

Abstract
Glioblastoma multiforme (GBM) are lethal brain tumors that are highly resistant to therapy. The only meaningful improvement in therapeutic response came from use of the SN1-type alkylating agent, temozolomide, in combination with ionizing radiation (IR). However, no genetic markers that might predict a better response to DNA alkylating agents have been identified in GBMs, except for loss of O6-methylguanine-DNA methyltransferase (MGMT) via promoter methylation. In this study, using genetically defined primary murine astrocytes as well as human glioma lines, we show that loss of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) confers sensitivity to N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), a functional analog of temozolomide. We find that MNNG induces replication-associated DNA double-strand breaks (DSBs) that are inefficiently repaired in PTEN-deficient astrocytes and trigger apoptosis. Mechanistically, this is because PTEN-null astrocytes are compromised in homologous recombination (HR), which is important for the repair of replication-associated DSBs. Our results suggest that reduced levels of Rad51 paralogs in PTEN-null astrocytes might underlie the HR deficiency of these cells. Importantly, the HR deficiency of PTEN-null cells renders them sensitive to the poly(ADP-ribose) polymerase (PARP) inhibitor ABT-888 due to synthetic lethality. In sum, our results tentatively suggest that patients with PTEN-null GBMs (about 36%) may especially benefit from treatment with DNA alkylating agents such as temozolomide. Significantly, our results also provide a rational basis for treating the sub-group of patients who are PTEN deficient with PARP inhibitors in addition to the current treatment regimen of radiation and temozolomide.
Keywords: glioblastoma multiforme (GBM), DNA double-strand break (DSB), temozolomide (TMZ), N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), PARP inhibitors, PTEN, homologous recombination (HR)
Introduction
Despite considerable work in recent years elucidating the molecular underpinnings of glioblastoma multiforme (GBM), the most deadly of brain cancers, little progress has been made in improving clinical outcomes. The most significant breakthrough in patient response to date emerged from the use of the SN1-type alkylating agent, temozolomide, in combination with ionizing radiation (IR) that increased the overall median survival from approximately 12 to 15 months (1-3). However, no genetic markers that might predict a better response to DNA alkylating agents have been identified for GBMs except for O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation (3, 4). The past year has seen unprecedented advances in the genomic analyses of adult GBM tumors by the Cancer Genome Atlas Network and other groups which reveal that these tumors have radically altered genomes with many mutations, gene copy number gains and losses, and methylation changes (5-7). Amongst the myriad of genetic alterations that populate the GBM genomic landscape, five genetic changes dominate: loss of Ink4a, Arf, p53, or phosphatase and tensin homolog deleted on chromosome 10 (PTEN) and amplification of epidermal growth factor receptor (EGFR). How these genetic aberrations confer therapeutic resistance remains unclear. Understanding the contribution of these lesions, singly and in combination, to GBM therapy resistance along with the underlying mechanism(s) will be of paramount importance in developing more effective therapeutic modalities.
In a previous study, we demonstrated that amplification of EGFRvIII confers radioresistance to GBM-relevant cells and tumors by promoting the repair of radiation-induced DNA double-strand breaks (DSBs) by non-homologous end joining (NHEJ) (8). In this study, using genetically defined primary murine astrocytes as well as human glioma lines, we focused on the role of PTEN in modulating sensitivity to the SN1-type alkylating agent, N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) (9). Loss of PTEN is a very prominent event during gliomagenesis, occurring in about 36% of GBMs (5, 6, 10). PTEN is a lipid phosphatase with a canonical role in dampening the phosphatidylinositol 3-kinase (PI3K)-Akt-1 signaling pathway; hence, loss of PTEN has oncogenic consequences during gliomagenesis (11). In addition, it is becoming increasingly clear that PTEN has novel nuclear functions, including transcriptional regulation of the Rad51 gene, whose product is essential for homologous recombination (HR) repair of DNA breaks (12, 13). We report here that loss of PTEN in astrocytes results in increased sensitivity to MNNG. We show that MNNG induces secondary DSBs that are poorly repaired in PTEN-null astrocytes due to compromised HR. The increased sensitivity of PTEN-null astrocytes to MNNG tentatively suggests that patients with PTEN-null GBMs may especially benefit from treatment with temozolomide. More importantly, the HR deficiency of PTEN-null astrocytes opens up the possibility of treating PTEN-deficient GBMs with poly(ADP-ribose) polymerase (PARP) inhibitors that are currently in clinical trials for treating HR-deficient breast and ovarian cancers (14-16).
Materials and Methods
Cell culture
Astrocytes were isolated from five day old pups as described (17) from littermates of an Ink4a/Arf-/- PTENf/+ × Ink4a/Arf-/- PTENf/+ cross (18, 19). Primary mouse astrocytes were maintained in DMEM media containing 10% FBS in a humidified 37°C incubator with 5% CO2. The floxed PTEN allele was deleted using an adenovirus expressing Cre. All cells were mycoplasma free.
Irradiation and drug treatment
A 137Cs source (JL Shepherd and Associates, CA) was used for γ-ray irradiation of cells. MNNG (Sigma) and CPT (Sigma) were dissolved in DMSO and stored at -20°C in aliquots of 100mM. ABT-888 (Alexis Biochemicals) was dissolved in cell culture grade water and stored at -20°C. MNNG treatments were given as a 1 h pulse, while CPT and ABT-888 were added continuously at the indicated concentrations.
Colony formation assays
Cells were plated in triplicate onto 60 mm dishes (300 cells per dish) and irradiated with graded doses of radiation or treated with increasing concentrations of MNNG, CPT, or ABT-888. Surviving colonies were stained with crystal violet about 7 days later as described (20).
Immunofluorescence (IF) staining and Western analyses
IF staining of cells and Western blot analyses of whole-cell extracts were performed as described (21). Antibodies used were anti-Rad51 (Santa Cruz), anti-γH2AX (Upstate), anti-53BP1 (Cell Signaling), anti-actin (Sigma), anti-Akt, anti-phospho-Akt(Ser473) (Cell Signaling), anti-MGMT (Santa Cruz), rhodamine red-conjugated goat anti-rabbit, and FITC-conjugated goat anti-mouse (Molecular Probes).
Metaphase chromosome preparations and Sister Chromatid Exchange (SCE) assay
To examine chromosome aberrations, astrocytes were treated with MNNG and, 24 hours later, 1μg/ml colcemid (Sigma) was added for 3 hours. Metaphase chromosome spreads were then prepared using standard procedures. Aberrations were counted and categorized as breaks or asymmetric exchanges (tri-radials and quadri-radials). To visualize SCEs, cells were incubated in the presence of BrdU (BD Biosciences, 10μM) for two cell divisions, after which metaphases were prepared according to the above protocol. For drug treatments, MNNG was added as a 1 h pulse immediately prior to BrdU, while CPT was added concurrently. Aberrations and SCEs were quantified by analyzing 100 metaphase spreads and differences were statistically analyzed as described below.
Statistical analyses
P values for experiments were calculated using GraphPad Prism. SCE and chromosome aberration data were analyzed by a two-tailed t-test, while qRT-PCR data and repair kinetics were analyzed using two-way ANOVA.
Additional methods
Please see Supplement.
Results
For this study, primary astrocytes were generated from Ink4a/Arf-/-PTEN+/+ or Ink4a/Arf-/-PTENf/f transgenic littermates. Once in culture, floxed PTEN alleles were deleted by adenoviral expression of Cre recombinase, generating a set of matched astrocytes with the following genotypes: Ink4a/Arf-/-PTEN+/+ and Ink4a/Arf-/-PTEN-/-. Because primary mouse astrocytes senesce within a couple of passages after extraction, the Ink4a/Arf-/- background ensures that the astrocytes are immortal and provides a tumor-suppressor background that is very relevant to GBMs (5, 6). PTEN deletion upon adenovirus infection was confirmed by Western blotting (Fig. 1a), as well as by PCR analysis (Supplement; Fig. S1). As expected, PTEN loss strongly activated PI3K signaling as evidenced by increased levels of phosphorylated Akt-1 (phospho-serine 473) (11). In accordance with previous reports (22, 23), we found that loss of PTEN resulted in increased resistance to IR as assayed by colony survival (Fig. 1b). In contrast, PTEN loss resulted in sensitization to MNNG (Fig. 1c). Flow cytometric analyses revealed a significant increase in the sub-G0 population in MNNG-treated PTEN-deficient cultures indicating that the sensitivity of these astrocytes to MNNG was due to an increase in cell death (Fig. 1d).
Figure 1. PTEN loss sensitizes astrocytes to MNNG.

(A) Loss of PTEN and activation of Akt in PTEN+/+ and -/- astrocytes were analyzed by Western blotting with α-PTEN and α-phospho-Akt(ser473) antibodies. (B) Radiation survival of astrocytes was quantified by colony formation assays. The fraction of surviving colonies (y-axis) was plotted against corresponding radiation dose (x-axis). (C) Sensitivity of astrocytes to MNNG was quantified by colony formation assays. Please note increased sensitivity of PTEN-null cells to MNNG. Error bars represent standard error of the mean of experiments performed three or more times. (D) Induction of cell death by MNNG was assessed by quantifying the sub-G0 population in MNNG-treated cultures by flow cytometry (numbers in red).
While the cytoplasmic role of PTEN in squelching the PI3K-Akt-1 pathway is an established concept, recent reports clearly indicate novel nuclear functions for this protein, including roles in transcription regulation (11-13). The sensitivity of PTEN-null cells to MNNG was probably not due to hyperactivation of Akt-1 as astrocytes expressing constitutively active myristylated Akt-1 (8) did not display an increased sensitivity to MNNG compared to parental cells (Supplement; Fig. S2). Therefore, it is plausible that the sensitivity to MNNG observed in PTEN-/- astrocytes was due to loss of a nuclear function of PTEN.
Toxicity from SN1-type alkylating agents results mainly from a specific type of DNA lesion, methylation of the O6 position of guanine (O6meG) (24), that can be reversed by the suicide repair enzyme, MGMT (25). It has been previously reported that MGMT promoter silencing by methylation corresponds to a better therapeutic response to temozolomide (3, 4). Therefore, we investigated whether a decrease in MGMT levels due to PTEN loss might underlie the sensitivity of these cell lines to MNNG. However, Western blot analyses of basal MGMT protein levels did not show a significant difference between PTEN+/+ and PTEN-/- astrocytes (Fig. 2a). MGMT transcription is induced in response to various DNA damaging agents, including MNNG (24, 26). However, quantitative real-time PCR (qRT-PCR) analyses revealed that both lines were capable of inducing MGMT transcription upon MNNG treatment (Fig. 2b). These data strongly suggest that the sensitivity of PTEN-null cells to MNNG was not due to attenuation of MNNG transcript or protein levels.
Figure 2. MGMT regulation is not affected by PTEN loss.
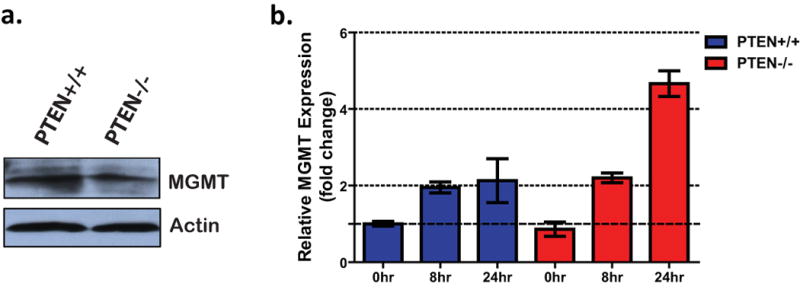
(A) MGMT protein levels in PTEN+/+ and -/- astrocytes were analyzed by Western blotting with α-MGMT antibody. (B) MGMT transcript levels in mock-treated and MNNG-treated astrocytes were quantified by qRT-PCR. Values were normalized to GAPDH levels and expressed as fold change relative to mock-treated PTEN+/+ cells. Error bars represent standard error of the mean.
The cytotoxicity of O6meG lesions is attributed to the recognition of O6-meG/C or O6-meG/T mispairs by the mismatch repair (MMR) system (27), with two opposing models proposed: (a) DNA damage signal transduction by the MMR complex engaged at the mismatch sites directly triggering apoptosis (direct signaling model) or (b) reiterative and futile repair attempts by MMR resulting in single-strand and double-strand DNA breaks (futile cycle model) (28). Since DNA double-strand breaks (DSBs) are the most lethal of DNA lesions, we investigated whether such breaks were induced in MNNG-treated astrocytes and whether these breaks were more persistent in PTEN-deficient cells. We analyzed the formation and dissolution of γH2AX and 53BP1 foci upon pulse-treatment with MNNG (8, 29), these foci being bona fide surrogate markers for DSBs (30, 31). Interestingly, MNNG induced equivalent levels of DSBs in both lines; however, PTEN-deficient astrocytes exhibited higher levels of DSBs at 24 h post-treatment indicating a deficiency in repair of these breaks (Fig. 3a).
Figure 3. PTEN loss compromises homologous recombination repair.

(A) Induction and repair of DSBs in astrocytes treated with a 1 hour pulse of 5μM MNNG. Cells were co-immunostained for γH2AX (red) and 53BP1 (green) foci at various times post-MNNG treatment. Representative pictures are shown. Foci were scored at the indicated times (average of 100 nuclei) and, after subtracting background (number of foci in untreated nuclei), average foci per nucleus was plotted against time. Statistical significance was determined by two-way ANOVA with a Bonferroni post-test: *p<0.05; ***p<0.001. (B) Sensitivity of astrocytes to camptothecin (CPT) was quantified by colony formation assays. Please note increased sensitivity of PTEN-null cells to CPT. (C) To quantify sister chromatid exchanges (SCEs), metaphase spreads were prepared from astrocytes treated with MNNG or CPT as indicated. Reciprocal exchange events (see arrows, inset) were counted and plotted as average SCE/metaphase. At least 40 metaphases were counted per treatment. Statistical significance was determined by a two-tailed t test; ***p<0.0001. (D) Metaphase spreads from astrocytes treated with 10μM MNNG were analyzed for chromatid breaks and asymmetric exchanges (1. triradials, 2. quadriradials, and 3. complex exchanges). At least 40 metaphases were counted per treatment and average aberrations per metaphase were plotted. Representative pictures of aberrations are shown. Statistical significance was determined by a two-tailed t test; **p=.0027, *** p=.0004. Error bars represent standard error of the mean for all plots.
DSBs are repaired by NHEJ or HR in mammalian cells. While NHEJ is operative in all phases of the cell cycle, HR is limited to S/G2 and is particularly important for resolving replication-associated breaks (32). MNNG-induced breaks are presumed to occur in the S/G2 phases as DNA replication is required for mispairing, and this is borne out by our observations indicating that MNNG-induced H2AX phosphorylation occurs only in S/G2 cells (Supplement; Fig. S3). Therefore, it is likely that these breaks may be resolved by HR rather than by NHEJ. Indeed, we observed no further sensitization upon treating these cells with NU7026, a potent inhibitor of the major NHEJ repair enzyme, DNA-PKcs (33) (Supplement; Fig. S4), thereby implicating HR in repair. In support of this idea, a recent report demonstrated that MNNG induces DSBs and that cells defective in HR (XRCC2 and Brca2 mutants), but not cells defective in NHEJ (Ku80 and DNA-PKcs mutants), were sensitive to MNNG, similar to our PTEN-null cells (34).
Cells deficient in various HR components show a decrease in the number of sister chromatid exchanges (SCEs) after treatment with DNA damaging agents (35), especially agents that induce replication-associated DSBs such as camptothecin (CPT) (36). Also, HR-deficient cells are sensitive to CPT (36) and we found that PTEN-deficient astrocytes were more sensitive to this drug compared to their PTEN-proficient counterparts (Fig. 3b). We quantified the number of SCEs in PTEN+/+ and PTEN-/- astrocytes after treatment with CPT or MNNG to determine relative HR proficiencies of these lines. A statistically significant reduction in SCE events was observed in PTEN-/- astrocytes relative to PTEN+/+ astrocytes indicating a defect in HR (Fig. 3c). Consequently, PTEN-null cells surviving MNNG treatment exhibited greater numbers of chromosome breaks and radial chromosomes (Fig. 3d), similar to that seen in HR-deficient cells, particularly those deficient in Brca1 or Brca2 (37). These aberrations are indicative of a diminished capacity to repair MNNG-induced DSBs by error-free HR and subsequent repair of these lesions by error-prone pathways such as NHEJ.
Interestingly, Shen et al. recently demonstrated that PTEN is important for maintaining basal levels of transcription of the Rad51 gene in mouse embryonic fibroblasts (12), providing a potential mechanism to explain the reduced HR capability of PTEN-null astrocytes. However, no significant changes in Rad51 protein or mRNA levels in mouse astrocytes upon PTEN loss were noted (Fig. 4 a and b). While Rad51 forms a presynaptic nucleofilament that is critical for HR, ancillary proteins such as BRCA1, BRCA2, Rad52, and the Rad51 paralogs (Rad51B, Rad51C, Rad51D, XRCC2, and XRCC3) facilitate multiple steps during the repair process (38). Because there are numerous recent reports of PTEN acting as a transcriptional regulator (13), we screened several of these “recombination mediators” for expression changes upon PTEN loss by qRT-PCR and observed decreases in the transcript levels of Rad51B, C, and D (Fig. 4b). As these proteins are known to exist in complexes facilitating Rad51 nucleofilament formation (38), it is plausible that reduced levels of these proteins could result in attenuated HR upon PTEN loss.
Figure 4. PTEN-null astrocytes express lower levels of Rad51 paralogs and are sensitive to PARP inhibitors.
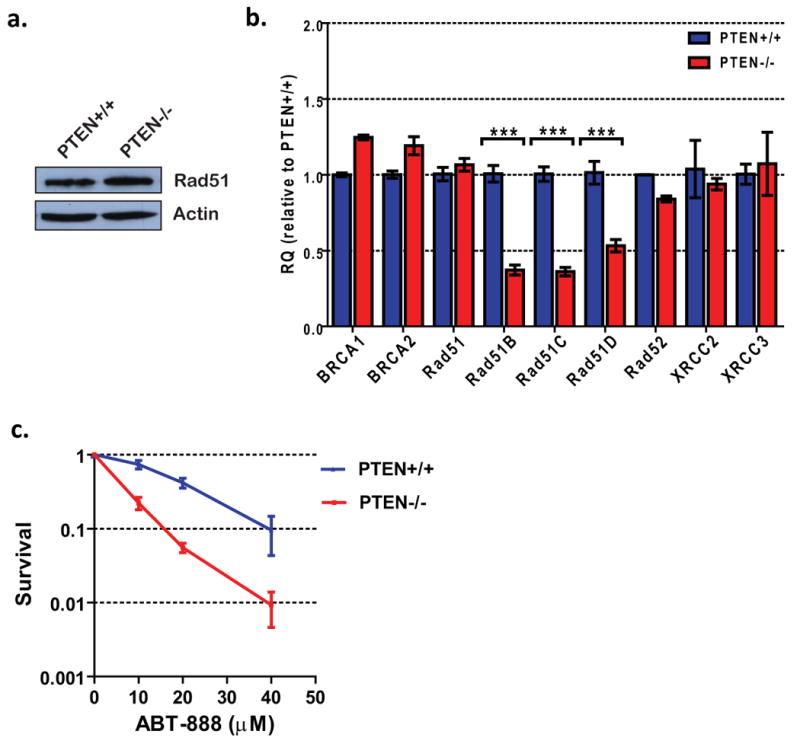
(A) Rad51 levels in PTEN+/+ and PTEN-/- astrocytes was analyzed by Western blotting with α-Rad51 antibody. (B) Transcript levels of critical HR genes were analyzed by qRT-PCR. Values were normalized to GAPDH levels and expressed as fold change relative to PTEN+/+ astrocytes. Statistical significance was determined by two-way ANOVA with a Bonferroni post-test; ** p<0.01, ***p<0.001. (C) Sensitivity of astrocytes to the PARP inhibitor ABT-888 was quantified by colony formation assays. Please note increased sensitivity of PTEN-null cells to ABT-888. Error bars represent standard error of the mean for all plots.
A very important prediction from the observed deficiency in HR is that PTEN-null astrocytes should be sensitive to PARP inhibitors that induce replication-associated DSBs. This phenomenon of “synthetic lethality” was originally identified in the context of BRCA1 and BRCA2 mutations in breast cancer (14, 15), and PARP inhibitors are now in clinical trials for treating HR-deficient breast and ovarian cancers (16). We found that PTEN-null cells were significantly more sensitive to the PARP inhibitor, ABT-888 (39), compared to PTEN-proficient cells (Fig. 4c). The sensitivity to ABT-888 is consistent with a HR-deficiency in PTEN-null cells, and suggests that it might be logical to treat PTEN-deficient GBMs with PARP inhibitors in the future.
The isogenic murine astrocytes used in this study are ideal for analyzing the effect of a single genetic change (PTEN loss) on MNNG sensitivity. However, in the context of human GBMs, the effect of a single genetic lesion could be modulated by innumerable background genetic changes (5-7). To explore the relevance of our findings in human GBMs, we compared two commonly used glioma lines (U87MG and U251MG) with a normal human astrocyte line (NHA) that had been immortalized by expression of human telomerase catalytic component (hTERT) and human papillomavirus 16 E6/E7 proteins (40). Both glioma lines are PTEN-null (41) and were more sensitive to MNNG compared to the NHA line, which has an intact PTEN gene (Supplement; Fig. S5). These results tentatively suggest that PTEN-deficient glioblastoma cells may be more sensitive to DNA alkylating agents compared to PTEN-proficient normal human astrocytes and this could confer a selective advantage to DNA alkylating agents for the treatment of PTEN-null GBMs. Importantly, siRNA-mediated depletion of PTEN rendered the NHA line sensitive to MNNG as quantified by the colony formation assay (Supplement; Fig. S6). This was possibly due to attenuated HR, as we observed a reduced induction of SCEs upon PTEN depletion. Interestingly, SCE induction in PTEN-null U87 cells were also reduced compared to the PTEN-proficient NHA line. More importantly, PTEN depletion could also sensitize transformed, gliomagenic NHAs (expressing E6, E7, hTERT, H-Ras, and myristylated Akt-1) (42), indicating that PTEN loss might result in sensitivity to DNA alkylating agents in the context of human gliomas. In sum, these results confirm that, as observed in murine astrocytes, PTEN loss plays an important role in modulating MNNG sensitivity of normal human astrocytes and gliomagenic derivatives.
Discussion
The data presented in this paper provide the basis for exciting new therapeutic options for GBMs. Thus far, the only meaningful improvement in GBM therapy came from the addition of temozolomide to radiation, with MGMT promoter methylation associated with a better outcome (3, 43). However, MGMT promoter methylation was also associated with an improvement in survival with radiation alone (4), indicating that this may be a general prognostic marker rather than a specific predictive marker for temozolomide treatment. We demonstrate here that PTEN-deficient astrocytes are impaired in HR and, therefore, sensitive to replication-associated DSBs generated by DNA alkylating agents or by PARP inhibitors. This has novel therapeutic implications: GBM patients with PTEN loss (about 36%) may: 1) specially benefit from temozolomide treatment, and 2) also benefit from the addition of PARP inhibitors to therapeutic regimens. At the time when this manuscript was in preparation, a report from the Ashworth group demonstrated that PTEN-null cells and tumors are indeed sensitive to PARP inhibitors (44) thereby bolstering the conclusions drawn from our study. Interestingly, a recent report indicates that acquired resistance of cancer cells to combinatorial treatment with temozolomide and PARP inhibitors is linked to upregulation of HR (45). These results complement our data showing that downregulation of HR due to PTEN loss results in sensitivity to DNA alkylating agents or PARP inhibitors. Both temozolomide and PARP inhibitors are in clinical trials to treat GBMs and have been used in combination to treat other cancers (46). In light of our results and other recent reports, we posit that: 1) PARP inhibitors may have a broader applicability outside of Brca1/2-null breast and ovarian cancers, and could be used to target other tumors with HR deficiencies due to specific mutations such as PTEN loss, and 2) GBM patients may perhaps be stratified for therapy with temozolomide or PARP inhibitors not just based upon their MGMT status but also upon their PTEN status.
While our results with genetically defined models suggest that PTEN-null tumors may be more responsive to temozolomide treatment, it is important to point out the caveat that glioblastomas are genetically very heterogeneous (5, 6, 10). Therefore, it is plausible that treatment with DNA alkylating agents could actually select for resistant clones, ultimately precipitating tumor resurgence and therapeutic failure. This possibility is tentatively indicated by in vitro studies where we find that U87 or U251 clones surviving MNNG treatment are more MNNG resistant (to varying degrees) compared to the parental lines (Supplement; Fig. S7). This possibility is also borne out by the Cancer Genome Atlas Research Network study, which suggests that treatment of MGMT-deficient glioblastomas with alkylating agents may result in the selection of mutations in mismatch repair genes which would lead to therapy resistance (5). It would be interesting to determine if the HR defect due to PTEN loss may actually increase the likelihood of development of resistant clones due to increased genomic instability.
While the results described in this paper come from proof-of-principle experiments carried out with cells growing in monolayer, it will be important to extend these results to organotypic 3D models to verify whether our conclusions are valid in a more “tumor-like” setting. In preliminary studies, we do find that both monolayer cultures as well as “spheroids” of U87 cells (generated by the “hanging-drop” method (47)) are similarly sensitive to MNNG (Supplement; Fig. S8). Eventually, extensive research with pre-clinical mouse models, especially orthotopic glioblastoma models, will be required to firmly establish the nascent, but highly novel, concepts formulated by this study.
Supplementary Material
Acknowledgments
We are grateful to Prof. David A. Boothman for critically reading the manuscript. We thank Dr. Steve Cho for help with designing qRT-PCR primers. This work was supported by grants from NASA (NNA05CS97G and NNX10AE08G to SB), the Cancer Prevention and Research Institute of Texas (RP100644 to SB), the Goldhirsh Foundation (RMB), and NCI (T32CA124334 to CVC).
References
- 1.Friedman HS, Kerby T, Calvert H. Temozolomide and treatment of malignant glioma. Clin Cancer Res. 2000;6:2585–97. [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 4.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. The New England journal of medicine. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 5.McLendon R, Friedman A, Bigner D, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008 doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mukherjee B, McEllin B, Camacho CV, et al. EGFRvIII and DNA double-strand break repair: a molecular mechanism for radioresistance in glioblastoma. Cancer research. 2009;69:4252–9. doi: 10.1158/0008-5472.CAN-08-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stojic L, Brun R, Jiricny J. Mismatch repair and DNA damage signalling. DNA Repair (Amst) 2004;3:1091–101. doi: 10.1016/j.dnarep.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 10.Furnari FB, Fenton T, Bachoo RM, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 11.Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–14. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 12.Shen WH, Balajee AS, Wang J, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–70. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 13.Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27:5443–53. doi: 10.1038/onc.2008.241. [DOI] [PubMed] [Google Scholar]
- 14.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 15.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 16.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. The New England journal of medicine. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 17.Bachoo RM, Maher EA, Ligon KL, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1:269–77. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 18.Lesche R, Groszer M, Gao J, et al. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32:148–9. doi: 10.1002/gene.10036. [DOI] [PubMed] [Google Scholar]
- 19.Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 20.Mukherjee B, Camacho CV, Tomimatsu N, Miller J, Burma S. Modulation of the DNA-damage response to HZE particles by shielding. DNA Repair (Amst) 2008;7:1717–30. doi: 10.1016/j.dnarep.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 21.Mukherjee B, McEllin B, Camacho CV, et al. EGFRvIII and DNA Double-Strand Break Repair: A Molecular Mechanism for Radioresistance in Glioblastoma. Cancer Res. 2009 doi: 10.1158/0008-5472.CAN-08-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kao GD, Jiang Z, Fernandes AM, Gupta AK, Maity A. Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J Biol Chem. 2007;282:21206–12. doi: 10.1074/jbc.M703042200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wick W, Furnari FB, Naumann U, Cavenee WK, Weller M. PTEN gene transfer in human malignant glioma: sensitization to irradiation and CD95L-induced apoptosis. Oncogene. 1999;18:3936–43. doi: 10.1038/sj.onc.1202774. [DOI] [PubMed] [Google Scholar]
- 24.Kaina B, Christmann M, Naumann S, Roos WP. MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair (Amst) 2007;6:1079–99. doi: 10.1016/j.dnarep.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 25.Gerson SL. MGMT: its role in cancer aetiology and cancer therapeutics. Nature reviews. 2004;4:296–307. doi: 10.1038/nrc1319. [DOI] [PubMed] [Google Scholar]
- 26.Fritz G, Tano K, Mitra S, Kaina B. Inducibility of the DNA repair gene encoding O6-methylguanine-DNA methyltransferase in mammalian cells by DNA-damaging treatments. Mol Cell Biol. 1991;11:4660–8. doi: 10.1128/mcb.11.9.4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7:335–46. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 28.Wang JY, Edelmann W. Mismatch repair proteins as sensors of alkylation DNA damage. Cancer Cell. 2006;9:417–8. doi: 10.1016/j.ccr.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 29.Mukherjee B, Camacho CV, Tomimatsu N, Miller J, Burma S. Modulation of the DNA-damage response to HZE particles by shielding. DNA Repair (Amst) 2008 doi: 10.1016/j.dnarep.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 30.Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair (Amst) 2004;3:959–67. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 31.Mochan TA, Venere M, DiTullio RA, Jr, Halazonetis TD. 53BP1, an activator of ATM in response to DNA damage. DNA Repair (Amst) 2004;3:945–52. doi: 10.1016/j.dnarep.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 32.Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008;9:297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]
- 33.Veuger SJ, Curtin NJ, Richardson CJ, Smith GC, Durkacz BW. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer research. 2003;63:6008–15. [PubMed] [Google Scholar]
- 34.Roos WP, Nikolova T, Quiros S, et al. Brca2/Xrcc2 dependent HR, but not NHEJ, is required for protection against O(6)-methylguanine triggered apoptosis, DSBs and chromosomal aberrations by a process leading to SCEs. DNA Repair (Amst) 2009;8:72–86. doi: 10.1016/j.dnarep.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 35.Sonoda E, Sasaki MS, Morrison C, Yamaguchi-Iwai Y, Takata M, Takeda S. Sister chromatid exchanges are mediated by homologous recombination in vertebrate cells. Mol Cell Biol. 1999;19:5166–9. doi: 10.1128/mcb.19.7.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol. 2001;307:1235–45. doi: 10.1006/jmbi.2001.4564. [DOI] [PubMed] [Google Scholar]
- 37.Venkitaraman AR. Linking the cellular functions of BRCA genes to cancer pathogenesis and treatment. Annu Rev Pathol. 2009;4:461–87. doi: 10.1146/annurev.pathol.3.121806.151422. [DOI] [PubMed] [Google Scholar]
- 38.San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–57. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 39.Donawho CK, Luo Y, Penning TD, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13:2728–37. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- 40.Sonoda Y, Ozawa T, Hirose Y, et al. Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer research. 2001;61:4956–60. [PubMed] [Google Scholar]
- 41.Ishii N, Maier D, Merlo A, et al. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999;9:469–79. doi: 10.1111/j.1750-3639.1999.tb00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sonoda Y, Ozawa T, Aldape KD, Deen DF, Berger MS, Pieper RO. Akt pathway activation converts anaplastic astrocytoma to glioblastoma multiforme in a human astrocyte model of glioma. Cancer research. 2001;61:6674–8. [PubMed] [Google Scholar]
- 43.Stupp R, Dietrich PY, Ostermann Kraljevic S, et al. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J Clin Oncol. 2002;20:1375–82. doi: 10.1200/JCO.2002.20.5.1375. [DOI] [PubMed] [Google Scholar]
- 44.Mendes-Pereira AM, Martin SA, Brough R, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1:315–22. doi: 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu X, Han EK, Anderson M, et al. Acquired Resistance to Combination Treatment with Temozolomide and ABT-888 Is Mediated by Both Base Excision Repair and Homologous Recombination DNA Repair Pathways. Mol Cancer Res. 2009 doi: 10.1158/1541-7786.MCR-09-0299. [DOI] [PubMed] [Google Scholar]
- 46.Plummer ER. Inhibition of poly(ADP-ribose) polymerase in cancer. Curr Opin Pharmacol. 2006;6:364–8. doi: 10.1016/j.coph.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 47.Del Duca D, Werbowetski T, Del Maestro RF. Spheroid preparation from hanging drops: characterization of a model of brain tumor invasion. J Neurooncol. 2004;67:295–303. doi: 10.1023/b:neon.0000024220.07063.70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.