

Abstract
Background & Aims
Heat shock proteins (HSPs) are highly conserved and serve a multitude of functions that mediate cell survival. HSP70, the only inducible form of the 70-kilodalton subfamily of HSPs, is overexpressed in pancreatic cancer cells and has been shown to inhibit caspase-dependent apoptosis. We aimed to elucidate the mechanism by which HSP70 inhibits apoptosis in cancer cells.
Methods
HSP70 expression was down-regulated in cultured pancreatic cancer cells by exposure to quercetin, triptolide, or short interfering RNAs. Intracellular Ca2+, cytosolic cathepsin B activity, caspase-3 activity, cell viability, and lysosome integrity were measured using colorimetric assays. Immunofluorescence assays were used to localize cathepsin B and Lamp2. BAPTA-AM was used to chelate intracellular Ca2+.
Results
Inhibition of HSP70 increased intracellular Ca2+ levels in pancreatic and colon cancer cell lines and led to loss of lysosome integrity in pancreatic cancer cells. The release of intracellular Ca2+ and lysosomal enzymes activated caspase-dependent apoptosis independently and simultaneously.
Conclusions
HSP70 inhibits apoptosis in cancer cells by 2 mechanisms: attenuation of cytosolic calcium and stabilization of lysosomes. HSP70-mediated cell survival might occur in other types of cancer cells.
Apoptosis or programmed cell death is a naturally occurring and very useful process that organisms use to remove unwanted cells during development and for tissue homeostasis in adults.1 Dysregulation of apoptosis has been implicated in the pathogenesis of cancer.2 Thus, an understanding of the initiation and regulation of apoptosis is of cardinal importance in the biology of cancer. Apoptosis-inducing stimuli activate various intracellular pathways that culminate in cell death. Among these, both calcium and lysosomal enzymes have been cited as regulators and activators of caspase-dependent apoptotic pathways in various cell systems.3–5 In this study we provide evidence for a hitherto unsuspected mechanism by which heat shock protein 70 (HSP70) inhibits apoptosis by 2 modes: attenuating cytosolic Ca2+ ( ) and stabilizing lysosomes, both operating simultaneously but independently of each other.
Increased has been observed during both early and late stages of apoptosis.6,7 Inhibiting this increase by Ca2+ chelators prevents apoptosis.6,8 In addition, the sensitivity of cells to apoptosis can be affected by modulating calcium homeostasis (eg, by proteins of the bcl-2 family).9 Although increasing during apoptosis can stimulate many signaling pathways, the widely held opinion is that Ca2+ movement from the endoplasmic reticulum to the mitochondria leads to mitochondrial membrane permeabilization, cytochrome c release, and caspase activation.7
A growing body of evidence suggests that lysosomal enzymes involved in apoptosis are released from the lysosomes into the cytosol by a wide variety of stimuli, including tumor necrosis factor-α,10,11 Fas,12 and oxidative stress.12 Lysosomal permeabilization during apoptosis leads to release of cathepsins into the cytosol, these then permeabilize mitochondrial membranes, leading to cytochrome c release and caspase activation.10,13 The cathepsins also may activate the caspases directly14,15 by proteolysis or even may act independently of caspases. Whether the 2 modes of activation of the apoptotic cascade, calcium- and lysosomal-dependent, operate simultaneously, and whether they interact or are independent, is unknown.
We previously have shown that HSP70 expression is increased in pancreatic cancer cells as compared with normal pancreatic ductal cells,16 and that its down-regulation with quercetin (a bioflavonoid) or short interfering RNA (siRNA) leads to caspase-dependent apoptosis.16 We recently used triptolide, another inhibitor of HSP70 expression, and have confirmed our previous findings that HSP70 is important for the survival of pancreatic cancer cells, and that its down-regulation induces cell death and apoptosis.17 HSP70 overexpression is observed in a multitude of cancers and is believed to play a role in tumorigenesis because it confers resistance to both apoptotic and necrotic cell death.18 Studies suggest that HSP70 can reduce apoptosis by inhibiting caspase activation; various probable sites of action, both downstream and upstream of mitochondrial cytochrome c release, have been proposed.19–22 However, the exact mechanism by which HSP70 inhibits apoptosis remains uncertain. In this study we have evaluated the mechanism by which HSP70 inhibits apoptosis in cancer cells.
Materials and Methods
Quercetin and triptolide were purchased from Fluka (Buchs, Switzerland) and Calbiochem (San Diego, CA), respectively. HSP70 siRNA as well as nonsilencing siRNA were from Dharmacon Inc (Lafayette, CO) and cathepsin B siRNA was from Qiagen (Valencia, CA). Lipofectamine 2000 reagent, Opti-MemI, Dulbecco’s modified Eagle medium, and McCoy’s 5A tissue culture media were from Invitrogen Corporation (Carlsbad, CA). Lysotracker green, BAPTA-AM, and calcium orange-1 were from Molecular Probes (Carlsbad, CA). CA074me was from Peptides International (Louisville, KY). The WST-8 viability assay was from Dojindo Molecular Technologies (Gaithersburg, MD); the Guava Nexin Apoptosis Kit was from Guava Technologies Inc (Hayward, CA); the Caspase-Glo 3/7 assay kit was from Promega (San Luis Obispo, CA). The BCA protein assay kit was purchased from Pierce (Rockford, IL). All other reagents were from Sigma Aldrich (St. Louis, MO). Pancreatic cancer cells (Panc-1 and MiaPaCa-2) were a kind gift from Dr Edward E. Whang (Brigham and Women’s Hospital, Harvard Medical School, Boston, MA). The HT29 colon cancer cell line was purchased from ATCC (Manassas, VA).
Cell Culture
Pancreatic cancer cells were cultured in Dulbecco’s modified Eagle medium containing 10% fetal bovine serum and 1% penicillin-streptomycin. HCT 116 was cultured in McCoy’s 5A medium (modified) with 10% fetal bovine serum and 1% penicillin-streptomycin. All cells were maintained at 37°C in a humidified air atmosphere with 5% CO2.
Drug Treatment
Cancer cells were treated with quercetin and triptolide in serum-free media at a final concentration of 100 and 0.2 μmol/L, respectively. For intracellular calcium chelation, cells were treated with 10 μmol/L BAPTA-AM in serum-free Dulbecco’s modified Eagle medium for 30 minutes, followed by appropriate treatment with quercetin or triptolide. For cathepsin B inhibition, cells were treated with CA074me 10 μmol/L in serum-free medium followed by appropriate treatment with quercetin or triptolide.
Transfection With siRNA
To inhibit HSP70 and cathepsin B expression by siRNA, transfection was performed as described by us previously.16
Western Blot
Western Blot for protein measurement was performed as described by us in our previous publication.17
Measurement of Intracellular Ca2+ Levels
After inhibition of HSP70 expression by triptolide, quercetin, or HSP70 siRNA, cells were collected, washed in phosphate-buffered saline, and incubated with 1 μmol/L calcium orange-1-AM (Molecular Probes, Eugene, OR) in phosphate-buffered saline for 30 minutes at 37°C. Stained cells subsequently were analyzed using a Guava Easycyte flow cytometer.
Measurement of Lysosomal Integrity by Lysotracker Green
After appropriate treatment, cells were harvested, washed twice with phosphate-buffered saline, and incubated with 50 nmol/L Lysotracker Green in phosphate-buffered saline for 30 minutes at 37°C. Stained cells again were analyzed using a Guava Easycyte flow cytometer.
Measurement of Cytosolic Cathepsin B Activity
To measure cytosolic cathepsin B activity, the cytosolic fraction was extracted by incubating cells with an extraction buffer (250 mmol/L sucrose, 20 mmol/L HEPES, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L ethylenediaminetetraacetic acid, 1 mmol/L ethylene glycolbis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid, and 1 mmol/L pefabloc, pH 7.5) containing 12.5–20 μg/mL digitonin for 12–15 minutes on ice. The digitonin concentration and treatment times were optimized to result in the total release of cytosolic lactate dehydrogenase activity without disrupting the lysosomes. Cathepsin B activity was determined using N-carbobenzoxy-arginyl-arginine+-naphthylamide as the substrate, according to the method of McDonald and Ellis.23 The cathepsin B activity in the cytosolic fraction was normalized to lactate dehydrogenase activity in the sample, as measured by the Cytox 96 assay (Promega).
Immunostaining for Cathepsin B and Lamp2
Pancreatic cancer cells grown in chamber slides were treated with triptolide for 24 hours or treated with nonsilencing siRNA or HSP70 siRNA for 72 hours, fixed with 2% paraformaldehyde for 30 minutes, and permeabilized with 0.2% saponin for 5 minutes. The cells were incubated with a 1/200 dilution of mouse monoclonal anti–cathepsin B antibody (Santa Cruz, Santa Cruz, CA) and 1/300 dilution of rabbit polyclonal anti-Lamp2 antibody (Santa Cruz) for 2 hours at 4°C. After three 5-minute washes with phosphate-buffered saline containing 0.02% Tween-20, cells were incubated with secondary antibodies: 1/1200 dilution of Alexa-488–conjugated donkey anti-mouse IgG (Molecular Probes) and 1/1500 dilution of Alexa-555–conjugated donkey anti-rabbit IgG (Molecular Probes) for 50 minutes at 4°C. The slides then were washed and mounted using Prolong Gold anti-fade agent (Molecular Probes). Immunofluorescence images were obtained on a Nikon Eclipse Ti confocal microscope (Nikon, Melville, NY) using a 60× oil immersion objective.
Determination of Cell Viability
Cell viability was determined by the Dojindo Cell Counting Kit-8 according to the manufacturer’s protocol.
Measurement of Annexin V–Positive Cells
Phosphatidylserine externalization was analyzed using the Guava Nexin kit and a Guava personal cell analysis flow cytometer using the manufacturer’s protocol.
Caspase-3 Activity Assays
Caspase-3 activity was measured by the Caspase Glo 3/7 assay from Promega according to the manufacturer’s protocol.
Statistical Analysis
Values are expressed as the mean ± SEM. All experiments with cells were repeated at least 3 times. The significance of the difference between the control and each experimental test condition was analyzed by the unpaired Student t test, and a P value of less than.05 was considered statistically significant.
Results
HSP70 Attenuates Cytosolic Calcium
We have shown previously that quercetin16 and triptolide17 treatment inhibit HSP70 expression in both MiaPaCa-2 and Panc-1 cells at 12 and 24 hours. Treatment with either quercetin or triptolide increases cytosolic calcium in both MiaPaCa-2 and Panc-1 at 12 and 24 hours (Figure 1A and 1B). To confirm that the effect of triptolide and quercetin on is in fact owing to inhibition of HSP70 expression, we next examined the effect of down-regulating HSP70 by siRNA (designated siRNA1) on . Cells transfected with nonsilencing siRNA were used as the appropriate control in the siRNA experiments. As shown in Figure 1F, treatment with HSP70 siRNA leads to marked inhibition of HSP70 expression at 48 hours. The down-regulation of HSP70 by siRNA1 increased in both MiaPaCa-2 (Figure 1C) and Panc-1 at 48 hours (Figure 1D), as compared with the control. To rule out the theoretical possibility that the effect of HSP70 siRNA1 on might be owing to its off-target effects, we ascertained the effect of an additional HSP70 siRNA sequence (designated siRNA216) on . The down-regulation of HSP70 by siRNA2 in MiaPaCa-2 affected in the same way as observed with the siRNA1 sequence (Figure 1C). To ascertain the generalizability of this phenomenon to cancer cell lines other than pancreatic cancer, we examined the effect of down-regulating HSP70 expression on in the colon cancer cell line HT-29, which also overexpresses HSP70. Quercetin and triptolide treatment inhibit HSP70 expression in the HT29 cell line at 12 hours (data not shown). The inhibition of HSP70 expression by quercetin or triptolide treatment in the HT29 colon cancer cell line also increased cytosolic calcium (Figure 1E).
Figure 1.

Effect of inhibition of HSP70 expression on cytosolic calcium. (A) Inhibition of HSP70 expression by quercetin (Q) in both MiaPaCa-2 and Panc-1 pancreatic cancer cell lines leads to increased cytosolic calcium at 12 and 24 hours. (B) Inhibition of HSP70 expression by triptolide (T) increases cytosolic calcium. (C) Inhibition of HSP70 expression with 2 different siRNA sequences, siRNA1 and 2, in MiaPaCa-2 increased cytosolic calcium. (D) Inhibition of HSP70 expression by HSP70 siRNA1 leads to increased cytosolic calcium levels in Panc-1. (E) Inhibition of HSP70 expression in colon cancer cell line HT29 by triptolide- and quercetin-induced cytosolic calcium increase. Data are expressed as a percentage of control. Cells transfected with nonsilencing siRNA served as controls in siRNA experiments. Data are expressed as mean ± SEM of 3 independent experiments. *P < .05 (t test) as compared with controls. Cells transfected with nonsilencing siRNA served as controls in siRNA experiments. (F) Treatment of MiaPaCa-2 with siRNA1 and siRNA2 for 48 hours and of Panc-1 with siRNA1 for 48 hours leads to marked reduction in HSP70 levels. Nonsilencing siRNA (NS) and treatment with Lipofectamine 2000 (L2K) did not influence HSP70 levels. Actin was used as the loading control.
HSP70 Stabilizes Lysosomes
We next examined the effect of HSP70 down-regulation on lysosomal integrity by using Lysotracker green, which concentrates inside intact lysosomes and emits a green fluorescence; decreased green fluorescence therefore suggests lysosomal permeabilization. The inhibition of HSP70 expression by quercetin (Figure 2A), triptolide (Figure 2B), or HSP70 siRNA (Figure 2C and D) leads to decreased Lysotracker green fluorescence, which suggests lysosomal permeabilization. Results were confirmed in MiaPaCa-2 with an additional siRNA sequence (siRNA2) (Figure 2C). As in pancreatic cancer cell lines, the inhibition of HSP70 expression by quercetin and triptolide in HT29 colon cancer cells induced lysosomal permeabilization at 12 hours as compared with the untreated controls (Figure 2E). The effect of HSP70 down-regulation on lysosomal integrity was confirmed further by evaluating the effect of inhibition of HSP70 expression on the release of lysosomal enzyme cathepsin B into the cytosol. HSP70 down-regulation by quercetin (Figure 3A), triptolide (Figure 3B), or HSP70 siRNA1 (Figure 3C) significantly increased cytosolic cathepsin B activity as compared with control, suggesting release of cathepsin B from the lysosomes into the cytosol. The effect of inhibition of HSP70 expression on lysosomal permeabilization with release of cathepsin B into the cytosol also was evaluated by confocal microscopy. As seen in Figure 3D, immunostaining for cathepsin B (green) shows punctuate staining in control cells that colocalizes with Lamp-2 (red), a lysosomal marker, hence appearing yellow in the merged images. This suggests an intralysosomal localization of cathepsin B in untreated MiaPaCa-2 cells and in the cells treated with nonsilencing siRNA. The Pearson correlation, an index that describes if the 2 colors overlap, was 0.670 in the untreated MiaPaCa-2 cells and is 0.756 for MiaPaCA-2 cells treated with nonsilencing siRNA (Pearson correlation: 1, complete overlap; 0, no overlap between the 2 colors). Inhibition of HSP70 expression either by triptolide (Figure 3D, lower panel) or by siRNA (Figure 3E, lower panel) leads to diffuse staining of cathepsin B with decreased colocalization with Lamp-2, suggesting release of cathepsin B into the cytosol. The Pearson coefficient for cells treated with triptolide is 0.034, whereas that for cells treated with HSP70 siRNA is 0.045.
Figure 2.

Effect of inhibition of HSP70 expression on lysosomal integrity. (A) Inhibition of HSP70 expression by quercetin (Q) in both MiaPaCa-2 and Panc-1 pancreatic cancer cell lines leads to decreased Lysotracker green uptake at 12 hours, suggesting lysosomal permeabilization. (B) Inhibition of HSP70 expression by triptolide (T) also leads to decreased Lysotracker green uptake at 12 hours, suggesting lysosomal permeabilization. (C) Inhibition of HSP70 expression with 2 different siRNA sequences, siRNA1 and 2, in MiaPaCa-2 leads to lysosomal permeabilization. (D) Inhibition of HSP70 expression by siRNA1 leads to lysosomal permeabilization in Panc-1. (E) Inhibition of HSP70 expression in colon cancer cell line HT29 by triptolide and quercetin induced a decreased Lysotracker green uptake. Data are expressed as a percentage of control. Cells transfected with nonsilencing siRNA served as controls in siRNA experiments. Data are expressed as the mean ± SEM of 3 independent experiments. *P < .05 (t test) as compared with controls.
Figure 3.
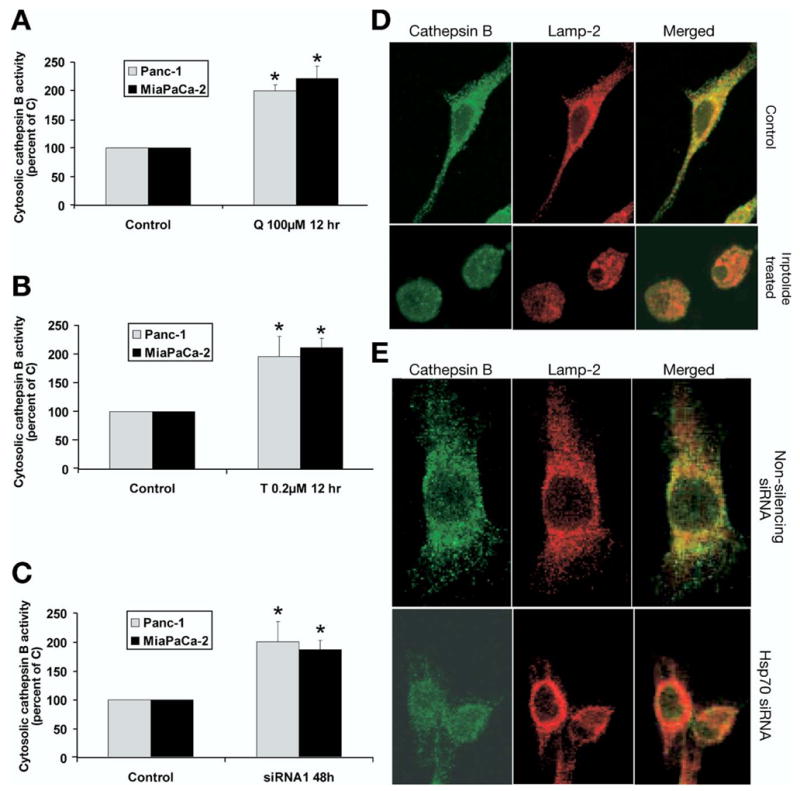
Effect of inhibition of HSP70 expression on cytosolic cathepsin B activity and its localization. Inhibition of HSP70 expression by (A ) quercetin, (B) triptolide, or (C) HSP70 siRNA in both MiaPaCa-2 and Panc-1 pancreatic cancer cell lines leads to increased cytosolic cathepsin B activity, which suggests lysosomal permeabilization. Cytosolic cathepsin B values are expressed as a percentage of control. Cells transfected with nonsilencing siRNA served as controls in siRNA experiments. Data are expressed as the mean ± SEM of 3 independent experiments. *P < .05 (t test) when compared with controls. (D and E) Inhibition of HSP70 leads to release of cathepsin B into the cytosol as evaluated with confocal microscopy. Cathepsin (green) colocalizes with Lamp-2 (lysosomal-associated membrane protein) in a punctuate fashion in control cells and as well as those transfected with nonsilencing siRNA, suggesting intralysosomal location. Inhibition of HSP70 expression by (D) triptolide as well as by (E) siRNA leads to diffuse staining of cathepsin B and reduced colocalization with Lamp-2, suggesting release of cathepsin B into the cytosol.
Role of and Cathepsin B in Cell Death, Apoptosis, and Caspase Activation Induced by Inhibition of HSP70 Expression
We next evaluated the role of the increased and lysosomal permeabilization in cell death induced by down-regulation of HSP70 expression. To better define the role of the increase in cell death induced by HSP70 down-regulation, we evaluated the effect of the intracellular calcium chelator BAPTA-AM on cell viability. BAPTA-AM was used at a dose (10 μmol/L) that prevented increase in response to HSP70 down-regulation (data not shown). The chelation of by BAPTA-AM pretreatment provided statistically significant protection against cell death induced by inhibiting HSP70 expression with quercetin (Figure 4A), triptolide (Figure 4B), and HSP70 siRNA (Figure 4C and D) in both Panc-1 and MiaPaCa-2 cell lines.
Figure 4.

Effect of chelating intracellular calcium and cathepsin B inhibition on cell death induced by inhibition of HSP70 expression. Inhibition of HSP70 expression by (A) quercetin, (B) triptolide, and (C and D) HSP70 siRNA induces cell death in both MiaPaCa-2 and Panc-1 pancreatic cancer cell lines. Chelation of cytosolic calcium by BAPTA protects against cell death induced by the down-regulation HSP70 by (A) quercetin, (B) triptolide, and (C and D) HSP70 siRNA. Similarly, the inhibition of cathepsin B by CA074me reduces cell death induced by (A) quercetin, (B) triptolide, and (C and D) HSP70 siRNA. Pretreatment with both BAPTA and CA074 provided significantly more protection against cell death than that offered by BAPTA and CA074me alone. Cell viability values (mean ± SEM) are expressed as a percentage of control. *p ≤ .05 (t test) as compared with inhibited HSP70 expression alone. #p ≤ .05 (t test) as compared with pretreatment with BAPTA or CA074me alone. (E) Western blot showing silencing of cathepsin B by siRNA in MiaPaCa-2. Actin was used as loading control. (F) Inhibition of cathepsin B by siRNA protects against cell death induced by inhibition of HSP70 expression by triptolide 200 nmol/L (T200). MiaPaCa-2 pancreatic cancer cell line was transfected with 2 different cathepsin B siRNA sequences for 48 hours followed by treatment with triptolide 200 nmol/L for 24 hours. Nonsilencing siRNA was used as control. The effect of cathepsin B inhibition by CA074me has been shown for comparison. *P < .05 when compared with treatment with NS siRNA and triptolide 200 nmol/L.
Next, we evaluated the effect of pretreatment with CA074me on cell death induced by HSP70 down-regulation. CA074me is a cell-permeable–specific cathepsin B inhibitor that, at a dose of 10 μmol/L, completely inhibited total cathepsin B activity in the cell lysate at 24 hours. Inhibiting cathepsin B with CA074me significantly reduced cell death from quercetin- (Figure 4A), triptolide- (Figure 4B), or HSP70 siRNA- (Figure 4C and D) induced HSP70 down-regulation in both the Panc-1 and MiaPaCa-2 cancer cell lines. We next evaluated the effect of pretreatment with a combination of BAPTA-AM and CA074me on viability. Pretreatment with the BAPTA-CA074me combination significantly reduced cell death induced by down-regulation of HSP70 (Figure 4A–D). Interestingly, protection against cell death conferred by the combination of BAPTA-AM and CA074me was significantly greater than that conferred by BAPTA-AM or CA074me alone (Figure 4A–D). To ensure that these effects observed with the cathepsin B inhibitor CA074 were in fact owing to inhibition of cathepsin B, we used cathepsin B siRNA to confirm our findings. As seen in Figure 4E, both the cathepsin B siRNA sequences that we used led to marked reduction of cathepsin B levels. As shown in Figure 4F, inhibition of cathepsin B expression by siRNA also provides protection against the cell death induced by HSP70 down-regulation, and this protection is very similar to that provided by CA074me.
We then evaluated the effect of intracellular calcium chelation and cathepsin B inhibition on apoptosis induced by HSP70 down-regulation. Apoptosis was monitored by measuring Annexin V staining, which is an early marker of apoptosis. Confirming our previous data,16 HSP70 down-regulation with quercetin (Figure 5A) or triptolide (Figure 5B) leads to apoptosis, suggested by increased Annexin V staining. Similar to its effect on viability, pretreatment with either intracellular calcium chelator BAPTA-AM or cathepsin B inhibitor CA074me significantly reduced the apoptosis induced by HSP70 down-regulation (Figure 5A and 5B) in both MiaPaCa-2 and Panc-1 pancreatic cancer cell lines. Remarkably, BAPTA-AM and CA074me in combination provided significantly greater protection (Figure 5A and 5B) against apoptosis as compared with that provided by BAPTA-AM or CA074me alone.
Figure 5.
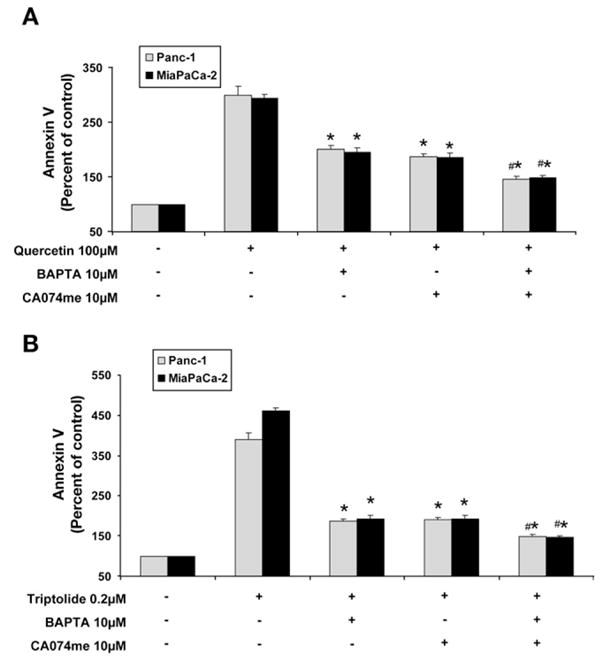
Effect of chelating intracellular calcium and inhibiting cathepsin B on apoptosis induced by inhibition of HSP70 expression. Inhibition of HSP70 expression by (A) quercetin and (B) triptolide induces apoptosis in both MiaPaCa-2 and Panc-1 pancreatic cancer cell lines as evidenced by Annexin V staining. Chelation of cytosolic calcium by BAPTA protects against apoptosis induced by the down-regulation of HSP70 by (A) quercetin and (B) triptolide. Similarly, the inhibition of cathepsin B by CA074me reduces apoptosis induced by (A) quercetin and (B) triptolide. Pretreatment with both BAPTA and CA074 provided significantly more protection against apoptosis than that of BAPTA and CA074me alone. Annexin V positivity (mean ± SEM) is expressed as a percentage of control. *p ≤ .05 (t test) as compared with inhibited HSP70 expression alone. #p ≤ .05 (t test) as compared with pretreatment with BAPTA or CA074me alone.
We have shown previously that apoptosis induced by inhibition of HSP70 expression involves caspase 3 activation.16 We next evaluated the role of and cathepsin B in caspase 3 activation. In accordance with our previous data, inhibition of HSP70 expression by quercetin (Figure 6A), triptolide (Figure 6B), and siRNA (Figure 6C and 6D) in both MiaPaCa-2 and Panc-1 led to the activation of caspase-3. Pretreatment with BAPTA-AM or CA074me significantly reduced caspase-3 activation induced by inhibition of HSP70 expression. A combination of BAPTA-AM and CA074me further decreased caspase-3 activation, which was statistically significant when compared with the effect of BAPTA-AM or CA074me alone. This result corroborates the cell viability and Annexin V data.
Figure 6.

Effect of chelating intracellular calcium and cathepsin B inhibition on caspase 3 activation induced by inhibition of HSP70 expression. Inhibition of HSP70 expression by (A) quercetin, (B) triptolide, and (C and D) HSP70 siRNA induces caspase 3 activation in both MiaPaCa-2 and Panc-1 pancreatic cancer cell lines. Chelation of cytosolic calcium by BAPTA protects against caspase 3 activation induced by the down-regulation of HSP70 by (A) quercetin, (B) triptolide, and (C and D) HSP70 siRNA. Similarly, the inhibition of cathepsin B by CA074me reduces caspase 3 activation induced by (A) quercetin, (B) triptolide, and (C and D) HSP70 siRNA. Pretreatment with both BAPTA and CA074me provided significantly more protection against caspase activation than that conferred by BAPTA and CA074me alone. Caspase 3 activity (mean ± SEM) is expressed as a percentage of control. *p ≤ .05 (t test) as compared with inhibited HSP70 expression alone. #p ≤ .05 (t test) as compared with pretreatment with BAPTA or CA074me alone.
Discussion
We have shown previously that HSP70 is overexpressed in pancreatic cancer cells and that its down-regulation induces caspase activation and cell death by apoptosis.16 We recently showed that triptolide, another inhibitor of HSP70 expression, remarkably reduces the growth of pancreatic tumors in the orthotopic mouse model and induces apoptosis not only in pancreatic cancer cells but also in HT29 colon cancer cells.17 This suggests that HSP70 is important for the survival not just of pancreatic cancer cells but also of other cancer cells that express HSP70, an observation made in a prior study24 as well. Many probable mechanisms have been proposed for the pro-survival function of HSP70. In vitro studies have suggested that HSP70 may interfere directly with the apoptosis-signaling machinery downstream of mitochondria by preventing apoptosome formation and caspase activation.25 Recent studies using cellular death models have pointed out that HSP70-mediated inhibition of caspase-dependent apoptosis occurs upstream of mitochondrial outer membrane permeabilization and cytochrome c release.21,26,27 Our studies suggest that the inhibition of HSP70 expression by quercetin, triptolide, or HSP70 siRNA leads to caspase activation and apoptosis, suggesting that HSP70 inhibits caspase-dependent apoptosis in pancreatic cancer cells. Because and lysosomal enzymes are 2 regulators of caspase-dependent apoptosis,5 we examined how HSP70 interacts with them.
HSP70 down-regulation by quercetin or triptolide in pancreatic cancer cell lines was associated with increased . In our previous studies16,17 we have shown that one of the major mechanisms by which quercetin and triptolide induce cell death in pancreatic cancer cells is by inhibition of HSP70 expression. However, these natural inhibitors may influence pathways other than HSP70 expression. To rule out the possibility that the effect of quercetin or triptolide on was the result of their effect on a pathway other than the inhibition of HSP70 expression, we confirmed our findings with HSP70 siRNA. We previously had used both the HSP70 siRNA sequences used in this study and found that they inhibited HSP70 protein expression consistently in pancreatic cancer cell lines at 48 and 72 hours.16 Furthermore, the inhibition of HSP70 expression increases not only in pancreatic cancer cell lines but also in the colon cancer cell line HT29, suggesting that this phenomenon can be generalized to other cancer cells with HSP70 overexpression. These findings suggest that HSP70 attenuates calcium response in cells, and that when HSP70 expression is inhibited, levels increase.
To evaluate whether the increase observed in pancreatic cancer cells contributes in any way to cell death and apoptosis induced by HSP70 down-regulation, we studied the effect of the intracellular calcium chelator BAPTA-AM on viability, Annexin V staining, and caspase-3 activation. BAPTA-AM was used at the lowest possible concentration that prevented an increase in in response to HSP70 down-regulation. Chelation of provided significant but incomplete protection against cell death induced by HSP70 down-regulation, which suggests that increase plays a role in cell death, but that it is not the sole mediator. Similarly, intracellular calcium chelation also reduced caspase-3 activation and the Annexin V positivity induced by HSP70 down-regulation, which suggests that plays a significant role in caspase-3 activation and apoptosis. Taken together, these facts indicate that at least one of the mechanisms by which HSP70 inhibits apoptosis is attenuation; HSP70 down-regulation leads to increased , which in turn induces caspase-3 activation, apoptosis, and cell death. However, intracellular calcium chelation provides incomplete protection against cell death, apoptosis, and caspase-3 activation, suggesting that in addition to intracellular calcium attenuation, HSP70 inhibits apoptosis by other mechanisms as well.
Because lysosomal enzymes also can lead to caspase-dependent apoptosis, we next examined the interaction between HSP70 and lysosomes. HSP70 down-regulation by quercetin, triptolide, or HSP70 siRNA resulted in lysosomal membrane permeabilization, as evidenced by decreased Lysotracker green uptake into the lysosomes, as well as cathepsin B release into the cytosol. This was confirmed by 2 different techniques, namely, cellular fractionation followed by biochemical measurement of cathepsin B levels in the cytosol and by confocal microscopy. These results in turn suggest that HSP70 stabilizes the lysosomes and prevents cathepsin release into the cytosol. This phenomenon was not limited to pancreatic cancer cell lines, but also was observed in the colon cancer cell line HT29, and thus can be generalized to other cancers expressing HSP70. To elucidate the role of cathepsin B in apoptosis induced by HSP70 inhibition, we evaluated the effect of cathepsin B inhibition on viability, apoptosis, and caspase-3 activation using CA074me, which is a cell-permeable methyl ester of the specific cathepsin B inhibitor CA074.28 CA074me was used at the lowest dose that would inhibit cathepsin B. Similar to the effect observed with calcium chelation, inhibiting cathepsin B by CA074me provided significant but incomplete protection against cell death, caspase-3 activation, and apoptosis induced by HSP70 down-regulation. Similar protective effects against cell death were obtained when cathepsin B expression was inhibited with cathepsin B siRNA. These observations taken together suggest that down-regulation of HSP70 expression leads to lysosomal membrane permeabilization, hence a release of cathepsin B into the cytosol, which then activates caspase-3 and in turn results in apoptosis.
Because both intracellular calcium chelation and cathepsin B inhibition provide significant but incomplete protection against caspase-3 activation, apoptosis, and cell death, we next examined the possibility that both the increase in and in lysosomal enzymes play a role in apoptosis simultaneously and independently. Pretreatment with BAPTA and CA074me combined provided significantly greater protection against caspase activation, apoptosis, and cell death induced by HSP70 down-regulation than that conferred by BAPTA or CA074me alone. This additive effect suggests that both the increase of intracellular calcium and lysosomal permeabilization with release of cathepsin B operate simultaneously and independently in the activation of HSP70 down-regulation–induced apoptosis. Conversely, HSP70 inhibits apoptosis by 2 independent and simultaneous mechanisms: by attenuating intracellular calcium and stabilizing lysosomes. It should be noted that although preventing an increase in cytosolic calcium and blocking cathepsin B markedly reduces the cell death induced by HSP70 down-regulation, cell death is not abrogated completely, suggesting that there may be mechanisms in addition to these 2 mechanisms that lead to cell death after inhibition of HSP70 expression. Because HSP70 is a chaperone protein and helps in proper folding of intracellular proteins, it is possible that HSP70 could protect the cells from the harmful effects of unfolded protein response, and this could be one of the additional mechanisms by which HSP70 protects against cell death. It will be interesting to evaluate this aspect in future studies.
Some other pro- and anti-apoptotic proteins, the most prominent of which is the Bcl-2 family of proteins, modulate the sensitivity of cancer cells to apoptosis by interacting with calcium homeostasis.9,29 Bax and Bak, the pro-apoptotic members of the Bcl-2 family, maintain the calcium stores in the endoplasmic reticulum that can be released in response to apoptotic stimuli and also augment calcium uptake by mitochondria, thereby regulating apoptosis.30 On the other hand, Bcl-2, which is the anti-apoptotic member of the Bcl-2 family, negatively regulates the endoplasmic reticulum calcium-filling state and thus negatively regulates apoptosis.3 In this study, we observed the effect of anti-apoptotic protein HSP70 on calcium homeostasis. We have shown that attenuating is one of the mechanisms by which HSP70 inhibits apoptosis in pancreatic cancer cells. How HSP70 attenuates is not known. A delicate balance between calcium release from the internal stores, calcium influx, efflux, and reuptake of calcium into the internal stores is central to regulation below a certain threshold because high levels are uniformly toxic to the cells. HSP70 could directly or indirectly influence one of these processes by acting on other proteins to attenuate and thus inhibit apoptosis. The delineation of the exact site will be of great importance.
We also have shown that HSP70 stabilizes lysosomes. Inhibition of HSP70 expression is associated with lysosomal permeabilization and concomitant release of cathepsin B into the cytosol, which then plays a role in caspase activation, apoptosis, and cell death. Various cathepsins, including cathepsin B, D, and L, are involved prominently in the execution of apoptosis, depending on the cell type and the apoptosis-inducing stimulus.4 We used cathepsin B as a surrogate for the lysosomal proteases released on lysosomal permeabilization. As discussed earlier, pretreatment with CA074me, a specific cathepsin B inhibitor, provided protection against cell death and against the apoptosis induced by HSP70 down-regulation, which suggests that cathepsin B plays a role in these processes.27 All the same, we cannot rule out the theoretical possibility that other cathepsins and lysosomal proteases also may be involved in apoptosis induced by HSP70 down-regulation in cancer cells. However, there is reason to believe that cathepsin B is the dominant lysosomal protease involved in apoptosis in pancreatic cancer cells because its inhibition along with the inhibition of the other calcium-dependent apoptosis activation pathway restores their viability to nearly that of untreated cells.
The mechanism by which HSP70 stabilizes the lysosomes currently is unknown. Various mechanisms by which lysosomes are permeabilized in apoptosis have been proposed, including one involving Bax, reactive oxygen species, and sphingosine generation. Other studies have proposed that HSP70 localizes to the lysosomal membrane and thus might stabilize it directly.25,31 Given the known role of Bax in lysosomal permeabilization and the ability of HSP70 to inhibit Bax translocation to mitochondria, it is possible that HSP70 prevents lysosomal permeabilization by preventing Bax activation and translocation to lysosomes.27,32 Because the improvements in viability, apoptosis, and caspase activation observed on pretreatment with CA074me and BAPTA combined are additive, the lysosomal enzyme-dependent and calcium-dependent activation of apoptosis seem to occur simultaneously yet independently of each other.
In sum, we have shown that HSP70 inhibits apoptosis by 2 mechanisms: by attenuating and by stabilizing lysosomes. Our findings also suggest that calcium and the lysosomal cathepsin-dependent activation of apoptosis operate simultaneously but independently of each other. This is a new paradigm that can be generalized to cancers other than pancreatic cancer.
Acknowledgments
The authors would like to thank Dr Ashok Sengupta and Mara Oikonomou for their critical readings of this manuscript.
Funding These studies were supported in part by National Institutes of Health grants DK58694, CA124723, CA131663 (to A.K.S.), grants from the Hirshberg Foundation for Pancreatic Cancer Research and Robert and Katherine Goodale Foundation (to A.K.S.), and by intramural funding from the University of Minnesota’s Surgery Department.
Abbreviations used in this paper
- HSP70
heat shock protein 70
- siRNA
short interfering RNA
Footnotes
Conflicts of interest The authors disclose no conflicts.
References
- 1.Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- 2.Hetts SW. To die or not to die: an overview of apoptosis and its role in disease. JAMA. 1998;279:300–307. doi: 10.1001/jama.279.4.300. [DOI] [PubMed] [Google Scholar]
- 3.Ferrari D, Pinton P, Szabadkai G, et al. Endoplasmic reticulum, Bcl-2 and Ca2+ handling in apoptosis. Cell Calcium. 2002;32:413–420. doi: 10.1016/s0143416002002014. [DOI] [PubMed] [Google Scholar]
- 4.Guicciardi ME, Leist M, Gores GJ. Lysosomes in cell death. Oncogene. 2004;23:2881–2890. doi: 10.1038/sj.onc.1207512. [DOI] [PubMed] [Google Scholar]
- 5.Kim R, Emi M, Tanabe K. Role of mitochondria as the gardens of cell death. Cancer Chemother Pharmacol. 2006;57:545–553. doi: 10.1007/s00280-005-0111-7. [DOI] [PubMed] [Google Scholar]
- 6.Kruman I, Guo Q, Mattson MP. Calcium and reactive oxygen species mediate staurosporine-induced mitochondrial dysfunction and apoptosis in PC12 cells. J Neurosci Res. 1998;51:293–308. doi: 10.1002/(SICI)1097-4547(19980201)51:3<293::AID-JNR3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 7.Rizzuto R, Pinton P, Ferrari D, et al. Calcium and apoptosis: facts and hypotheses. Oncogene. 2003;22:8619–8627. doi: 10.1038/sj.onc.1207105. [DOI] [PubMed] [Google Scholar]
- 8.Kim JA, Kang YS, Lee YS. A phospholipase C-dependent intracellular Ca2+ release pathway mediates the capsaicin-induced apoptosis in HepG2 human hepatoma cells. Arch Pharm Res. 2005;28:73–80. doi: 10.1007/BF02975139. [DOI] [PubMed] [Google Scholar]
- 9.Pinton P, Rizzuto R. Bcl-2 and Ca2+ homeostasis in the endoplasmic reticulum. Cell Death Differ. 2006;13:1409–1418. doi: 10.1038/sj.cdd.4401960. [DOI] [PubMed] [Google Scholar]
- 10.Guicciardi ME, Deussing J, Miyoshi H, et al. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest. 2000;106:1127–1137. doi: 10.1172/JCI9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foghsgaard L, Lademann U, Wissing D, et al. Cathepsin B mediates tumor necrosis factor-induced arachidonic acid release in tumor cells. J Biol Chem. 2002;277:39499–39506. doi: 10.1074/jbc.M206669200. [DOI] [PubMed] [Google Scholar]
- 12.Brunk UT, Svensson I. Oxidative stress, growth factor starvation and Fas activation may all cause apoptosis through lysosomal leak. Redox Rep. 1999;4:3–11. doi: 10.1179/135100099101534675. [DOI] [PubMed] [Google Scholar]
- 13.Boya P, Andreau K, Poncet D, et al. Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J Exp Med. 2003;197:1323–1334. doi: 10.1084/jem.20021952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishisaka R, Utsumi T, Kanno T, et al. Participation of a cathepsin L-type protease in the activation of caspase-3. Cell Struct Funct. 1999;24:465–470. doi: 10.1247/csf.24.465. [DOI] [PubMed] [Google Scholar]
- 15.Katunuma N, Matsui A, Le QT, et al. Novel procaspase-3 activating cascade mediated by lysoapoptoses and its biological significances in apoptosis. Adv Enzyme Regul. 2001;41:237–250. doi: 10.1016/s0065-2571(00)00018-2. [DOI] [PubMed] [Google Scholar]
- 16.Aghdassi A, Phillips P, Dudeja V, et al. Heat shock protein 70 increases tumorigenicity and inhibits apoptosis in pancreatic adenocarcinoma. Cancer Res. 2007;67:616–625. doi: 10.1158/0008-5472.CAN-06-1567. [DOI] [PubMed] [Google Scholar]
- 17.Phillips PA, Dudeja V, McCarroll JA, et al. Triptolide induces pancreatic cancer cell death via inhibition of heat shock protein 70. Cancer Res. 2007;67:9407–9416. doi: 10.1158/0008-5472.CAN-07-1077. [DOI] [PubMed] [Google Scholar]
- 18.Jaattela M. Multiple cell death pathways as regulators of tumour initiation and progression. Oncogene. 2004;23:2746 –2756. doi: 10.1038/sj.onc.1207513. [DOI] [PubMed] [Google Scholar]
- 19.Beere HM, Wolf BB, Cain K, et al. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- 20.Saleh A, Srinivasula SM, Balkir L, et al. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat Cell Biol. 2000;2:476 –483. doi: 10.1038/35019510. [DOI] [PubMed] [Google Scholar]
- 21.Creagh EM, Carmody RJ, Cotter TG. Heat shock protein 70 inhibits caspase-dependent and -independent apoptosis in Jurkat T cells. Exp Cell Res. 2000;257:58–66. doi: 10.1006/excr.2000.4856. [DOI] [PubMed] [Google Scholar]
- 22.Mosser DD, Caron AW, Bourget L, et al. The chaperone function of hsp70 is required for protection against stress-induced apoptosis. Mol Cell Biol. 2000;20:7146–7159. doi: 10.1128/mcb.20.19.7146-7159.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garrido C, Brunet M, Didelot C, et al. Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell Cycle. 2006;5:2592–2601. doi: 10.4161/cc.5.22.3448. [DOI] [PubMed] [Google Scholar]
- 24.Beere HM, Green DR. Stress management—heat shock protein-70 and the regulation of apoptosis. Trends Cell Biol. 2001;11:6–10. doi: 10.1016/s0962-8924(00)01874-2. [DOI] [PubMed] [Google Scholar]
- 25.Bivik C, Rosdahl I, Ollinger K. Hsp70 protects against UVB induced apoptosis by preventing release of cathepsins and cytochrome c in human melanocytes. Carcinogenesis. 2007;28:537–544. doi: 10.1093/carcin/bgl152. [DOI] [PubMed] [Google Scholar]
- 26.Stankiewicz AR, Lachapelle G, Foo CP, et al. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J Biol Chem. 2005;280:38729 –38739. doi: 10.1074/jbc.M509497200. [DOI] [PubMed] [Google Scholar]
- 27.Buttle DJ, Murata M, Knight CG, et al. CA074 methyl ester: a proinhibitor for intracellular cathepsin B. Arch Biochem Biophys. 1992;299:377–380. doi: 10.1016/0003-9861(92)90290-d. [DOI] [PubMed] [Google Scholar]
- 28.Motyl T. Regulation of apoptosis: involvement of Bcl-2-related proteins. Reprod Nutr Dev. 1999;39:49–59. doi: 10.1051/rnd:19990103. [DOI] [PubMed] [Google Scholar]
- 29.Scorrano L, Oakes SA, Opferman JT, et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 30.Nylandsted J, Gyrd-Hansen M, Danielewicz A, et al. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med. 2004;200:425–435. doi: 10.1084/jem.20040531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feldstein AE, Werneburg NW, Li Z, et al. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1339 – G1346. doi: 10.1152/ajpgi.00509.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McDonald JK, Ellis S. On the substrate specificity of cathepsins B1 and B2 including a new fluorogenic substrate for cathepsin B1. Life Sci. 1975;17:1269–1276. doi: 10.1016/0024-3205(75)90137-x. [DOI] [PubMed] [Google Scholar]