

Abstract
Heat shock proteins (HSPs) are a highly conserved family of proteins which inhabit almost all subcellular locations and cellular membranes. Depending on their location, these proteins perform a variety of chaperoning functions including folding of newly synthesised polypeptides. HSPs also play a major role in the protection of cells against stressful and injury-inciting stimuli. By virtue of this protective function, HSPs have been shown to prevent acinar cell injury in acute pancreatitis. Also, the levels of HSPs have been shown to be markedly elevated in various forms of cancers when compared with non-transformed cells. Further, inhibition of HSPs has been shown to induce apoptotic cell death in cancer cells suggesting that inhibition of HSPs has a potential to emerge as novel anti-cancer therapy, either as monotherapy or in combination with other chemotherapeutic agents. Several studies have suggested that HSPs can interact with and inhibit both intrinsic and extrinsic pathways of apoptosis at multiple sites. Besides the anti-apoptotic role of HSPs, recent studies suggest that they play a role in the generation of anti-cancer immunity, and attempts have been made to utilise this property of HSPs in the generation of anti-cancer vaccines. The anti-apoptotic function and mechanism of various subtypes of HSPs as well as the current status of anti-HSP therapy are discussed in this review.
Heat shock proteins (HSPs) are a set of evolutionarily conserved proteins. They were discovered in 1962 by Ritossa1 while investigating why a sublethal thermal stress protected against a subsequent lethal thermal injury. This initial study brought to light a new family of proteins and also underscored the role of HSPs in protection against injury-inducing stimuli. Although HSPs were initially shown to be induced by thermal stress, it is now known that they are expressed in response to an array of stresses including hyperthermia, oxygen-derived free radicals, amino acid analogues, ethanol and heavy metals.2–8 Further, several clinical situations like ischaemia/reperfusion injury and various inflammatory diseases also induce the heat shock response.
HSPs have been classified into various subfamilies based on their molecular weight. Broadly, in mammals, four major families of HSPs are recognised: HSP90 (HSP90α, HSP90β, GRP94), HSP70 (HSP70, HSC70, mHSP70, GRP78), HSP60 and small HSPs9 (table 1). Some HSPs (eg, HSP90, HSP60) are expressed constitutively in various intracellular compartments of the cell and play important house-keeping functions aimed at preventing aggregation and assisting refolding of misfolded intracellular proteins and translocation of newly synthesised polypeptides across membranes.10 On the other hand, HSPs like HSP70 and HSP27 are upregulated in response to injury-inciting stimuli and protect against programmed cell death or apoptosis. In this regard HSPs are over-expressed in a variety of tumours and are believed to play a role in carcinogenesis by virtue of their anti-apoptotic functions.11–13 In this review the role of heat shock proteins in imparting protection against cell death in pancreatitis, a disease in which acinar cell injury leading to cell death is an integral part of the disease process, has been discussed followed by the discussion of the current understanding of the role of HSPs, mainly HSP70, in the pathogenesis of the cancer. A distinction should be made between HSP70 and HSC70 at this point. Both are the members of the HSP70 superfamily of HSPs. However, HSC70 is expressed constitutively and plays a role in protein homeostasis of cells. On the other hand, HSP70 is over-expressed in response to stress and protects against stress-induced injury. The protein HSP70 discussed further in the manuscript refers to this latter, inducible member of the HSP70 superfamily.
Table 1.
Location and expression mechanism of heat shock proteins (HSPs)
| Family | Name | Location | Expression (constitutive/inducible) |
|---|---|---|---|
| Small HSPs | HSP27, α-crystallins | Cytosol | Constitutive, inducible |
| HSP60 | HSP60 | Mitochondria | Constitutive, inducible |
| HSP70 | HSP70 | Cytosol/nucleus | Inducible |
| HSC70 | Cytosol/nucleus | Constitutive | |
| HSP90 | HSP90 | Cytosol | Constitutive, inducible |
Some of the HSPs, like HPS27 and HSP90, are expressed constitutively in the cells but also demonstrate increased expression in response to stress.
HEAT SHOCK PROTEINS IN ACUTE PANCREATITIS
Pancreatitis is an inflammatory disease of the pancreas. Acinar cells are believed to be the seat of initiation of events culminating in acinar cell injury and death in pancreatitis.14, 15 Acinar cells synthesise, store and secrete various digestive enzymes. Several protective mechanisms have evolved to prevent the auto-digestion of the acinar cells by these enzymes.14, 15 It is generally believed that during pancreatitis these protective mechanisms are overwhelmed leading to activation of the digestive enzyme zymogens inside acinar cells, which then induce acinar cell injury.14, 15 In fact activation of digestive enzyme zymogens has been observed in both in vitro and in vivo models of pancreatitis.15 Further, activated digestive enzymes have been detected within the pancreatic parenchyma, pancreatic juice and ascitic fluid of some patients with severe acute pancreatitis.16, 17 The mechanism by which these digestive enzyme zymogens become activated inside the acinar cells has been the focus of intense research. Co-localisation of the digestive enzyme zymogens and the lysosomal enzyme has been proposed as one of the mechanisms leading to intra-acinar cell activation of digestive enzyme zymogens and has been tested extensively and validated in various models of pancreatitis.15, 18 According to this theory, early on in pancreatitis there is redistribution of lysosomal enzymes leading to the co-localisation of digestive enzyme zymogens and lysosomal enzymes. This “co-localisation” brings the lysosomal enzyme cathepsin B into contact with trypsinogen, which then activates trypsinogen into trypsin thus initiating the cascade of digestive enzyme zymogen activation and injury. The role of cathepsin B and thus co-localisation in the pathogenesis of pancreatitis is proved by the fact that inhibition of cathepsin B prevents intra-acinar cell activation of trypsinogen and reduces acinar cell injury both in vitro and in animal model of pancreatitis.15, 19, 20 Also, cathepsin B knock-out mice demonstrate reduced severity of caerulein-induced pancreatitis.21 The role of cathepsin B in the activation of trypsinogen is further validated by the experiments in cell-free systems where it has been shown that cathepsin B can activate trypsinogen.22
The role of HSPs in protection against pancreatitis has been explored in various in vitro and in vivo models of pancreatitis. In accordance with the protective role of HSPs against injury, it has been shown that prior induction of HSP60, which is induced in rats by the stress of water immersion, protects against pancreatitis.23 In fact, all the markers of severity of pancreatitis — hyperamylasaemia, pancreatic oedema and acinar cell necrosis — were reduced by this prior induction of HSP60.23 Also, intra-acinar cell trypsinogen activation, which, as discussed previously, is the hallmark of pancreatitis, is also reduced.23 The protective effect of HSPs in pancreatitis is not limited to HSP60, but is also observed with HSP70 induction. Thus, prior HSP70 induction by thermal or non-thermal stresses (beta-adrenergic stimulation or sodium arsenite administration)24, 25 protect against acinar cell injury in pancreatitis. Since cellular injury in pancreatitis is mediated by both necrosis and apoptosis pathways and since HSP70 can protect cells against both necrotic and apoptotic programmed cell death, it is probable that HSP70 protects against both necrotic and apoptotic cell death during pancreatitis. However, the exact mechanism by which it operates needs further exploration.
Some studies have suggested that the protective effects of thermal stress against pancreatitis may not be due to HSP70 expression.26 To rule out the possibility that thermal stress protects against pancreatitis by mechanisms other than induction of HSP70 and to conclusively prove the role of HSP70 in protection against pancreatitis, Bhagat et al27 have used anti-sense technology. Prior administration of the anti-sense HSP70 oligonucleotide sequence but not the sense HSP70 sequence prevents thermal stress-induced HSP70 expression.27 Remarkably, prior administration of the anti-sense but not the sense HSP70 sequence abrogates the protective effect of thermal stress against acinar cell injury in pancreatitis.27 Thus, the animals in which thermal stress-induced HSP70 expression is prevented by anti-sense HSP70 administration, demonstrate similar severity of caerulein-induced pancreatitis when compared to animals without thermal stress, clearly suggesting that the protective effects of thermal stress on the severity of pancreatitis are in fact mediated by HSP70 expression.27 Also, it is worth noting that caerulein-induced pancreatitis by itself induces HSP70 expression in the pancreas. It appears that this induction of HSP70 in the pancreas by the stress of pancreatitis is protective against pancreatitis, since prevention of this increase in HSP70 by administration of the anti-sense HSP70 in itself aggravates the severity of pancreatitis.27
The specific role of HSP70 in protection against pancreatitis is further proved by the use of heat shock factor-1 (HSF-1) knock-out mice and HSP70 transgenic mice.28 HSF-1 is the transcription factor for HSP70. Thus, HSF-1 knock-out animals lack the ability to express HSP70. As demonstrated in fig 1, HSF-1 knock-out animals demonstrate enhanced severity of caerulein-induced pancreatitis. Similarly, HSP70 transgenic mice, which demonstrate markedly increased expression of HSP70, have reduced severity of pancreatitis.29
Figure 1.

Heat shock factor-1 (HSF1) knock-out mice have increased severity of pancreatitis. In the absence of caerulein administration, HSF1 knock-out mice (B) demonstrate similar pancreatic morphology as wild-type mice (A). HSF1 knock-out mice (D) demonstrate increased severity of pancreatitis in response to caerulein administration as compared to HSF1 wild-type mice (C).
Some progress has been made in elucidating how HSPs protect against pancreatitis. It has been shown that HSP70 induction prevents activation of trypsinogen to trypsin, a hallmark event of pancreatitis.25, 27 Rakonczay et al have shown that HSP70 induction does not protect against the pancreatitis induced by trypsin injection into the pancreas, again suggesting that HSP70 mediates protection against pancreatitis by influencing the events before trypsin activation.30 Also, evidence suggests that HSP70 influences the trypsinogen activation and the other downstream events during pancreatitis by preventing the co-localisation of lysosomal enzyme cathepsin B and the digestive enzyme zymogen.27 The events regulating co-localisation are less clear. However, evidence strongly suggests that cytosolic calcium is required for this process.31 Stimulation of pancreatic acini with a supramaximal dose of caerulein leads to marked elevation of cytosolic calcium followed by prolonged sustained elevation of cytosolic calcium maintained by calcium influx into the cell. It appears that these cytosolic calcium changes are important for co-localisation to occur since chelation of intracellular calcium prevents co-localisation.31 Remarkably, HSP60 induction by water immersion stress attenuates cytosolic calcium response to supramaximal caerulein stimulation32 (fig 2). Some other studies in non-pancreatic acinar cells have also suggested that HSPs could influence calcium homeostasis in the cell.33, 34 Thus, one of the mechanisms by which HSPs could influence co-localisation and the subsequent downstream events in pancreatitis is by attenuating cytosolic calcium (fig 3). Given the importance of calcium in regulation of cellular processes including cell death and apoptosis, the finding that HSPs could influence calcium homeostasis is of much broader importance than just of significance to the pathophysiology of pancreatitis.
Figure 2.
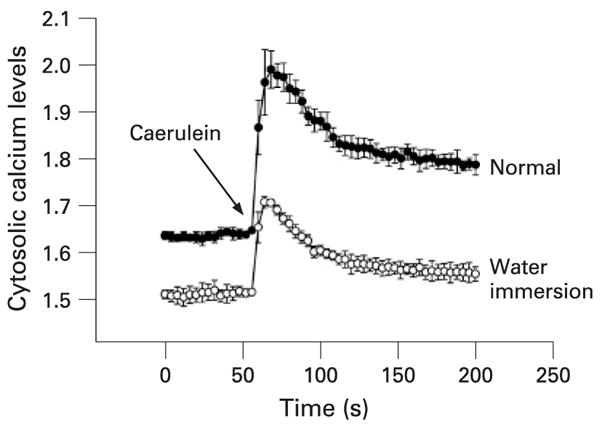
Heat shock protein 60 (HSP60) attenuates cytosolic calcium response. Prior water immersion stress-induced HSP60 induction attenuates the cytosolic calcium signal in response to caerulein administration.30 Cytosolic calcium levels were measured by Fura-2 AM by using spectrophotometry.
Figure 3.

A schematic diagram of the possible mechanism by which heat shock protein 70 (HSP70) protects against acinar cell injury in pancreatitis. The events that initiate pancreatitis also increase cytosolic calcium in acinar cells, which in turn causes lysosomes and digestive enzyme zymogen to co-localise. These events bring the inactive trypsinogen into contact with cathepsin B, which then activates trypsinogen. Active trypsin in turn activates other digestive enzyme zymogens, and those active enzymes cause acinar cell injury. HSP70 possibly protects the acinar cell from injury in pancreatitis by attenuating cytosolic calcium increase and thus abrogating all the downstream events in the injury pathway.
HEAT SHOCK PROTEINS IN CHRONIC PANCREATITIS AND OTHER INFLAMMATORY DISEASES OF THE GASTROINTESTINAL TRACT
A few studies have evaluated the expression level of various HSPs in chronic pancreatitis. HSP70 over-expression has been observed in the connective tissue cells as well as the residual acinar cells in chronic pancreatitis.35 Similarly, levels of HSP90β have been found to be increased in chronic pancreatitis.36 However, information on the expression of various HSPs in chronic pancreatitis and their significance is lacking in literature. Given the role of HSPs in protection against cell death, this area is worth pursuing in future studies.
Studies examining HSP expression in the gastrointestinal tract have received considerable attention, especially with reference to inflammatory bowel disease (IBD). Overall, in mouse models, inducible HSP staining (HSP27 and HSP70) in the colon appears to be predominantly localised to the surface epithelial cells, thus supporting the premise that this HSP expression may be protecting the surface epithelial cells from injury.37 Increased expression of HSP70 has been observed in the epithelial cells of both ulcerative colitis and Crohn’s disease.38 Increased expression of HSP60 but not HSP90 has also been observed in IBD.39, 40 However, no correlation between the level of expression and the disease activity was observed. Whether or not these HSPs are over-expressed in response to stress and thus play a role in protecting the epithelial cells against stress-induced death is unclear. Autoantibodies to HSPs are found IBD.41 The reason that these antibodies develop is not clear. One theory suggests that patients may develop antibodies to mycobacterial 65 kDa HSP (HSP60), which then cross-react with their own HSPs and help induce inflammation.41 An alternative theory suggests that autoantibodies could result from the release of native HSPs into the extracellular space following injury to the gut epithelium. Overall, the pathophysiological significance of autoantibodies to HSPs in IBD remains unknown.
HSP70 has been shown to have protect against the progression of atrophic gastritis in animal models.42 Also, HSP70 has been shown to protect against gastritis induced by Helicobacter pylori.43 The importance of HSPs in protection against gastritis in human subjects remains to be seen.
HEAT SHOCK PROTEIN 70 IN PANCREATIC AND OTHER CANCERS
Dysregulation of apoptosis is a hallmark of carcinogenesis and since HSPs protect against apoptosis, they are believed to contribute to the development and progression of cancer. Attempts have been made to elucidate the role of HSPs in cancer by quantifying the levels of various HSPs in cancer cells and comparing them with the non-transformed cells. Aghdassi et al11 have shown that HSP70 levels are markedly high in various pancreatic cancer cells lines as compared to normal pancreatic ductal cells, the cells of origin of pancreatic cancer (fig 4A). The levels of HSP70 were increased at both the protein and mRNA level suggesting that increased transcription of HSP70 is responsible for the increased protein levels.11 This finding is clinically relevant since HSP70 over-expression was also observed in human pancreatic cancer specimens when the levels were compared with normal pancreas margins11 (fig 4B). Similar over-expression of HSP70 has been observed in other cancers also. HSP70 is over-expressed in hepatocellular carcinoma as compared to non-neoplastic hepatocytes44 and the levels of HSP70 have a positive correlation with poor outcome.44, 45 HSP70 over-expression has also been observed in colorectal cancers and has been associated with advanced stage, poor differentiation and poor prognosis.46 Besides evidence from the studies correlating the level of HSPs in human cancer tissue, a variety of in vitro and animal studies have provided evidence for the role of HSPs, especially HSP70 in the pathogenesis of cancer. It has been demonstrated that permanent over-expression of HSP70 in a variety of cell lines imparts the ability to form tumours in nude mice and anchorage-independent growth with strong acceleration of the growth.47 Similarly, over-expression of HSP70 can suppress apoptosis in response to a variety of stimuli.47 These data together strongly point towards the role of HSP70 in tumorigenesis.
Figure 4.

Heat shock protein 70 (HSP70) is over-expressed in pancreatic cancer as compared with non-transformed cells. (A) HSP70 is markedly over-expressed in four human pancreatic cancer cell lines when compared with non-transformed pancreatic ductal cells. (B) HSP70 mRNA levels are markedly increased in human pancreatic cancer specimen when compared with normal tissue margins (normal). *p<0.002, n = 7.11
Based on the role of HSP70 in the pathogenesis of cancer, inhibition of HSP70 expression has been evaluated as a strategy to treat cancer with much success. Inhibition of HSP70 expression in pancreatic cancer cells by quercetin, a well-known inhibitor of HSP70 expression,11 as well as by triptolide12 (a novel inhibitor of HSP70 expression) has been shown to induce apoptotic cell death in pancreatic cancer cells. Both these inhibitors of HSP70 expression are very effective in reducing the growth of pancreatic tumours in a mouse model of pancreatic cancer (fig 5A,B).11, 12 Remarkably, triptolide markedly reduced the loco-regional spread of pancreatic cancer in an orthotopic model of pancreatic cancer (fig 5C). Since triptolide is very effective in animal models of pancreatic cancer at a very low dose (0.2 mg/kg/day) as compared to quercetin (50 mg/kg/day) and also does not have any toxic effects in the animals, it offers an exciting opportunity to develop inhibition of HSP70 expression as a novel therapy for pancreatic cancer.12 To rule out the possibility that the effects of these pharmacological inhibitors are due to effects other than inhibition of HSP70 expression, the phenotypical effect of inhibition of HSP70 expression on pancreatic cancer cells has been confirmed by using siRNA technology. Thus, inhibition of HSP70 expression by HSP70 siRNA also induces caspase-dependent apoptotic cell death in pancreatic cancer cells (fig 6), similar to that observed with either quercetin or triptolide.11
Figure 5.

Inhibition of heat shock protein 70 (HSP70) expression is highly effective as a therapeutic strategy in animal models of pancreatic cancer. (A) Inhibition of HSP70 expression by triptolide markedly reduces the growth of pancreatic tumours in an orthotopic model of pancreatic cancer in nude mice. Triptolide was administered at a dose of 0.2 mg/kg/day for 60 days. Animals were killed at the 60th day and the tumour volumes in the triptolide group were compared with the control group (treated with vehicle alone). n = 8 in each group, *p<0.05. (B) Representative picture of tumour tissue in the control and triptolide treatment group. (C) Graph demonstrating reduced loco-regional spread in the triptolide treatment group. (p<0.001, n = 8, χ2 analysis).12
Figure 6.

Inhibition of heat shock protein 70 (HSP70) expression by HSP70 siRNA induces caspase-dependent apoptotic cell death in pancreatic cancer cells. (A) Inhibition of HSP70 expression by HSP70 siRNA leads to decreased viability in the MiaPaCa-2 pancreatic cancer cell line at 72 h. Viability was measured by the MTT assay. Two different sequences of HSP70 siRNA were used to rule out any off-target effects of siRNA. n = 3, *p<0.05. (B) Inhibition of HSP70 expression by two different sequences leads to caspase 3 activation. n = 3, *p<0.05. MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
Remarkably, inhibition of HSP70 expression by quercetin or triptolide does not induce apoptosis in normal pancreatic ductal cells.11, 12 Although the reason for this specificity of anti-HSP70 therapy to the cancer cells is unclear, the data point towards certain mechanisms. As shown in fig 4A, the level of HSP70 in normal ductal cells is very low as compared to pancreatic cancer cells. Thus, it seems that HSP70 expression is important for the survival of pancreatic cancer cells but may not be required for the survival of normal non-neoplastic cells under unstressed conditions. Similar observations have been reported by others48 where in contrast to cancer cells, non-transformed cells do not undergo cell death on depletion of HSP70, suggesting that tumour cells but not non-transformed cells cannot survive endogenously activated cell death if they lack HSP70.47 This, along with the fact that triptolide is non-toxic to experimental animals, clearly suggests that inhibition of HSP70 expression can emerge as tumour-selective therapy.
Similar efficacy of anti-HSP70 therapy has been demonstrated in other gastrointestinal and non-gastrointestinal tumours. Inhibition of HSP70 expression by quercetin (50 mg/kg) decreased tumour growth and tumour angiogenesis in mice bearing lung carcinoma, hepatoma and colon cancer.49 Similarly, inhibition of HSP70 by transfection with anti-sense HSP70 construct sensitises cancer cells to apoptosis and eradicates tumours of breast, colon and gliomas.13 Phillips et al have shown that besides inducing apoptosis in pancreatic cancer cells, triptolide induces cell death in colon cancer cell lines as well, suggesting that anti-HSP70 therapy will be of broader value rather than for pancreatic cancer only.12 Thus, development of effective and bio-available inhibitors of HSP70 expression will be useful in a wide variety of cancers as mono-therapy or in combination with other chemotherapeutic agents. In this regard triptolide12 appears to be a very promising molecule which at very low doses (0.2 mg/kg) is able to inhibit HSP70 expression in mice models and also have optimal anti-cancer effects with inhibition of both local tumour growth and locoregional spread.
MECHANISM OF THE ANTI-APOPTOTIC ACTION OF HEAT SHOCK PROTEIN 70
The mechanism by which HSP70 inhibits apoptosis has been evaluated extensively and studies suggest that HSP70 can inhibit both death receptor-mediated extrinsic pathways as well as intrinsic pathways initiated by intracellular stress signals. Although initial studies suggested that HSP70 inhibits intrinsic pathway-mediated apoptosis mainly downstream of mitochondrial cytochrome c release, recent studies suggest that HSP70 can interact and inhibit pre-mitochondrial events as well, since inhibition of HSP70 leads to release of cytochrome c from the mitochondria.12 As a matter of fact, HSP70 may inhibit apoptosis at multiple points simultaneously. Once released into the cytosol, cytochrome c interacts with cytosolic apoptosis protease-activating factor-1 (APAF-1) and pro-caspase 9 to form apoptosome. This leads to activation of caspase 9, which then activates caspase 3.50 Besides release of cytochrome c, various other factors (eg, apoptosis-inducing factor (AIF), Smac/Diablo and Htra2/Omi) are released into the cytosol by mitochondrial membrane permeabilisation. HSP70 has been shown to modulate the function of apoptosome by directly interacting with APAF-1.51, 52 However, recent studies have suggested that the contribution of these effects of HSP70 downstream of mitochondria, in the inhibition of apoptosis is small, if any.53 Besides these possible post-mitochondrial sites of action, HSP70 has been shown to influence mitochondrial and pre-mitochondrial events in apoptosis. HSP70 prevents the translocation of Bax to the mitochondria during heat-induced apoptosis, thus preventing formation of pores in the mitochondrial membrane and release of cytochrome c and AIF.54 HSP70 has been shown to be a natural inhibitor of cJun N-terminal kinase (JNK) activation55 and this JNK inhibitory action appears to mediate the protection against caspase-dependent apoptosis.56 It appears that JNK is required for Bid-dependent release of cytochrome c from the mitochondria and that, by inhibiting JNK, HSP70 prevents Bid-dependent cytochrome c release and downstream activation of caspases.57 Recent studies have shown that lysosomal enzymes play a very important role in the activation of apoptosis and may become all the more important in tumour cells where the other mechanisms of apoptosis are compromised.58 Once released into the cytosol these lysosomal enzymes can then activate multiple pathways of cell death including the mitochondrial pathway.58 Studies suggest that HSP70 may protect cancer cells from apoptosis by stabilising the lysosomes thus preventing the release of lysosomal enzymes into the cytosol.59, 60 Thus interaction of HSP70 and lysosomes could be one of the sites where HSP70 inhibits apoptosis upstream of mitochondria. HSP70 could also play an anti-apoptotic role in caspase-independent apoptosis as well. For example, HSP70 binds to AIF, which induces apoptosis in a caspase-independent fashion, and prevents its translocation to the nucleus thus preventing activation of apoptosis.61 HSP70 also interacts with and inhibits extrinsic pathways of apoptosis by binding to the death receptors DR4 and DR5, thus inhibiting the assembly induced by the TNF-related apoptosis-inducing ligand (TRAIL) and the activation of the death-inducing signalling complex (DISC).62
Calcium has been shown to play a major role in the regulation of apoptosis.63 Hietaranta et al32 have shown that HSP60 could attenuate cytosolic calcium signalling, thus suggesting that HSPs could interact with and modulate cytosolic calcium signalling. Creagh et al have suggested that HSP70 could modulate the increase in cytosolic calcium during H2O2-induced apoptosis in jurkat cells.64 Whether HSP70 could interact with calcium homeostasis in cancer cells and thus inhibit apoptosis needs further investigation.
In summary, HSP70 is an inhibitor of the mitochondrial pathway of apoptosis which can interact with and inhibit the components of this pathway at the pre-mitochondrial, mitochondrial and post-mitochondrial levels (fig 7). In fact, it may act at multiple levels simultaneously.
Figure 7.

A schematic diagram of the possible sites where heat shock protein 70 (HSP70) inhibits intrinsic pathway of apoptosis. It is believed that mitochondrial permeabilisation and subsequent cytochrome c release, along with other apoptosis-inducing factors, constitute central steps in apoptotic cell death. Multiple mechanisms of this mitochondrial permeabilisation have been proposed, including Bax, increased cytosolic calcium, lysosomal enzymes, and cJun N-terminal kinase (JNK). Once cytochrome c is released from the mitochondria it interacts with apoptosis protease-activating factor-1 (APAF-1) and pro-caspase 9 in the cytosol leading to formation of caspase 9 which then activates the effector caspases. These events result ultimately in apoptosis. HSP70 has been shown to inhibit apoptosis by participating in events both before and after mitochondrial membrane permeabilisation, as depicted in the figure.
ROLE OF HEAT SHOCK PROTEIN 27 IN CANCERS
Small HSPs including HSP27 and α-crystallins are ATP-independent chaperones whose functions are regulated by oligomerisation and phosphoryla-tion.13 Studies measuring the levels of HSP27 in various tumour tissues have suggested that HSP27 may play a role in the pathogenesis of cancer. HSP27 expression has been observed in gastric carcinoma specimens and its expression has been correlated with a poor prognosis.65 HSP27 over-expression has been observed with step-wise progression of hepatic carcinogenesis in hepatitis B-associated hepatocellular carcinoma44, 66 and its expression portends poor prognosis.44 In a small study over-expression of HSP27 has been observed in pancreatic and colon cancer as compared to non-transformed cells. HSP27 over-expression has also been observed in a number of non-gastrointestinal cancers — breast, prostate and ovarian cancers — as compared to the non-transformed cells. Also, it has been observed that over-expression of HSP27 in various cell lines can suppress apoptosis in response to a variety of stimuli.47 Generally, HSP27 expression in tumours is considered a poor prognostic factor.67
Various studies have attempted to elucidate the mechanism by which HSP27 exerts its anti-apoptotic and tumorigenic action. It has been demonstrated that, similar to HSP70, HSP27 can interact and inhibit the apoptotic cascade at multiple sites. HSP27 has been demonstrated to inhibit caspase-dependent apoptosis downstream of mitochondrial cytochrome c release by interacting with and sequestering cytochrome c.13, 67 Also, HSP27 has been shown to interact with pro-caspase 3 and prevent its activation.13, 67 HSP27 has also been shown to exert its anti-apoptotic action at the mitochondrial level by blocking the release of cytochrome c from the mitochondria.68 It has been suggested that HSP27 can also inhibit apoptosis at the pre-mitochondrial level, by stabilising the actin cytoskeleton and thus prevent the translocation of pro-apoptotic factors, such as Bid, to the mitochondria.13, 67 The anti-oxidant properties of HSP27 are also believed to contribute to its anti-apoptotic properties.13, 67 HSP27 could also possibly modulate apoptosis by influencing cell signalling pathways. HSP27 has been shown to bind to protein kinase B/Akt and plays a role in its activation in stressed cells. Once activated Akt inhibits apoptosis and protects the cells from stress-induced cell death.13, 67 HSP27 can also inhibit the Fas-induced extrinsic pathway of apoptosis by interacting with Daxx, a protein which is considered a mediator in Fas-mediated cell death. Importantly, treatment with commonly used chemotherapeutic agents including doxorubicin leads to an increase in HSP27 levels, which could counteract the anti-cancer effects of these chemotherapeutic agents.69 Thus, combination of these agents with anti-HSP agents may be more effective.
HEAT SHOCK PROTEIN 90 AND CANCER
HSP90 inhibitors have gained much attention in the treatment of cancers. Interest in HSP90 as a therapeutic target began when it was identified as the target of the antibiotic geldanamycin with anti-tumour properties. HSP90 is believed to play a more global role in the facilitating neoplastic transformation by virtue of its interaction with multiple pro-survival proteins. Thus inhibition of HSP90 function, by an expanding list of agents (eg, geldanamcyin, ansamycin, 17-allyamino-demethoxy geldanamycin), induces degradation of the HSP90 client proteins that require HSP90 for maturation and stability. The HSP90 client proteins include a myriad of proteins including various kinases, such as human epidermal growth factor receptor-2 (HER-2), insulin-like growth factor-IR (IGF-1R), Raf-1, steroid hormone receptors like androgen receptor and oestrogen receptor, and other proteins like HIF-1α.70 In this regard it is noteworthy that HSP90 binds to HSF1 and has been shown to play a role in its regulation.71 In unstressed cells HSF1, the primary transcription factor of HSP70, resides in the cytosol in inactive monomer form bound to HSP90 which plays a major role in repressing its function. Inhibition of HSP90 expression or function leads to release and activation of HSF1 with increased expression of HSP70.72 In fact activation of stress response in the tumour tissue which is reflected in the increased expression of HSP70 in peripheral blood mononuclear cells has been used as the pharmacodynamic endpoint.73 However, this increased expression of HSP70 may counteract the anti-neoplastic activity of HSP90 inhibitors and may lead to resistance of cancer cells to these drugs. For example, inhibition of HSP70 expression by stable transfection with siRNA, which inhibits 17-allylamino-demethoxy geldanamycin (17-AAG)-induced HSP70 expression, sensitises leukaemia cells to 17-AAG-induced apoptosis. The fact that 17-AAG is an inhibitor of HSP90 function suggests that HSP90 inhibitors may be more efficacious if used with inhibitors of HSP70 or stress response. It has also been suggested that HSP90 inhibition may lead to activation of the anti-apoptotic and pro-survival pathways. For example, HSP90 inhibitors may activate the src kinase which may promote tumour growth.74 Thus, HSP90 inhibitors may be more useful in combination with other anti-tumour therapies which counteract these untoward effects. Furthermore, a recent report by Kang et al75 has suggested that, in tumour cells, the HSP90 chaperone network may be compartmentalised into sub-cellular locations (eg, mitochondria) and may play important anti-apoptotic roles. Currently used HSP90 inhibitors may not permeate into these sub-cellular locations and thus may not be able to inhibit the HSP90 activity in these locations.75 Thus, newer agents which are able to achieve enough concentration in these sub-cellular compartments and are able to inhibit HSP90 in these locations will be more effective.75
HSP90 inhibitors have been shown to be effective in tumours which depend on the HSP90 client proteins. For example, it has been suggested that HER2 is the HSP90 client protein most sensitive to 17-AAG induced degradation. Since HER2 plays a major role in the pathogenesis of breast cancer, it forms the basis for the evaluation of 17-AAG in combination with cytotoxic therapies in patients with HER2-amplified breast cancer.70 Studies in various gastrointestinal tumours have shown that HSP90 inhibitors may have a role in the treatment of gastrointestinal cancers. In a small study, constitutive expression of HSP90 has been observed in gastric, colon and pancreatic tumours.76 17-AAG has been shown to inhibit epidermal growth factor receptor (EGFR)-dependent signalling in gastric tumours and thus reduce the growth and vascularisation of gastric tumours in vivo.77 HSP90 inhibitors have been shown to not only sensitise colon cancer cell lines to oxaliplatin and TRAIL-induced cell death but also induce cell death when administered alone.78, 79 Clinical trials of HSP90 inhibitors in combination with various chemotherapeutic agents in solid tumours are currently under way.70
IS HEAT SHOCK PROTEIN 60 PRO-APOPTOTIC OR ANTI-APOPTOTIC?
Of all the HSPs the role of HSP60, which is largely but not exclusively compartmentalised in mitochondria, in the pathogenesis of cancer is least understood. Early reports suggested that HSP60 has a pro-apoptotic role and facilitates the activation of pro-caspase 3 to active caspase 3 during apoptosis.80, 81 However, recent reports have suggested that HSP60 may have an anti-apoptotic effect, at least in certain cell systems, by sequestering the pro-apoptotic protein Bax.82 The data on the expression of HSP60 in various cancers has been somewhat ambiguous. HSP60 expression has been found to be increased in some cancers83, 84 and decreased in others.85 In a recent report, Ghosh et al86 have proposed a potential mechanism for the anti-apoptotic property of HSP60. They have proposed that HSP60 inhibits apoptosis in cancer cells by playing a dual role: (1) binding to and stabilising the anti-apoptotic protein survivin in mitochondria; and (2) binding to and inhibiting the function of the pro-apoptotic protein p53.
HEAT SHOCK PROTEINS AND IMMUNOLOGY
Although intracellular HSP expression has been associated with facilitation of tumorigenesis, it has been suggested that the extracellular and membrane-associated HSPs facilitate tumour rejection by the immune system.87 Thus, on the one hand, downregulation of intracellular HSPs has been attempted in order to induce apoptosis in cancer cells and, on the other hand, the extracellular and membrane-associated HSPs have been used to develop cancer immunotherapy.
HSPs (eg, HSP90, HSP70, GP96, calreticulin, HSP110) have been shown to bind non-covalently to peptides generated inside cells. Thus, inside tumour cells these HSPs bind to tumoural antigens. On cell damage, these HSP–peptide complexes are released into the extracellular milieu and gain access to antigen-presenting cells (APCs) and perform their immune function. The HSP–peptide complex binds to receptors on APCs and is taken up by receptor endocytosis. Once inside the APCs, the antigenic peptides carried by HSPs are processed by the antigen-presenting pathways and are presented on the surface of APCs, where they are recognised by CD8+ T cells, thus initiating the immune response cascade. Several molecules on the surface of APCs, such as CD91, LOX-1, CD40, TLR2/4, have been recognised as the receptors for the HSP–peptide complex.88 Thus the immune response generated by these HSP–peptide complexes is primarily effected by the T cell pathway.
In animal models the HSP–tumour peptide vaccines have been effective, with a reduction in the size of primary tumours, increased survival and elimination of metastases. It is worth noting that, in animal studies, the doses of HSPs required for immunity fell within a very narrow window88 and inoculation of mice with relatively large doses of HSPs led to the enhancement of disease. This point is of immense importance in considering the use of HSP vaccines in human studies since the optimal doses can not be extrapolated from animal studies. Also, by the inherent nature of the vaccine, each HSP vaccine must be produced from each patient’s tumours thus imposing novel technical considerations. The quantity of tissue available for the generation of vaccine is limited and determined by the amount of tissue removed surgically. If a large amount of tissue is required then the generation of the vaccine will not be practical. Further, it has also been demonstrated that certain HSP–peptide complexes are more immunogenic then others; for example, the HSP90–peptide complex is only 10% as potent as GP96–peptide or HSP70–peptide complexes.89
Currently, HSP vaccines are under trial in metastatic melanoma, renal cell carcinoma and colorectal cancer. Preliminary results have suggested mixed clinical response with some cases of complete regression but mainly some prolongation of survival and disease stabilisation. Future studies are needed to make this therapy more effective and also for identifying the subset of the patients which will respond most favourably to this form of therapy.
SUMMARY
HSPs have a broad range of functions which play a major role in cellular homeostasis and protect cells against apoptosis and cell death.
In diseases where pathogenesis involves cell death, eg, pancreatitis, HSPs protect against cell death and reduce the severity of disease by virtue of their protective functions. In these situations expression of HSPs is helpful in the remission of disease.
Dysregulation and inhibition of apoptosis is a hallmark of carcinogenesis and HSPs contribute to the pathogenesis of cancer by inhibiting apoptosis. In cancer, HSP expression is associated with poor prognosis and resistance to chemotherapy. Inhibition of HSPs is emerging as a novel anti-cancer therapy.
HSPs can inhibit apoptosis and thus contribute to carcinogenesis by multiple mechansisms. HSP70 and HSP27 have been demonstrated to interact with and inhibit apoptosis at the pre-mitochondrial, mitochondrial and post-mitochondrial levels. HSP90 is believed to chaperone and stabilise various tumorigenic proteins and kinases and thus contribute to carcinogenesis.
In contrast to intracellular HSPs, extracellular HSPs can present peptide from tumour cells to the antigen-presenting cells and thus augment the development of anti-tumour immunity. This approach is being utilised in anti-cancer vaccine development and is under investigation in various malignancies.
Acknowledgments
Funding: AS would like to acknowledge support from NIH grants DK-58694, CA-124723, CA-131663 and a generous contribution from the Dr Robert and Katherine Goodale Foundation.
Footnotes
Competing interests: None.
References
- 1.Ritossa FM. A new puffing pattern induced by temperature shock and DNP in Drosophila. Experientia. 1962;18:571–3. [Google Scholar]
- 2.Welch WJ. Mammalian stress response: cell physiology, structure/function of stress proteins, and implications for medicine and disease. Physiol Rev. 1992;72:1063–81. doi: 10.1152/physrev.1992.72.4.1063. [DOI] [PubMed] [Google Scholar]
- 3.Wong HR, Wispe JR. The stress response and the lung. Am J Physiol. 1997;273(1 Pt 1):L1–9. doi: 10.1152/ajplung.1997.273.1.L1. [DOI] [PubMed] [Google Scholar]
- 4.Benjamin IJ, McMillan DR. Stress (heat shock) proteins: molecular chaperones in cardiovascular biology and disease. Circ Res. 1998;83:117–32. doi: 10.1161/01.res.83.2.117. [DOI] [PubMed] [Google Scholar]
- 5.Kiang JG, Tsokos GC. Heat shock protein 70 kDa: molecular biology, biochemistry, and physiology. Pharmacol Ther. 1998;80:183–201. doi: 10.1016/s0163-7258(98)00028-x. [DOI] [PubMed] [Google Scholar]
- 6.Mathew A, Morimoto RI. Role of the heat-shock response in the life and death of proteins. Ann N Y Acad Sci. 1998;851:99–111. doi: 10.1111/j.1749-6632.1998.tb08982.x. [DOI] [PubMed] [Google Scholar]
- 7.De Maio A. Heat shock proteins: facts, thoughts, and dreams. Shock. 1999;11:1–12. doi: 10.1097/00024382-199901000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Schafer C, Williams JA. Stress kinases and heat shock proteins in the pancreas: possible roles in normal function and disease. J Gastroenterol. 2000;35:1–9. doi: 10.1080/003655200750024443. [DOI] [PubMed] [Google Scholar]
- 9.Garrido C, Gurbuxani S, Ravagnan L, et al. Heat shock proteins: endogenous modulators of apoptotic cell death. Biochem Biophys Res Commun. 2001;286:433–42. doi: 10.1006/bbrc.2001.5427. [DOI] [PubMed] [Google Scholar]
- 10.Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–66. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 11.Aghdassi A, Phillips P, Dudeja V, et al. Heat shock protein 70 increases tumorigenicity and inhibits apoptosis in pancreatic adenocarcinoma. Cancer Res. 2007;67:616–25. doi: 10.1158/0008-5472.CAN-06-1567. [DOI] [PubMed] [Google Scholar]
- 12.Phillips PA, Dudeja V, McCarroll JA, et al. Triptolide induces pancreatic cancer cell death via inhibition of heat shock protein 70. Cancer Res. 2007;67:9407–16. doi: 10.1158/0008-5472.CAN-07-1077. [DOI] [PubMed] [Google Scholar]
- 13.Garrido C, Brunet M, Didelot C, et al. Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell Cycle. 2006;5:2592–601. doi: 10.4161/cc.5.22.3448. [DOI] [PubMed] [Google Scholar]
- 14.Pandol SJ, Saluja AK, Imrie CW, Saluja AK, Imrie CW, et al. Acute pancreatitis: bench to the bedside. Gastroenterology. 2007;133:1127–51. doi: 10.1053/j.gastro.2007.01.055. [DOI] [PubMed] [Google Scholar]
- 15.Saluja AK, Lerch MM, Phillips PA, et al. Why does pancreatic overstimulation cause pancreatitis? Annu Rev Physiol. 2007;69:249–69. doi: 10.1146/annurev.physiol.69.031905.161253. [DOI] [PubMed] [Google Scholar]
- 16.Geokas MC, Murphy R, McKenna RD. The role of elastase in acute pancreatitis. I. Intrapancreatic elastolytic activity in bile-induced acute pancreatitis in dogs. Arch Pathol. 1968;86:117–26. [PubMed] [Google Scholar]
- 17.Geokas MC, Rinderknecht H. Free proteolytic enzymes in pancreatic juice of patients with acute pancreatitis. Am J Dig Dis. 1974;19:591–8. doi: 10.1007/BF01073012. [DOI] [PubMed] [Google Scholar]
- 18.Saluja A, Saluja M, Villa A, et al. Pancreatic duct obstruction in rabbits causes digestive zymogen and lysosomal enzyme colocalization. J Clin Invest. 1989;84:1260–6. doi: 10.1172/JCI114293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saluja AK, Donovan EA, Yamanaka K, et al. Cerulein-induced in vitro activation of trypsinogen in rat pancreatic acini is mediated by cathepsin B. Gastroenterology. 1997;113:304–10. doi: 10.1016/s0016-5085(97)70108-2. [DOI] [PubMed] [Google Scholar]
- 20.Van Acker GJ, Saluja AK, Bhagat L, et al. Cathepsin B inhibition prevents trypsinogen activation and reduces pancreatitis severity. Am J Physiol Gastrointest Liver Physiol. 2002;283:G794–800. doi: 10.1152/ajpgi.00363.2001. [DOI] [PubMed] [Google Scholar]
- 21.Halangk W, Lerch MM, Brandt-Nedelev B, et al. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest. 2000;106:773–81. doi: 10.1172/JCI9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Figarella C, Miszczuk-Jamska B, Barrett AJ. Possible lysosomal activation of pancreatic zymogens. Activation of both human trypsinogens by cathepsin B and spontaneous acid. Activation of human trypsinogen 1. Biol Chem Hoppe Seyler. 1988;369(Suppl):293–8. [PubMed] [Google Scholar]
- 23.Lee HS, Bhagat L, Frossard JL, et al. Water immersion stress induces heat shock protein 60 expression and protects against pancreatitis in rats. Gastroenterology. 2000;119:220–9. doi: 10.1053/gast.2000.8551. [DOI] [PubMed] [Google Scholar]
- 24.Bhagat L, Singh VP, Dawra RK, et al. Sodium arsenite induces heat shock protein 70 expression and protects against secretagogue-induced trypsinogen and NF-kappaB activation. J Cell Physiol. 2008;215:37–46. doi: 10.1002/jcp.21286. [DOI] [PubMed] [Google Scholar]
- 25.Frossard JL, Bhagat L, Lee HS, et al. Both thermal and non-thermal stress protect against caerulein induced pancreatitis and prevent trypsinogen activation in the pancreas. Gut. 2002;50:78–83. doi: 10.1136/gut.50.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rakonczay Z, Jr, Mandi Y, Kaszaki J, et al. Induction of HSP72 by sodium arsenite fails to protect against cholecystokinin–octapeptide-induced acute pancreatitis in rats. Dig Dis Sci. 2002;47:1594–603. doi: 10.1023/a:1015883522648. [DOI] [PubMed] [Google Scholar]
- 27.Bhagat L, Singh VP, Song AM, et al. Thermal stress-induced HSP70 mediates protection against intrapancreatic trypsinogen activation and acute pancreatitis in rats. Gastroenterology. 2002;122:156–65. doi: 10.1053/gast.2002.30314. [DOI] [PubMed] [Google Scholar]
- 28.Bhagat L, Van Acker GJD, Manzoor R, et al. Targeted deletion of heat shock factor (HSF-1) exacerbates the severity of secretagogue-induced pancreatitis. Pancreas. 2002;25:421. [Google Scholar]
- 29.Bhagat L, Dawra R, Dhaulakhandi D, et al. Overexpression of heat shock protein 70 protects against secretagogue-induced pancreatitis. Pancreas. 2003;27:371. [Google Scholar]
- 30.Rakonczay Z, Jr, Takacs T, Ivanyi B, et al. Induction of heat shock proteins fails to produce protection against trypsin-induced acute pancreatitis in rats. Clin Exp Med. 2002;2:89–97. doi: 10.1007/s102380200012. [DOI] [PubMed] [Google Scholar]
- 31.Bhagat L, Saluja AK, Akella U, et al. Role of calcium in lysosomal enzyme redistribution and intra-acinar cell activation of trypsinogen in an in-vitro model of pancreatitis. Gastroenterology. 2000;118:A164. [Google Scholar]
- 32.Hietaranta AJ, Singh VP, Bhagat L, et al. Water immersion stress prevents caerulein-induced pancreatic acinar cell NF-kappa b activation by attenuating caerulein-induced intracellular Ca2+ changes. J Biol Chem. 2001;276:18742–7. doi: 10.1074/jbc.M009721200. [DOI] [PubMed] [Google Scholar]
- 33.Kiang JG, Ding XZ, McClain DE. Overexpression of HSP-70 attenuates increases in [Ca2+]i and protects human epidermoid A-431 cells after chemical hypoxia. Toxicol Appl Pharmacol. 1998;149:185–94. doi: 10.1006/taap.1997.8364. [DOI] [PubMed] [Google Scholar]
- 34.Mortaz E, Redegeld FA, van der Heijden MW, et al. Mast cell activation is differentially affected by heat shock. Exp Hematol. 2005;33:944–52. doi: 10.1016/j.exphem.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 35.Gress TM, Muller-Pillasch F, Weber C, et al. Differential expression of heat shock proteins in pancreatic carcinoma. Cancer Res. 1994;54:547–51. [PubMed] [Google Scholar]
- 36.Ogata M, Naito Z, Tanaka S, et al. Overexpression and localization of heat shock proteins mRNA in pancreatic carcinoma. J Nippon Med Sch. 2000;67:177–85. doi: 10.1272/jnms.67.177. [DOI] [PubMed] [Google Scholar]
- 37.Kojima K, Musch MW, Ren H, et al. Enteric flora and lymphocyte-derived cytokines determine expression of heat shock proteins in mouse colonic epithelial cells. Gastroenterology. 2003;124:1395–407. doi: 10.1016/s0016-5085(03)00215-4. [DOI] [PubMed] [Google Scholar]
- 38.Ludwig D, Stahl M, Ibrahim ET, et al. Enhanced intestinal expression of heat shock protein 70 in patients with inflammatory bowel diseases. Dig Dis Sci. 1999;44:1440–7. doi: 10.1023/a:1026616221950. [DOI] [PubMed] [Google Scholar]
- 39.Stahl M, Ludwig D, Fellermann K, et al. Intestinal expression of human heat shock protein 90 in patients with Crohn’s disease and ulcerative colitis. Dig Dis Sci. 1998;43:1079–87. doi: 10.1023/a:1018847205420. [DOI] [PubMed] [Google Scholar]
- 40.Winrow VR, Mojdehi GM, Ryder SD, et al. Stress proteins in colorectal mucosa. Enhanced expression in ulcerative colitis. Dig Dis Sci. 1993;38:1994–2000. doi: 10.1007/BF01297075. [DOI] [PubMed] [Google Scholar]
- 41.Maunder R. Mediators of stress effects in inflammatory bowel disease: not the usual suspects. J Psychosom Res. 2000;48:569–77. doi: 10.1016/s0022-3999(00)00098-2. [DOI] [PubMed] [Google Scholar]
- 42.Liu WL, Chen SJ, Chen Y, et al. Protective effects of heat shock protein70 induced by geranylgeranylacetone in atrophic gastritis in rats. Acta Pharmacol Sin. 2007;28:1001–6. doi: 10.1111/j.1745-7254.2007.00589.x. [DOI] [PubMed] [Google Scholar]
- 43.Yeo M, Park HK, Kim DK, et al. Restoration of heat shock protein70 suppresses gastric mucosal inducible nitric oxide synthase expression induced by Helicobacter pylori. Proteomics. 2004;4:3335–42. doi: 10.1002/pmic.200400951. [DOI] [PubMed] [Google Scholar]
- 44.Joo M, Chi JG, Lee H. Expressions of HSP70 and HSP27 in hepatocellular carcinoma. J Korean Med Sci. 2005;20:829–34. doi: 10.3346/jkms.2005.20.5.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Sui C, Kong F, et al. Expression of HSP70 and JNK-related proteins in human liver cancer: Potential effects on clinical outcome. Dig Liver Dis. 2007;39:663–70. doi: 10.1016/j.dld.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 46.Wang XP, Qiu FR, Liu GZ, et al. Correlation between clinicopathology and expression of heat shock protein 70 and glucose-regulated protein 94 in human colonic adenocarcinoma. World J Gastroenterol. 2005;11:1056–9. doi: 10.3748/wjg.v11.i7.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sherman M, Multhoff G. Heat shock proteins in cancer. Ann N Y Acad Sci. 2007;1113:192–201. doi: 10.1196/annals.1391.030. [DOI] [PubMed] [Google Scholar]
- 48.Nylandsted J, Brand K, Jaattela M. Heat shock protein 70 is required for the survival of cancer cells. Ann N Y Acad Sci. 2000;926:122–5. doi: 10.1111/j.1749-6632.2000.tb05605.x. [DOI] [PubMed] [Google Scholar]
- 49.Yuan ZP, Chen LJ, Fan LY, et al. Liposomal quercetin efficiently suppresses growth of solid tumors in murine models. Clin Cancer Res. 2006;12:3193–9. doi: 10.1158/1078-0432.CCR-05-2365. [DOI] [PubMed] [Google Scholar]
- 50.Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 51.Beere HM, Wolf BB, Cain K, et al. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–75. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- 52.Li CY, Lee JS, Ko YG, et al. Heat shock protein 70 inhibits apoptosis downstream of cytochrome c release and upstream of caspase-3 activation. J Biol Chem. 2000;275:25665–71. doi: 10.1074/jbc.M906383199. [DOI] [PubMed] [Google Scholar]
- 53.Steel R, Doherty JP, Buzzard K, et al. Hsp72 inhibits apoptosis upstream of the mitochondria and not through interactions with Apaf-1. J Biol Chem. 2004;279:51490–9. doi: 10.1074/jbc.M401314200. [DOI] [PubMed] [Google Scholar]
- 54.Stankiewicz AR, Lachapelle G, Foo CP, et al. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J Biol Chem. 2005;280:38729–39. doi: 10.1074/jbc.M509497200. [DOI] [PubMed] [Google Scholar]
- 55.Park HS, Lee JS, Huh SH, et al. Hsp72 functions as a natural inhibitory protein of c-Jun N-terminal kinase. Embo J. 2001;20:446–56. doi: 10.1093/emboj/20.3.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gabai VL, Yaglom JA, Volloch V, et al. Hsp72-mediated suppression of c-Jun N-terminal kinase is implicated in development of tolerance to caspase-independent cell death. Mol Cell Biol. 2000;20:6826–36. doi: 10.1128/mcb.20.18.6826-6836.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gabai VL, Mabuchi K, Mosser DD, et al. Hsp72 and stress kinase c-jun N-terminal kinase regulate the bid-dependent pathway in tumor necrosis factor-induced apoptosis. Mol Cell Biol. 2002;22:3415–24. doi: 10.1128/MCB.22.10.3415-3424.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guicciardi ME, Leist M, Gores GJ. Lysosomes in cell death. Oncogene. 2004;23:2881–90. doi: 10.1038/sj.onc.1207512. [DOI] [PubMed] [Google Scholar]
- 59.Bivik C, Rosdahl I, Ollinger K. Hsp70 protects against UVB induced apoptosis by preventing release of cathepsins and cytochrome c in human melanocytes. Carcinogenesis. 2007;28:537–44. doi: 10.1093/carcin/bgl152. [DOI] [PubMed] [Google Scholar]
- 60.Nylandsted J, Gyrd-Hansen M, Danielewicz A, et al. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med. 2004;200:425–35. doi: 10.1084/jem.20040531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ravagnan L, Gurbuxani S, Susin SA, et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol. 2001;3:839–43. doi: 10.1038/ncb0901-839. [DOI] [PubMed] [Google Scholar]
- 62.Guo F, Sigua C, Bali P, et al. Mechanistic role of heat shock protein 70 in Bcr-Abl-mediated resistance to apoptosis in human acute leukemia cells. Blood. 2005;105:1246–55. doi: 10.1182/blood-2004-05-2041. [DOI] [PubMed] [Google Scholar]
- 63.Rizzuto R, Pinton P, Ferrari D, et al. Calcium and apoptosis: facts and hypotheses. Evaluation of the neuronal apoptotic pathways involved in cytoskeletal disruption-induced apoptosis. Oncogene. 2003;22:8619–27. doi: 10.1038/sj.onc.1207105. [DOI] [PubMed] [Google Scholar]
- 64.Creagh EM, Carmody RJ, Cotter TG. Heat shock protein 70 inhibits caspase-dependent and -independent apoptosis in Jurkat T cells. Exp Cell Res. 2000;257:58–66. doi: 10.1006/excr.2000.4856. [DOI] [PubMed] [Google Scholar]
- 65.Kapranos N, Kominea A, Konstantinopoulos PA, et al. Expression of the 27-kDa heat shock protein (HSP27) in gastric carcinomas and adjacent normal, metaplastic, and dysplastic gastric mucosa, and its prognostic significance. J Cancer Res Clin Oncol. 2002;128:426–32. doi: 10.1007/s00432-002-0357-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lim SO, Park SG, Yoo JH, et al. Expression of heat shock proteins (HSP27, HSP60, HSP70, HSP90, GRP78, GRP94) in hepatitis B virus-related hepatocellular carcinomas and dysplastic nodules. World J Gastroenterol. 2005;11:2072–9. doi: 10.3748/wjg.v11.i14.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Concannon CG, Gorman AM, Samali A. On the role of Hsp27 in regulating apoptosis. Apoptosis. 2003;8:61–70. doi: 10.1023/a:1021601103096. [DOI] [PubMed] [Google Scholar]
- 68.Samali A, Robertson JD, Peterson E, et al. Hsp27 protects mitochondria of thermotolerant cells against apoptotic stimuli. Cell Stress Chaperones. 2001;6:49–58. doi: 10.1379/1466-1268(2001)006<0049:hpmotc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vargas-Roig LM, Gago FE, Tello O, et al. Heat shock protein expression and drug resistance in breast cancer patients treated with induction chemotherapy. Int J Cancer. 1998;79:468–75. doi: 10.1002/(sici)1097-0215(19981023)79:5<468::aid-ijc4>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 70.Solit DB, Rosen N. Hsp90: a novel target for cancer therapy. Curr Top Med Chem. 2006;6:1205–14. doi: 10.2174/156802606777812068. [DOI] [PubMed] [Google Scholar]
- 71.Zou J, Guo Y, Guettouche T, et al. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94:471–80. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
- 72.Voellmy R. On mechanisms that control heat shock transcription factor activity in metazoan cells. Cell Stress Chaperones. 2004;9:122–33. doi: 10.1379/CSC-14R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bagatell R, Whitesell L, Whitesell L, et al. Altered Hsp90 function in cancer: a unique therapeutic opportunity. The stress response: implications for the clinical development of hsp90 inhibitors. Mol Cancer Ther. 2004;3:1021–30. [PubMed] [Google Scholar]
- 74.Koga F, Xu W, Karpova TS, et al. Hsp90 inhibition transiently activates Src kinase and promotes Src-dependent Akt and Erk activation. Proc Natl Acad Sci U S A. 2006;103:11318–22. doi: 10.1073/pnas.0604705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kang BH, Plescia J, Dohi T, et al. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell. 2007;131:257–70. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 76.Ehrenfried JA, Herron BE, Townsend CM, Jr, et al. Heat shock proteins are differentially expressed in human gastrointestinal cancers. Surg Oncol. 1995;4:197–203. doi: 10.1016/s0960-7404(10)80036-2. [DOI] [PubMed] [Google Scholar]
- 77.Lang SA, Klein D, Moser C, et al. Inhibition of heat shock protein 90 impairs epidermal growth factor-mediated signaling in gastric cancer cells and reduces tumor growth and vascularization in vivo. Mol Cancer Ther. 2007;6:1123–32. doi: 10.1158/1535-7163.MCT-06-0628. [DOI] [PubMed] [Google Scholar]
- 78.Moser C, Lang SA, Kainz S, et al. Blocking heat shock protein-90 inhibits the invasive properties and hepatic growth of human colon cancer cells and improves the efficacy of oxaliplatin in p53-deficient colon cancer tumors in vivo. Mol Cancer Ther. 2007;6:2868–78. doi: 10.1158/1535-7163.MCT-07-0410. [DOI] [PubMed] [Google Scholar]
- 79.Vasilevskaya IA, O’Dwyer PJ. 17-Allylamino-17-demethoxygeldanamycin overcomes TRAIL resistance in colon cancer cell lines. Biochem Pharmacol. 2005;70:580–9. doi: 10.1016/j.bcp.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 80.Samali A, Cai J, Zhivotovsky B, et al. Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. Embo J. 1999;18:2040–8. doi: 10.1093/emboj/18.8.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xanthoudakis S, Roy S, Rasper D, et al. Hsp60 accelerates the maturation of pro-caspase-3 by upstream activator proteases during apoptosis. Embo J. 1999;18:2049–56. doi: 10.1093/emboj/18.8.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shan YX, Liu TJ, Su HF, et al. Hsp10 and Hsp60 modulate Bcl-2 family and mitochondria apoptosis signaling induced by doxorubicin in cardiac muscle cells. J Mol Cell Cardiol. 2003;35:1135–43. doi: 10.1016/s0022-2828(03)00229-3. [DOI] [PubMed] [Google Scholar]
- 83.Tang D, Khaleque MA, Jones EL, et al. Expression of heat shock proteins and heat shock protein messenger ribonucleic acid in human prostate carcinoma in vitro and in tumors in vivo. Cell Stress Chaperones. 2005;10:46–58. doi: 10.1379/CSC-44R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thomas X, Campos L, Mounier C, et al. Expression of heat-shock proteins is associated with major adverse prognostic factors in acute myeloid leukemia. Leuk Res. 2005;29:1049–58. doi: 10.1016/j.leukres.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 85.Cappello F, Di Stefano A, David S, et al. Hsp60 and Hsp10 down-regulation predicts bronchial epithelial carcinogenesis in smokers with chronic obstructive pulmonary disease. Cancer. 2006;107:2417–24. doi: 10.1002/cncr.22265. [DOI] [PubMed] [Google Scholar]
- 86.Ghosh JC, Dohi T, Kang BH, et al. Hsp60 regulation of tumor cell apoptosis. J Biol Chem. 2008;283:5188–94. doi: 10.1074/jbc.M705904200. [DOI] [PubMed] [Google Scholar]
- 87.Suto R, Srivastava PK. A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science. 1995;269:1585–8. doi: 10.1126/science.7545313. [DOI] [PubMed] [Google Scholar]
- 88.Binder RJ. Heat shock protein vaccines: from bench to bedside. Int Rev Immunol. 2006;25:353–75. doi: 10.1080/08830180600992480. [DOI] [PubMed] [Google Scholar]
- 89.Wang HH, Mao CY, Teng LS, et al. Recent advances in heat shock protein-based cancer vaccines. Hepatobiliary Pancreat Dis Int. 2006;5:22–7. [PubMed] [Google Scholar]