10.1016/s0079-6107(97)00007-2. [DOI] [PubMed] [Google Scholar]
- 10.Bechinger B, Ruysschaert JM, Goormaghtigh E. Biophys J. 1999;76:552–563. doi: 10.1016/S0006-3495(99)77223-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradbury EM, Brown L, Downie AR, Elliot A, Fraser RDB, Hanby WE. J Mol Biol. 1962;5:230–247. doi: 10.1016/s0022-2836(62)80086-2. [DOI] [PubMed] [Google Scholar]
- 12.Fraser RDB. J Chem Phys. 1953;21:1511–1515. [Google Scholar]
- 13.Higgs PW. Proc R Soc Lond A. 1953;220:472–485. [Google Scholar]
- 14.Lee SH, Krimm S. Biopolymers. 1998;46:283–317. [Google Scholar]
- 15.Marsh D, Müller M, Schmitt FJ. Biophys J. 2000;78:2499–2510. doi: 10.1016/S0006-3495(00)76795-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marsh D. Methods Enzymol. 1999;294:59–92. doi: 10.1016/s0076-6879(99)94007-7. [DOI] [PubMed] [Google Scholar]
- 17.Marsh D. Biophys J. 1998;75:354–358. doi: 10.1016/S0006-3495(98)77519-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyazawa T, Blout ER. J Am Chem Soc. 1961;83:712–719. [Google Scholar]
- 19.Reisdorf W, Jr, Krimm S. Biophys J. 1995;69:271–273. doi: 10.1016/S0006-3495(95)79898-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamm LK, Tatulian SA. Q Rev Biophys. 1997;30:365–429. doi: 10.1017/s0033583597003375. [DOI] [PubMed] [Google Scholar]
- 21.Tsuboi M. J Polymer Sci. 1962;59:139–153. [Google Scholar]
- 22.Chen LX, Strauss HL, Snyder RG. Biophys J. 1993;64:1533–1541. doi: 10.1016/S0006-3495(93)81522-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dwivedi AM, Krimm S. Biopolymers. 1984;23:2025–2065. doi: 10.1002/bip.360230509. [DOI] [PubMed] [Google Scholar]
- 24.Fanconi B, Tomlinson B, Nafie LA, Small W, Peticolas WL. J Chem Phys. 1969;51:3993–4005. doi: 10.1063/1.1672620. [DOI] [PubMed] [Google Scholar]
- 25.Fanconi B. Biopolymers. 1973;12:2759–2776. doi: 10.1002/bip.1973.360121210. [DOI] [PubMed] [Google Scholar]
- 26.Koenig JL, Sutton PL. Biopolymers. 1969;8:167–171. [Google Scholar]
- 27.Lee S-H, Krimm S. J Raman Spectrosc. 1998;29:73–80. [Google Scholar]
- 28.Overman SA, Tsuboi M, Thomas GJ., Jr J Mol Biol. 1996;259:331–336. doi: 10.1006/jmbi.1996.0323. [DOI] [PubMed] [Google Scholar]
- 29.Tsuboi M, Benevides JM, Bondre P, Thomas GJ., Jr Biochemistry. 2005;44:3091–3100. [Google Scholar]
- 30.Tsuboi M, Ikeda T, Ueda T. J of Raman Spectrosc. 1991;22:619–626. [Google Scholar]
- 31.Wilser WT, Fitchen DB. J Chem Phys. 1975;62:720–724. [Google Scholar]
- 32.Axelsen PH, Kaufman BK, McElhaney RN, Lewis RN. Biophys J. 1995;69:2770–2781. doi: 10.1016/S0006-3495(95)80150-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bechinger B. Biochim Biophys Atca. 1999;1462:157–183. doi: 10.1016/s0005-2736(99)00205-9. [DOI] [PubMed] [Google Scholar]
- 34.Bechinger B, Aisenbrey C, Bertani P. Biochim Biophys Atca. 2004;1666:190–204. doi: 10.1016/j.bbamem.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 35.Durr UHN, Yamamoto K, Ramamoorthy A. J Am Chem Soc. 2007;129:6670–6671. doi: 10.1021/ja069028m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dvinskikh SV, Durr UHN, Yamamoto K, Ramamoorthy A. J Am Chem Soc. 2007;129:794–802. doi: 10.1021/ja065536k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandez C, Adeishvili K, Wuthrich K. Proc Nat Acad Sci USA. 98;98:2358–2363. doi: 10.1073/pnas.051629298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee D, Walter KFA, Bruckner AK, Hilty C, Becker S, Griesinger C. J Am Chem Soc. 2008;130:13822–13823. doi: 10.1021/ja803686p. [DOI] [PubMed] [Google Scholar]
- 39.Lindblom G, Grobner G. Curr Opin Colloid Interface Sci. 2006;11:24–29. [Google Scholar]
- 40.Naito A, Kawamura I. Biochim Biophys Atca. 2007;1768:1900–1912. doi: 10.1016/j.bbamem.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 41.Andronesi OC, Becker S, Seidel K, Heise H, Young HS, Baldus M. J Am Chem Soc. 2005;127:12965–12974. doi: 10.1021/ja0530164. [DOI] [PubMed] [Google Scholar]
- 42.Hwang PM, Choy WY, Lo EI, Chen L, Forman-Kay JD, Raetz CR, Privé GG, Bishop RE, Kay LE. Proc Natl Acad Sci USA. 2002;99:13560–13565. doi: 10.1073/pnas.212344499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kovacs F, Quine J, Cross TA. Proc Natl Acad Sci USA. 1999;96:7910–7915. doi: 10.1073/pnas.96.14.7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen YR. The Principles of Nonlinear Optics. John Wiley & Sons; New York: 1984. [Google Scholar]
- 45.Shen YR, Ostroverkhov V. Chem Rev. 2006;106:1140–1154. doi: 10.1021/cr040377d. [DOI] [PubMed] [Google Scholar]
- 46.Bain CD. J Chem Soc Faraday Trans. 1995:1281–1296. [Google Scholar]
- 47.Eisenthal KB. Chem Rev. 1996;96:1343–1360. doi: 10.1021/cr9502211. [DOI] [PubMed] [Google Scholar]
- 48.Gracias DH, Chen Z, Shen YR, Somorjai GA. Acc Chem Res. 1999;32:930–940. [Google Scholar]
- 49.Moore FG, Richmond GL. Acc Chem Res. 2008;41:739–748. doi: 10.1021/ar7002732. [DOI] [PubMed] [Google Scholar]
- 50.Gautam KS, Dhinojwala A. Phys Rev Lett. 2002;88:145501. doi: 10.1103/PhysRevLett.88.145501. [DOI] [PubMed] [Google Scholar]
- 51.Baldelli S. Acc Chem Res. 2008;41:421–431. doi: 10.1021/ar700185h. [DOI] [PubMed] [Google Scholar]
- 52.Anglin TC, Conboy JC. Biophys J. 2008;95:186–193. doi: 10.1529/biophysj.107.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gopalakrishnan S, Liu DF, Allen HC, Kuo M, Shultz MJ. Chem Rev. 2006;106:1155–1175. doi: 10.1021/cr040361n. [DOI] [PubMed] [Google Scholar]
- 54.Perry A, Neipert C, Space B, Moore PB. Chem Rev. 2006;106:1234–1258. doi: 10.1021/cr040379y. [DOI] [PubMed] [Google Scholar]
- 55.Li Q, Hua R, Cheah IJ, Chou KC. J Phys Chem B. 2008;112:694–697. doi: 10.1021/jp072147j. [DOI] [PubMed] [Google Scholar]
- 56.Stiopkin IV, Jayathilake HD, Bordenyuk AN, Benderskii AV. J Am Chem Soc. 2008;130:2271–2275. doi: 10.1021/ja076708w. [DOI] [PubMed] [Google Scholar]
- 57.Voges AB, Stokes GY, Gibbs-Davis JM, Lettan RB, Bertin PA, Pike RC, Nguyen ST, Scheidt KA, Geiger FM. J Phys Chem C. 2007;111:1567–1578. [Google Scholar]
- 58.Dreesen L, Sartenaer Y, Humbert C, Mani AA, Methivier C, Pradier CM, Thiry PA, Peremans A. Chem Phys Chem. 2004;5:1719–1725. doi: 10.1002/cphc.200400213. [DOI] [PubMed] [Google Scholar]
- 59.Dreesen L, Humbert C, Sartenaer Y, Caudano Y, Volcke C, Mani AA, Peremans A, Thiry PA, Hanique S, Frere JM. Langmuir. 2004;20:7201–7207. doi: 10.1021/la0488001. [DOI] [PubMed] [Google Scholar]
- 60.Rupprechter G, Weilach C. J Phys Condens Matter. 2008;20:184019. [Google Scholar]
- 61.Fourkas JT, Walker RA, Can SZ, Gershgoren E. J Phys Chem C. 2007;111:8902–8915. [Google Scholar]
- 62.Holman J, Ye S, Neivandt DJ, Davies PB. J Am Chem Soc. 2004;126:14322–14323. doi: 10.1021/ja046954x. [DOI] [PubMed] [Google Scholar]
- 63.Iwahashi T, Miyamae T, Kanai K, Seki K, Kim D, Ouchi Y. J Phys Chem B. 2008;112:11936–11941. doi: 10.1021/jp8021908. [DOI] [PubMed] [Google Scholar]
- 64.Ma G, Allen HC. Langmuir. 2007;23:589–597. doi: 10.1021/la061870i. [DOI] [PubMed] [Google Scholar]
- 65.Chen X, Wang J, Kristalyn CB, Chen Z. Biophys J. 2007;93:866–875. doi: 10.1529/biophysj.106.099739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen X, Chen Z. Biochim Biophys Acta. 2006;1758:1257–1273. doi: 10.1016/j.bbamem.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 67.Chen X, Clarke ML, Wang J, Chen Z. Int J Mod Phys B. 2005;19:691–713. [Google Scholar]
- 68.Clarke ML, Wang J, Chen Z. J Phys Chem B. 2005;109:22027–22035. doi: 10.1021/jp054456k. [DOI] [PubMed] [Google Scholar]
- 69.Evans-Nguyen KM, Fuierer RR, Fitchett BD, Tolles LR, Conboy JC, Schoenfisch MH. Langmuir. 2006;22:5115–5121. doi: 10.1021/la053070y. [DOI] [PubMed] [Google Scholar]
- 70.Kim J, Cremer PS. Chem Phys Chem. 2001;2:543–546. doi: 10.1002/1439-7641(20010917)2:8/9<543::AID-CPHC543>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 71.Kim J, Somorjai GA. J Am Chem Soc. 2003;125:3150–3158. doi: 10.1021/ja028987n. [DOI] [PubMed] [Google Scholar]
- 72.Kim G, Gurua MC, Lim SM, Cremer PS. J Phys Chem B. 2003;107:1403–1409. [Google Scholar]
- 73.Kim G, Gurau MC, Kim J, Cremer PS. Langmuir. 2002;18:2807–2811. [Google Scholar]
- 74.Knoesen A, Pakalnis S, Wang M, Wise WD, Lee N, Frank CW. IEEE J Sel Top Quantum Electron. 2004;10:1154–1163. [Google Scholar]
- 75.Mermut O, Phillips DC, York RL, McCrea KR, Ward RS, Somorjia GA. J Am Chem Soc. 2006;128:3598–3607. doi: 10.1021/ja056031h. [DOI] [PubMed] [Google Scholar]
- 76.Rocha-Mendoza I, Yankelevich DR, Wang M, Reiser KM, Frank CW, Knoesen A. Biophys J. 2007;93:4433–4444. doi: 10.1529/biophysj.107.111047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Weidner T, Breen NF, Drobny GP, Castner DG. J Phys Chem B. 2009;113:15423–15426. doi: 10.1021/jp908773c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang J, Even MA, Chen X, Schmaier AH, Waite JH, Chen Z. J Am Chem Soc. 2003;125:9914–9915. doi: 10.1021/ja036373s. [DOI] [PubMed] [Google Scholar]
- 79.Wang J, Buck SM, Chen Z. Analyst. 2003;128:773–778. doi: 10.1039/b212551j. [DOI] [PubMed] [Google Scholar]
- 80.Wang J, Clarke ML, Zhang YB, Chen X, Chen Z. Langmuir. 2003;19:7862–7866. [Google Scholar]
- 81.Wang J, Chen X, Clarke ML, Chen Z. Proc Nat Acad Sci USA. 2005;102:4978–4983. doi: 10.1073/pnas.0501206102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang J, Chen X, Clarke ML, Chen Z. J Phys Chem B. 2006;110:5017–5024. doi: 10.1021/jp0534683. [DOI] [PubMed] [Google Scholar]
- 83.York RL, Holinga GJ, Guyer DR, McCrea KR, Ward RS, Somorjai GA. Appl Spectrosc. 2008;62:937–1047. doi: 10.1366/000370208785793227. [DOI] [PubMed] [Google Scholar]
- 84.Chen X, Wang J, Sniadecki JJ, Even MA, Chen Z. Langmuir. 2005;21:2262–2264. doi: 10.1021/la050048w. [DOI] [PubMed] [Google Scholar]
- 85.Chen X, Boughton AP, Tesmer JJG, Chen Z. J Am Chem Soc. 2007;129:12658–12659. doi: 10.1021/ja075542w. [DOI] [PubMed] [Google Scholar]
- 86.Chen X, Wang J, Paszti Z, Wang F, Schrauben JN, Tarabara VV, Schmaier AH, Chen Z. Anal Bioanal Chem. 2007;388:65–72. doi: 10.1007/s00216-006-0999-8. [DOI] [PubMed] [Google Scholar]
- 87.Wang J, Chen C, Buck SM, Chen Z. J Phys Chem B. 2001;105:12118–12125. [Google Scholar]
- 88.Moad AJ, Simpson GJ. J Phys Chem A. 2005;109:1316–1323. doi: 10.1021/jp045866w. [DOI] [PubMed] [Google Scholar]
- 89.Moad AJ, Simpson GJ. J Phys Chem B. 2004;108:3548–3562. [Google Scholar]
- 90.Suzuki E. Spectrochim Acta A. 1967;23:2303–2308. [Google Scholar]
- 91.Fraser RDB, Suzuki E. Spectrochim Acta A. 1970;26:423–426. [Google Scholar]
- 92.Marsh D. Biophys J. 1997;72:2710–2716. doi: 10.1016/S0006-3495(97)78914-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Miyazawa T. J Chem Phys. 1960;32:1647–1652. [Google Scholar]
- 94.Tsuboi M, Kaneuchi F, Ikeda T, Akahane F. Can J Chem. 1991;69:1752–1757. [Google Scholar]
- 95.Tsuboi M, Kubo Y, Akahane K, Benevides JM, Thomas GJ., Jr J Raman Spectrosc. 2006;37:240–247. [Google Scholar]
- 96.Krimm S, Abe Y. Proc Nat Acad Sci USA. 1972;69:2788–2792. doi: 10.1073/pnas.69.10.2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rousseau ME, Lefevre T, Beaulieu L, Asakura T, Pezolet M. Biomacromolecules. 2004;5:2247–2257. doi: 10.1021/bm049717v. [DOI] [PubMed] [Google Scholar]
- 98.Chirgadze YN, Nevskaya NA. Biopolymers. 1976;15:607–625. doi: 10.1002/bip.1976.360150402. [DOI] [PubMed] [Google Scholar]
- 99.Choi J-H, Hahn S, Cho M. Biopolymers. 2006;83:519–536. doi: 10.1002/bip.20583. [DOI] [PubMed] [Google Scholar]
- 100.Belkin MA, Shen YR. Phys Rev Lett. 2003;91:213907(1)–213907(4). doi: 10.1103/PhysRevLett.91.213907. [DOI] [PubMed] [Google Scholar]
- 101.Haupert LM, Simpson GJ. Annu Rev Phys Chem. 2009;60:345–365. doi: 10.1146/annurev.physchem.59.032607.093712. [DOI] [PubMed] [Google Scholar]
- 102.Oh-e M, Yokoyama H, Yorozuya S, Akagi K, Belkin MA, Shen YR. Phys Rev Lett. 2004;93:267402(1)–267402(4). doi: 10.1103/PhysRevLett.93.267402. [DOI] [PubMed] [Google Scholar]
- 103.Simpson GJ. Chem Phys Chem. 2004;5:1301–1310. doi: 10.1002/cphc.200300959. [DOI] [PubMed] [Google Scholar]
- 104.Belkin MA, Kulakov TA, Ernst K-H, Yan L, Shen YR. Phys Rev Lett. 2000;85:4474–4477. doi: 10.1103/PhysRevLett.85.4474. [DOI] [PubMed] [Google Scholar]
- 105.Heinz TF, Tom HWK, Shen YR. Phys Rev A. 1983;28:1883–1885. [Google Scholar]
- 106.Heinz TF. Nonlinear Surface Electromagnetic Phenomena. Vol. 29. North-Holland; New York: 1991. p. 352. [Google Scholar]
- 107.Mazely TL, Hetherington WM., III J Chem Phys. 1987;86:3640–3647. [Google Scholar]
- 108.Simpson GJ, Perry JM, Ashmore-Good CL. Phys Rev B. 2002;66:65437(1)–65437(10). [Google Scholar]
- 109.Manella CA, Forte M, Columbini M. VDAC J Bioenerg Biomembr. 1992;24:7–19. doi: 10.1007/BF00769525. [DOI] [PubMed] [Google Scholar]
- 110.Kawano K, Yoneya T, Miyata T. J Biol Chem. 1990;265:15365–15367. [PubMed] [Google Scholar]
- 111.Nakamura T, Furunaka H, Miyata T. J Biol Chem. 1988;263:16709–16713. [PubMed] [Google Scholar]