

Abstract
Membrane type-I matrix metalloproteinase (MT1-MMP) plays an essential role in protease-mediated extracellular matrix (ECM)-degradation, but it also functions as a sheddase releasing non-ECM substrates such as Receptor activator of NF-κB ligand (RANKL), an osteoclastogenic factor typically confined to the surface of osteoblasts. We previously found high expression of MT1-MMP in skeletal metastasis of prostate cancer (PC) patients, in a pattern similar to RANKL expression. We also showed that overexpression of MT1-MMP in PC cells increases tumor growth and osteolysis in an intratibial mouse model of bone metastasis, and that soluble factor/s shed by tumor-derived MT1-MMP enhance osteoclast differentiation in a RANKL-dependent manner. Recent evidence indicates that the cognate receptor for RANKL, RANK, is expressed in PC cells, suggesting the presence of an autocrine pathway. In this study, we show that MT1-MMP-expressing LNCaP PC cells display enhanced migration. Moreover, conditioned medium from LNCaP cells expressing both RANKL and MT1-MMP stimulates the migration of MT1-MMP-deficient C42b PC cells. This enhanced chemotaxis can be abrogated by osteoprotegerin (soluble decoy receptor of RANKL), MIK-G2 (a selective inhibitor for MT1-MMP), and PP2 (a Src inhibitor). These findings indicate that tumor-derived MT1-MMP enhances tumor cell migration via initiation of an autocrine loop requiring ectodomain shedding of membrane-bound RANKL in PC cells, and that Src is a key downstream mediator of RANKL-induced migration of PC cells.
Keywords: Receptor Activator of NF-κB Ligand, Membrane Type-I Matrix Metalloproteinase, Prostate Cancer, Src Family Kinase, Migration
Introduction
Disseminated tumor cells must proteolytically degrade and then invade the surrounding extracellular matrix (ECM) in order to expand into a large metastatic deposit (1, 2). In bone metastasis, tumor cells must invade through type-I collagen, the main organic component of interstitial ECM. Matrix metalloproteinases (MMPs) are well known to play a significant role in ECM remodeling (3–5). Membrane type-I matrix metalloproteinase (MT1-MMP), in particular, plays a critical role in type-I collagen turnover, as demonstrated by skeletal defects seen in MT1-MMP-deficient mice (6). Furthermore, MT1-MMP has been shown to degrade type-I collagen directly (7), and it is essential for tumor cell invasion through three-dimensional type-I collagen matrices (8). Previously, we showed a heterogeneous pattern of MT1-MMP expression in carcinoma glands within the prostate of patients undergoing radical prostatectomy (9). In contrast, we found a homogeneous and intense expression by cancer cells within human prostate cancer (PC) bone metastases from a “rapid autopsy” program (10). Moreover, we found that PC cells ectopically expressing MT1-MMP display increased tumor growth and tumor-induced osteolysis in an intratibial mouse model of bone tumor growth (10).
Typically expressed on the surface of cells of the osteoblast lineage, the receptor activator of NF-κB ligand (RANKL) is a potent inducer of osteoclast maturation (11). RANKL is a homotrimeric transmembrane complex which can be shed from the cell surface via MMP-mediated proteolytic cleavage (12–14). Among the members of the MMP family, MT1-MMP is the most efficient sheddase of RANKL (15). In fact, knockdown of endogenous MT1-MMP proved to be sufficient to diminish levels of soluble RANKL in vitro and in vivo (16). Interestingly, recent reports show that RANKL is also expressed by PC cells in human bone metastasis (17–21) and in several human PC cell lines (22, 23). Keller and colleagues demonstrated that PC cells release soluble RANKL, promoting in vitro osteoclastogenesis independent of osteoblasts or bone stromal cells (22). These data suggest a role for tumor-derived RANKL in mediating some of the bone responses seen in metastatic PC. We subsequently found that conditioned media (CM) derived from PC cells expressing both RANKL and MT1-MMP enhanced in vitro differentiation of osteoclasts, an effect blocked by either osteoprotegerin (OPG) or a selective MT1-MMP inhibitor (10). These data highlight a mechanism for metastatic tumor expansion wherein tumor-associated MT1-MMP acts as a mediator of autocrine/paracrine signaling via solubilization of RANKL. Of course, this does not rule out direct degradation of the ECM by MT1-MMP.
Consistent with the “osteomimicry” theory (24–26), recent evidence indicates that RANK, the cognate receptor for RANKL, is also found on the surface of PC cells (27, 28). Activation of RANK in prostate tumor cells is associated with increased cell migration, invasion through a collagen matrix, stimulation of mitogen activated kinases (MAPKs), and augmented expression of osteoclast-related genes (27–29). Based on these considerations, we hypothesized that MT1-MMP may play a role in RANK activation and subsequent migration in tumor cells. Herein we show that tumor-associated MT1-MMP promotes tumor cell migration via a novel indirect mechanism involving solubilization of RANKL by tumor-associated MT1-MMP and subsequent autocrine activation of RANK. RANK-mediated migration proceeds via rapid activation of a Src-dependent pathway. This MT1-MMP/RANKL/RANK/Src axis may have important implications for the treatment of prostate cancer bone metastasis.
Materials and Methods
Cell culture
LNCaP and PC3 cells, obtained from American Type Culture Collection (ATCC), were maintained in RPMI 1640. C42b cells, an LNCaP variant isolated from castrated mice with preferential growth in bone (30) (courtesy of Dr. Leland Chung, Emory University, Atlanta, GA), were maintained in T-medium. DU145 cells were obtained from ATCC and maintained in DMEM. All culture media were purchased from Invitrogen (Carlsbad, CA), and supplemented with 10% fetal bovine serum (FBS). Pooled populations of LNCaP cells with ectopic expression of wild-type MT1-MMP (LNCaP-MT1) or control (LNCaP-Neo) were established and maintained as previously described (10). Cell lines obtained from ATCC are routinely authenticated through cell morphology monitoring, growth curve analysis, species verification by isoenzymology and karyotyping, identity verification using short tandem repeat profiling analysis, and contamination checks.
Immunoblot analysis
LNCaP, C42b, PC3, DU145, or LNCaP transfectants were cultured to 80–90% confluency. Whole cell lysates were resolved on 10% SDS-polyacrylamide gels or 4–12% Bis-Tris gradient gels (Invitrogen) under reducing conditions, and immunoblotted with antibodies targeted at the catalytic domain of human MT1-MMP (LEM-2/15 monoclonal antibody kindly provided by Dr. A. Arroyo, Hospital de la Princesa, Madrid, Spain), RANKL (R&D Systems, Minneapolis, MN), RANK (Cell Signaling Technology, Danvers, MA), or OPG (R&D Systems). Immunoreactive proteins were detected with horseradish peroxidase-conjugated anti-mouse IgG or anti-rabbit IgG antibodies, and enhanced chemiluminescence (Pierce, Rockford, IL). Blots were stripped and re-probed with an antibody to β-actin (Sigma-Aldrich, St. Louis, MO), used as loading control. Western blots were repeated under independent conditions at least twice; representative blots are shown.
Transwell migration assay
Serum-starved C42b cells (5×104) were allowed to migrate for 16 h through non-coated 8 μm pore-sized Transwell inserts (BD Falcon, San Jose, CA) toward human recombinant RANKL (rRANKL) (Leinco Technologies, Inc., St. Louis, MO) or protein-normalized 48-h CM obtained from LNCaP-Neo or LNCaP-MT1 cells (40% in culture medium at a final FBS concentration of 0.5 %). Cells that migrated through the filters were counted in 5 randomly picked 200x fields, or in the entire filter if the number of cells was low. Data were expressed as mean ± SE of average number of cells obtained in each filter (n ≥ 3), with experiments repeated under independent conditions at least twice. For migration studies under Src inhibition, cells were treated 45 min before the assay and during the assay with 5 μM 4-amino-5-(4-chlorophenyl)-7-(t-butyl)prazolo[3,4-d]pyrimidine (PP2). To inhibit RANKL-induced migration, RANKL or CM were incubated together with OPG for 45 min at 37°C prior to insertion of upper chamber containing cells into the 24-well plate. In experiments aimed at inhibiting the activity of tumor-associated MT1-MMP, LNCaP-Neo and LNCaPMT1 cells were incubated for 48 h with 10 μmol/L 4-[4-(methanesulfonamido) phenoxy] phenylsulfonyl methylthiirane (MIK-G2) (31), an MT1-MMP inhibitor, prior to collection of CM.
Experiments using LNCaP-transfectants alone were performed as described above, except that no chemoattractant was used.
In vitro cell viability assessment
C42b cells were incubated with different concentrations of recombinant RANKL, OPG, PP2, or MIK-G2 for 16 h, to parallel the conditions used in the migration assays. Cell viability was measured using the WST-1 assay following manufacturer’s instructions (Roche Applied Science, Indianapolis, IN), and the Trypan blue exclusion test.
Signaling studies
C42b cells at 90% confluency were serum-starved for 18h and then treated with 200ng/mL rRANKL in serum-free T-medium for different lengths of time or increasing concentrations of rRANKL. Whole-cell lysates were separated by SDS-polyacrylamide gels as described above. The following primary antibodies from Cell Signaling Technology were used: anti-phospho-Src (Y419), total Src (L4A1), polyclonal phospho-Akt (Ser473); polyclonal total Akt; monoclonal phospho-p44/42 MAPK (Thr202/Tyr204) (197G2); polyclonal total p44/p42 MAP Kinase; polyclonal phospho-SAPK/JNK (Thr183/Tyr185); polyclonal total SAPK/JNK; polyclonal phospho-p38 MAPK (Thr180/Tyr182); and polyclonal total p38 MAPK. The antigen-antibody complexes were detected as described above. For studies in which Src was inhibited, cells were pre-treated for 45 min in the presence of different concentrations of PP2, and then treated with 200ng/mL of rRANKL in the presence of PP2. Studies of phosphorylated and total Src were also performed in PC3 or LNCaP-transfectant cells as described above. Western blots were repeated under independent conditions at least twice; representative blots are shown.
Confocal microscopy
C42b cells grown in eight-well chamber Labtek II CC2™ slides (NUNC, Rochester, NY) were serum-starved for ~18 h, and then treated with 200ng/mL of rRANKL for 5 min or with vehicle control. Cells were fixed with 4% paraformaldehyde, permeabilized, and then incubated with fluorescent-labeled phalloidin conjugate (Millipore, Billerica, MA). After mounting with anti-fade Vectashield Mounting Media with DAPI (nuclear stain; Vector Laboratories Inc., Burlingame, CA), the cells were examined on a Zeiss LSM510 confocal microscopy system (Carl Zeiss, Inc., Thornwood, NY) at 63x magnification.
Statistical analysis
Differences among more than two groups were analyzed by one-way analysis of variance with Tukey-Kramer post-testing. Data comparing differences between two groups were statistically analyzed using unpaired Student’s t-test. Differences were considered significant when P < 0.05.
Results
LNCaP cells with ectopic expression of MT1-MMP display increased motility
To examine a role for tumor-derived MT1-MMP independent of ECM proteolytic degradation, we examined the effect of ectopic expression of MT1-MMP in PC cells in the absence of a restrictive ECM. LNCaP-transfectants with differential expression of ectopic MT1-MMP previously established by us (10) were assayed for cell motility in the absence of any chemoattractant. Notably, the number of LNCaP-MT1 cells traversing the pored filters was three times as many as that of control LNCaP-Neo cells (Fig. 1) despite comparable doubling times (10) and cell survival (data not shown), suggesting an intrinsic motile response.
Figure 1.

Increased cell migration in LNCaP cells with ectopic expression of MT1-MMP. A, Immunoblot analysis of LNCaP-MT1 and LNCaP-Neo cell lysates for MT1-MMP. B, Migration of LNCaP-Neo and LNCaP-MT1 transfectants in the absence of any chemoattractant. Columns, mean number of cells traversing the filters from triplicate determinations; bars, SE. *, P<0.01.
Conditioned medium from LNCaP cells transfected with wild-type MT1-MMP stimulates migration of C42b cells in an MT1-MMP - and RANKL-dependent manner
Given the ability of tumor-associated MT1-MMP to shed RANKL from the cell surface (10), we evaluated the possibility that the motility of MT1-MMP-expressing LNCaP cells was due to autocrine activation of RANK by tumor-derived soluble RANKL. To test this hypothesis, C42b cells were chosen as ‘reporter’ cells because, like LNCaP cells, they express RANK, but they do not express MT1-MMP or OPG (Fig. 2A). We found that CM derived from LNCaP-MT1 cells significantly enhanced C42b cell migration compared to control LNCaP-Neo-derived CM (Fig. 2B). Moreover, this migration was significantly reduced when the media was conditioned in the presence of MIK-G2, a selective MT1-MMP inhibitor, suggesting a role for MT1-MMP activity in the generation of the soluble factor responsible for the migratory response.
Figure 2.
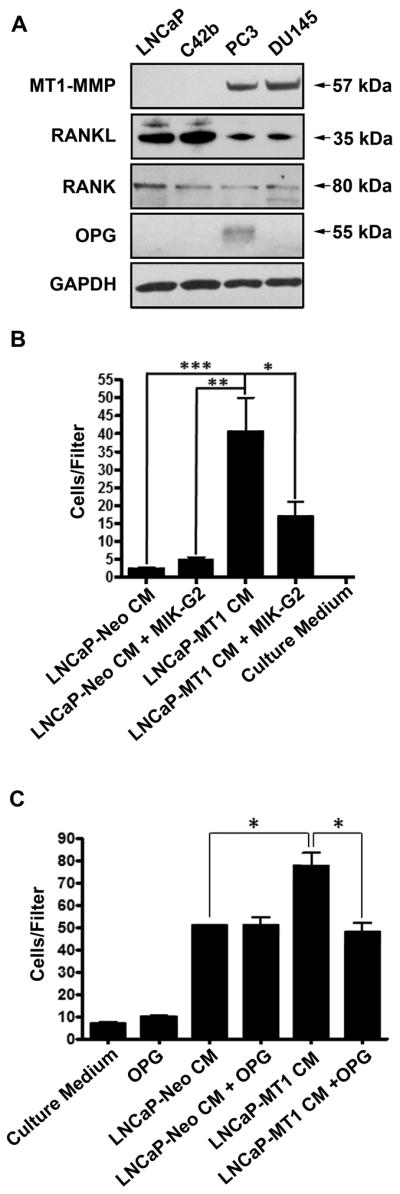
C42b cells express RANK, and show enhanced migration towards CM from LNCaP cells transfected with wild-type MT1-MMP. A, Immunoblot analysis for MT1-MMP, RANKL, RANK, and OPG in human PC cell lines. B, Treatment of LNCaP-MT1 cells for 48 h with the MT1-MMP inhibitor MIK-G2 (10 μM) reduces the chemotactic capacity of their CM on C42b cells. C, Migration of C42b cells towards CM from LNCaP-MT1 cells is inhibited when the CM is pre-incubated with 500ng/mL of OPG. Columns, mean (n ≥ 4); bars, SE. *, P<0.05; **, P<0.01; ***, P<0.001; Tukey-Kramer post-hoc applied to significant effect of group ANOVA. Conditioned Media were normalized by protein concentration, as measured using BCA method.
Given that the LNCaP-transfectants express RANKL (10), similar to the other human PC cell lines tested by us (Fig. 2A), we asked whether the enhanced migratory response of C42b cells was caused by soluble RANKL present in the CM of LNCaP-MT1 cells. Therefore, cell migration assays were conducted with CM in the presence or absence of OPG, a natural decoy receptor that competes with RANK for RANKL. As can be seen in Figure 2C, the presence of OPG significantly reduced the ability of the LNCaP-MT1-derived CM to stimulate the migration of C42b cells, reducing the total number of migrated cells to levels observed with media derived from LNCaP-Neo cells. Conversely, pre-incubation of LNCaP-Neo-derived CM with OPG did not alter the baseline cell migration rates of C42b cells. These results suggest that increased migration of the C42b cells was due to the presence of soluble RANKL present in the CM of LNCaP-MT1 cells.
We next sought to determine the chemotactic effect of rRANKL on C42b cells. We observed a dose-dependent increase in C42b cell migration (Fig. 3A) in response to soluble rRANKL, which was abrogated by pre-incubation with increasing concentrations of OPG in a dose-dependent manner (Fig. 3B). This result is consistent with the ability of CM derived from LNCaP-MT1 cells to stimulate cell migration in an OPG-sensitive manner, and it directly implicates RANK activation by soluble RANKL (Fig. 2C). Furthermore, we confirmed that RANKL has no mitogenic effect on C42b cells (data not shown). Therefore, the increased number of cells traversing the filter was due to migration.
Figure 3.

Dose-dependent migration of C42b cells in response to rRANKL. A, Chemotaxis of C42b cells towards increasing concentrations of rRANKL. B, Chemotaxis of C42b cells towards 100 ng/mL of rRANKL pre-incubated with increasing concentrations of OPG. Columns, mean (n ≥ 4); bars, SE. *, P<0.05; **, P<0.01; Tukey-Kramer post-hoc applied to significant effect of group ANOVA.
RANKL activates Src in a dose-dependent manner and initiates cytoskeletal rearrangement
Next, we examined downstream signaling as a consequence of RANK activation in C42b cells in response to soluble rRANKL. RANK lacks intrinsic kinase activity, requiring the recruitment of TNF Receptor Associated Factors (TRAFs) to the cell membrane for downstream signaling (32). Therefore, we looked downstream of TRAF binding to find a sensitive endpoint for RANK activation. We found that treatment of C42b cells with 200ng/mL of rRANKL induced activation of multiple signaling pathways (Fig. 4A). Of all the intracellular signaling molecules we studied, Src and Erk showed the strongest and most rapid activation. Src phosphorylation of tyrosine 419 was observed within 15 min of exposure to rRANKL (Fig. 4A), followed by a reduced activation at 30 min, while Erk phosphorylation was noticeable between 15 and 30 min after treatment with rRANKL. Although Src has been reported to be an upstream activator of Akt in osteoclasts (33), we did not see any effect on phosphorylated Akt levels in rRANKL-treated C42b cells. As previously reported (28), we found that RANK activation by rRANKL also promoted activation of MAPKs (Erk1/2, p38 MAPK, and JNK/SAPK).
Figure 4.

RANKL activates Src in a dose-dependent manner along with the MAPK pathway, and initiates cytoskeletal rearrangement. A, C42b cells were serum-starved overnight and treated with rRANKL (200ng/mL) for the period indicated. Activation of Src, Erk, p38, JNK, and AKT was determined by Western blotting analysis using phospho-specific antibodies. B, Immunoblot analyses for Src and phospho-Src in lysates obtained from C42b cells treated for 10 min with increasing concentrations of rRANKL (upper blot), and lysates from C42b cells treated with 200 ng/mL of rRANKL at the indicated time points (bottom blot). C, Src and p-Src detection by Western blotting analysis in PC3 cells treated with 200ng/mL of rRANKL for 10 min. D, Treatment of serum-starved C42b cells with 200ng/mL of rRANKL for 5 min results in cystoskeletal rearrangement, as revealed by phalloidin staining of F actin fibers (shown in red). Nuclei are stained with DAPI (shown in blue). Merge of F actin fibers (red) and nuclei (blue) images are shown. Original magnification 63x.
Based on a potential “osteomimetic” parallel between Src activation in C42b prostate cancer cells by rRANKL and osteoclast dependence on Src activity, we focused on Src activation in PC cells upon activation by RANKL. We observed that rRANKL had a dose-dependent effect on Src phosphorylation (Fig. 4B, upper panel), attesting to the specificity of rRANKL on regulation of Src. rRANKL consistently increased phosphorylation of tyrosine 419, the autophosphorylation site located within the Src catalytic domain, within 15 min of exposure to exogenous rRANKL, actually peaking at 5 min (Fig. 4B, lower panel). This rapid activation of Src is consistent with previous studies showing that Src is one of the immediate signaling molecules activated by the RANKL/RANK signaling axis (34). PC3 cells, which also express RANK (Fig. 2A), also responded to RANKL treatment by activating Src, showing that this effect is not specific to C42b cells (Fig. 4C). Based on our observation that rRANKL increased C42b cell migration, and the reported role of Src as a mediator of cellular migration (27, 29), we examined cytoskeletal rearrangements in C42b cells. TRITC-phalloidin staining of F-actin in C42b cells treated for 5 min with 200ng/mL of rRANKL revealed actin polymers beneath the plasma membrane, as compared to control (Fig. 4D).
rRANKL-induced C42b cell migration is dependent on Src activity
To establish the role of Src in rRANKL-induced C42b migration, we utilized the specific Src inhibitor PP2, which binds active Src within the ATP binding pocket inhibiting autoactivation (35). Exposure of C42b cells to increasing concentrations of PP2 inhibited the ability of rRANKL (200ng/mL for 5 min) to induce Src phosphorylation (Fig. 5A). At 1.25μM PP2, levels of active Src were decreased, and at 2.5μM PP2, phosphorylation at tyrosine 419 of Src was not detectable by immunoblotting. Concurrent examination of MAPKs revealed that PP2 did not affect RANKL-induced activation of these kinases, suggesting that the RANKL-RANK-Src pathway is independent of the RANKL-RANK-MAPK pathway. To implicate Src activation in RANKL-induced migration, we next analyzed chemotaxis of C42b cells towards rRANKL in the presence of PP2. As can be seen in Figure 5B, PP2 inhibited C42b cell migration in a dose-dependent fashion. Of note, at these concentrations, PP2 did not affect C42b cell viability (data not shown).
Figure 5.
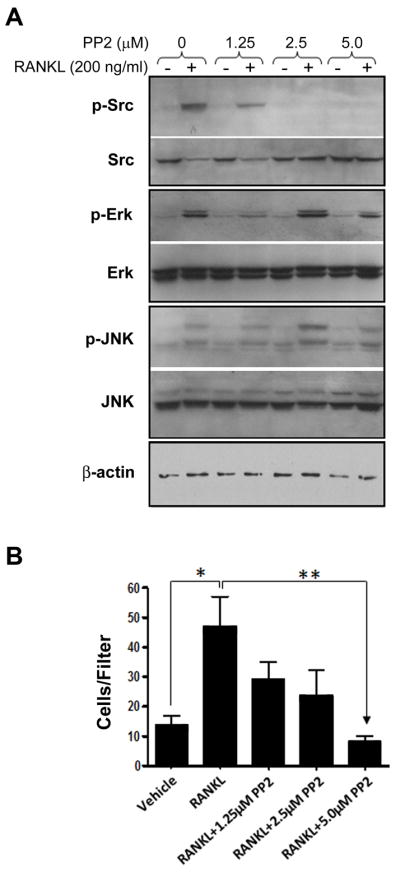
RANKL-induced C42b Cell Migration is Dependent on Src Activity. A, Immunoblot analysis for Src, Erk, JNK, and their respective phosphorylated forms in whole cell lysates from C42b cells pre-incubated with increasing concentrations of the Src inhibitor PP2 and treated for 15 min with 200ng/mL rRANKL. B, Chemotaxis of C42b cells pre-incubated with increasing concentrations of PP2 towards 200 ng/mL of rRANKL. Bars represent Mean ± SE number of cells traversing the filters. *, P<0.05; **, P<0.01; Tukey-Kramer post-hoc applied to significant effect of group ANOVA.
MT1-MMP expression by LNCaP cells stimulates cell migration in a Src-dependent manner
To this point, we showed only the effects of rRANKL on inducing Src-dependent cell migration in PC cells. It was therefore crucial to determine if CM from PC cells expressing MT1-MMP and RANKL produced these same effects. We first examined the basal levels of Src activity in our LNCaP-Neo and LNCaP-MT1 cells. By western blot, we observed an increase in phosphorylated Src in LNCaP-MT1 cells versus LNCaP-Neo (Fig. 6A), suggesting a correlation between tumor-associated MT1-MMP expression/activity and increased Src activity. We then examined the ability of LNCaP transfectant-derived CM to activate Src in C42b cells. Serum-starved C42b cells were treated for 15 min with protein-normalized CM from LNCaP-Neo and LNCaP-MT1 cells. We found that C42b treatment with LNCaP-MT1-derived CM increased phosphorylated Src levels relative to LNCaP-Neo derived CM (Fig. 6B). Moreover, the enhanced Src activation caused by LNCaP-MT1 CM was hindered by pre-treatment of C42b cells with 5 μM PP2, confirming effective Src kinase inhibition (Fig. 6B). To confirm that Src activation was due to RANKL signaling by soluble RANKL present in LNCaP-MT1-derived CM, we pre-incubated the CM with excess of OPG. As can be seen in Figure 6B, pre-incubation with OPG resulted in considerably reduced levels of phosphorylated Src in C42b treated with LNCaP-MT1-derived CM.
Figure 6.

Increase in Src phosphorylation and Src-dependent migration in LNCaP cells overexpressing MT1-MMP. A, Lysates from serum-starved LNCaP-MT1 or LNCaP-Neo cells probed for phospho-Src (Y419) and total Src. B, Activation of Src in C42b cells pre-incubated with 5 μM PP2 and then treated with normalized CM from LNCaP-Neo or LNCaP-MT1 cells, and in C42b cells treated with the same CM in the presence of OPG, as determined by Western blot. C, Chemotaxis of C42b cells pre-incubated with 5 μM PP2 towards CM from LNCaP transfectants. Columns, mean number from five replicates; bars, SE. *, P<0.05; **, P<0.01; ***, P<0.001; Tukey-Kramer post-hoc applied to significant effect of group ANOVA. D, model of autocrine prostate tumor-derived MT1-MMP/RANKL/RANK pathway mediated through Src. The details are described in the text.
We next sought to confirm that increased Src activation in C42b, as a result of treatment with LNCaP-MT1-derived CM, was responsible for the enhanced migration displayed by C42b cells, similarly to rRANKL. C42b cells were pretreated with 5 μM of the Src inhibitor PP2, and then assayed for cell migratory ability towards LNCaP-MT1-or LNCaP-Neo derived CM. The enhanced chemotactic response of C42b towards LNCaP-MT1-derived CM was significantly inhibited by PP2, whereas the migratory response to LNCaP-Neo-derived CM remained unchanged despite the presence or absence of the Src inhibitor (Fig. 6C). These data suggest that Src activity is a requirement for C42b migration in response to LNCaP-MT1-derived CM.
Discussion
Substantial evidence supports involvement of tumor-derived MMPs in several steps of the metastatic cascade, and it is now well accepted that MMPs have roles above and beyond degradation of ECM. However, the failure of broad-spectrum MMP inhibition in clinical trials has shifted the focus to specific members of the MMP family (36, 37). Functional redundancies within the MMP family have hindered these efforts; however, the unique properties of MT1-MMP in skeletal development (6) may offer an opportunity for specific MMP-targeted therapy. Indeed, MT1-MMP inhibition continues to be of interest to the pharmaceutical industry (38). The role of MT1-MMP in providing a mechanism for tumor cell movement through ECM is without a doubt an important function of this protease; however, in this study we examined an alternative role for tumor-associated MT1-MMP. Our data demonstrate the ability of tumor-derived MT1-MMP to enhance PC cell migration independent of protease-mediated ECM degradation via an MT1-MMP/RANKL/RANK autocrine pathway dependent on Src activation.
Initially, we observed increased motility of PC cells expressing MT1-MMP in an ECM-free system. We also saw RANKL-dependent enhancement of the migratory capacity of C42b cells toward CM from LNCaP cells expressing MT1-MMP. In agreement with others (27, 29), we found RANK to be expressed in all PC cells examined, and the RANK expressed was functional, since we observed a specific RANKL-mediated dose-dependent increase in C42b cell migration. The use of PC cell-derived CM in our studies (in addition to recombinant RANKL) opened the possibility of multiple soluble factors having redundant effects. Therefore, we systematically and independently inhibited MT1-MMP and RANKL to support the existence of a specific tumor-associated MT1-MMP/RANKL/RANK autocrine signaling axis. Our data supplement prior findings (27, 29) in which RANKL induced activation of MAPKs in PC cells.
Based on the “osteomimetic” properties displayed by tumor cell types with a preference for bone metastasis, we reasoned that signaling critical for osteoclasts would also be important for cancer cells growing within the bone. In particular, Src plays a pivotal role in osteoclast RANK signaling, affecting bone structure and function (34). In fact, the only major finding in c-Src knockout mice is an osteopetrotic phenotype, despite ubiquitous Src expression in many tissues (39, 40). Therefore, we investigated RANKL-induced Src activation in PC cells. We found that chemical inhibition of Src activity significantly inhibited RANKL-induced C42b cell migration, with no effect on MAPK signaling. Additionally, LNCaP-MT1 cells displayed a constitutive increase in Src activation when compared to LNCaP-Neo. This activation was also observed in C42b cells exposed to LNCaP-MT1-derived CM, and was inhibited when the CM was pre-incubated with OPG. These results clearly show an association between tumor-associated MT1-MMP activity and soluble RANKL derived from the same cells, resulting in activation of RANK expressed on the surface of PC cells and subsequent activation of Src. Moreover, inhibition of Src by PP2 in C42b cells suppressed the enhanced migration caused by LNCaP-MT1-derived CM, implying an essential role for Src in this mechanism. To our knowledge, we are the first to show that Src is a key downstream mediator of RANKL-induced migration in tumor cells.
The novelty of our findings is based on the involvement of tumor-derived MT1-MMP in proteolytic cleavage of a non-ECM substrate to enhance PC cell migration. Recently, osteoclast-derived MMP-7 has been implicated in prostate and mammary tumor-induced osteolysis, presumably due to processing of osteoblast-derived membrane-bound RANKL to its soluble form leading to osteoclast maturation and activation (14, 41). We are the first to describe a connection between tumor-derived RANKL and tumor-associated MT1-MMP, wherein an autocrine loop in PC cells triggered by RANKL proteolytically shed from the surface of the cancer cell leads to enhanced cell migration. Furthermore, our data imply that the migration induced by the MT1-MMP/RANKL/RANK axis in PC cells is dependent on Src activity.
Notably, in vivo studies performed by us with the same MT1-MMP-overexpressing PC cells used herein clearly showed an essential role of tumor-associated MT1-MMP in intraosseous growth and osteolysis (10), suggesting that MT1-MMP/RANKL/RANK-mediated migration of PC cells may also have in vivo implications on tumor expansion within bones. Of course, these findings do not exclude a role for tumor - or osteoblast-derived RANKL in stimulating osteoclasts. To address the actual role of tumor-derived RANKL and autocrine signaling, knock-down experiments are currently underway in our laboratory to suppress the expression of endogenous RANKL or RANK in PC cells with either high or absent MT1-MMP expression and activity. Using these cell variants and the intratibial model, we expect to dissect the contribution of tumor-derived soluble RANKL and autocrine RANKL/RANK signaling to intraosseous tumor expansion.
The potential clinical relevance of a MT1-MMP/RANKL/RANK pathway is supported by data from human tissues. Prior studies by us and others showed expression of MT1-MMP (10) and RANKL (17–21) in clinical PC bone metastases, as well as expression of RANK (28, 29) and Src (42, 43) in human PC cells. Moreover, in addition to the apparent trend for an enhanced expression of MT1-MMP in PC bone metastasis (10) with respect to primary PC (9) observed by us, it has recently been proposed that the interaction of PC and the bone microenvironment results in an increase in RANKL expression by PC cells (44). Figure 6D shows a model that incorporates our current data and published results and illustrates an autocrine prostate tumor-derived MT1-MMP/RANKL/RANK pathway mediated through Src. Thus, in addition to the effects of RANKL on bone stroma, our model suggests a role for RANKL in PC cells. According to this model, active tumor-associated MT1-MMP cleaves RANKL expressed on the surface of PC cells into a soluble form of RANKL by ectodomain shedding. The binding of soluble RANKL to RANK at the membrane of PC cells signals through a pathway that involves Src activation, resulting in cytoskeletal rearrangement and increased migration of tumor cells. Inhibition of MT1-MMP activation (MIK-G2), RANKL binding (OPG), or Src phosphorylation (PP2), abrogates the enhanced migration of PC cells exposed to soluble factors produced by MT1-MMP-expressing cancer cells, suggesting that these events constitute potential targets that could be used to improve the treatment of bone metastatic disease. Recently, clinical trials with a fully human anti-RANKL antibody have been initiated in patients with PC bone metastases with promising results (45). Also, Src inhibitors are currently under clinical investigation for patients with metastatic castration-resistant PC; available data demonstrate activity in bone (46–48). Based on our results, we propose that the use of Src inhibitors in combination with RANKL and/or selective MT1-MMP inhibitors may be of therapeutic benefit in prostate cancer patients with skeletal metastasis.
Acknowledgments
We thank Alan Bonfil for his meticulous work in preparing the figures, and Allen Saliganan for his technical assistance.
Grant support
National Institutes of Health (NIH), National Institute of Digestive and Kidney Diseases Grant DK67687 (Michael L. Cher), The Fund for Cancer Research Grant (R. Daniel Bonfil and Michael L. Cher), NIH, National Institute of Cancer Grants CA137280 (Michael L. Cher and Rafael Fridman) and CA61986 (Rafael Fridman), and NIH Ruth L. Kirschstein National Research Service Award (NRSA) T32-CA009531 (Aaron L. Sabbota).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Yamada KM. Cell biology: tumour jailbreak. Nature. 2003;424:889–90. doi: 10.1038/424889a. [DOI] [PubMed] [Google Scholar]
- 2.Sahai E. Mechanisms of cancer cell invasion. Curr Opin Genet Dev. 2005;15:87–96. doi: 10.1016/j.gde.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 3.Sabeh F, Ota I, Holmbeck K, et al. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J Cell Biol. 2004;167:769–81. doi: 10.1083/jcb.200408028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zaman MH, Trapani LM, Sieminski AL, et al. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc Natl Acad Sci U S A. 2006;103:10889–94. doi: 10.1073/pnas.0604460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Itoh Y, Seiki M. MT1-MMP: a potent modifier of pericellular microenvironment. J Cell Physiol. 2006;206:1–8. doi: 10.1002/jcp.20431. [DOI] [PubMed] [Google Scholar]
- 6.Holmbeck K, Bianco P, Caterina J, et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 1999;99:81–92. doi: 10.1016/s0092-8674(00)80064-1. [DOI] [PubMed] [Google Scholar]
- 7.Ohuchi E, Imai K, Fujii Y, Sato H, Seiki M, Okada Y. Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J Biol Chem. 1997;272:2446–51. doi: 10.1074/jbc.272.4.2446. [DOI] [PubMed] [Google Scholar]
- 8.Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, Weiss SJ. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell. 2003;114:33–45. doi: 10.1016/s0092-8674(03)00513-0. [DOI] [PubMed] [Google Scholar]
- 9.Upadhyay J, Shekarriz B, Nemeth JA, et al. Membrane type 1-matrix metalloproteinase (MT1-MMP) and MMP-2 immunolocalization in human prostate: change in cellular localization associated with high-grade prostatic intraepithelial neoplasia. Clin Cancer Res. 1999;5:4105–10. [PubMed] [Google Scholar]
- 10.Bonfil RD, Dong Z, Trindade Filho JC, et al. Prostate cancer-associated membrane type 1-matrix metalloproteinase: a pivotal role in bone response and intraosseous tumor growth. Am J Pathol. 2007;170:2100–11. doi: 10.2353/ajpath.2007.060720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kong YY, Yoshida H, Sarosi I, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–23. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 12.Lum L, Wong BR, Josien R, et al. Evidence for a role of a tumor necrosis factor-alpha (TNF-alpha)-converting enzyme-like protease in shedding of TRANCE, a TNF family member involved in osteoclastogenesis and dendritic cell survival. J Biol Chem. 1999;274:13613–8. doi: 10.1074/jbc.274.19.13613. [DOI] [PubMed] [Google Scholar]
- 13.Schlondorff J, Lum L, Blobel CP. Biochemical and pharmacological criteria define two shedding activities for TRANCE/OPGL that are distinct from the tumor necrosis factor alpha convertase. J Biol Chem. 2001;276:14665–74. doi: 10.1074/jbc.M010741200. [DOI] [PubMed] [Google Scholar]
- 14.Lynch CC, Hikosaka A, Acuff HB, et al. MMP-7 promotes prostate cancer-induced osteolysis via the solubilization of RANKL. Cancer Cell. 2005;7:485–96. doi: 10.1016/j.ccr.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 15.Hikita A, Kadono Y, Chikuda H, et al. Identification of an alternatively spliced variant of Ca2+-promoted Ras inactivator as a possible regulator of RANKL shedding. J Biol Chem. 2005;280:41700–6. doi: 10.1074/jbc.M507000200. [DOI] [PubMed] [Google Scholar]
- 16.Hikita A, Yana I, Wakeyama H, et al. Negative regulation of osteoclastogenesis by ectodomain shedding of receptor activator of NF-kappaB ligand. J Biol Chem. 2006;281:36846–55. doi: 10.1074/jbc.M606656200. [DOI] [PubMed] [Google Scholar]
- 17.Chen G, Sircar K, Aprikian A, Potti A, Goltzman D, Rabbani SA. Expression of RANKL/RANK/OPG in primary and metastatic human prostate cancer as markers of disease stage and functional regulation. Cancer. 2006;107:289–98. doi: 10.1002/cncr.21978. [DOI] [PubMed] [Google Scholar]
- 18.Perez-Martinez FC, Alonso V, Sarasa JL, et al. Receptor activator of nuclear factor-kappaB ligand (RANKL) as a novel prognostic marker in prostate carcinoma. Histol Histopathol. 2008;23:709–15. doi: 10.14670/HH-23.709. [DOI] [PubMed] [Google Scholar]
- 19.Huang L, Cheng YY, Chow LT, Zheng MH, Kumta SM. Tumour cells produce receptor activator of NF-kappaB ligand (RANKL) in skeletal metastases. J Clin Pathol. 2002;55:877–8. doi: 10.1136/jcp.55.11.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Odero-Marah VA, Wang R, Chu G, et al. Receptor activator of NF-kappaB Ligand (RANKL) expression is associated with epithelial to mesenchymal transition in human prostate cancer cells. Cell Res. 2008;18:858–70. doi: 10.1038/cr.2008.84. [DOI] [PubMed] [Google Scholar]
- 21.Brown JM, Corey E, Lee ZD, et al. Osteoprotegerin and rank ligand expression in prostate cancer. Urology. 2001;57:611–6. doi: 10.1016/s0090-4295(00)01122-5. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, Dai J, Qi Y, et al. Osteoprotegerin inhibits prostate cancer-induced osteoclastogenesis and prevents prostate tumor growth in the bone. J Clin Invest. 2001;107:1235–44. doi: 10.1172/JCI11685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inoue H, Nishimura K, Oka D, et al. Prostate cancer mediates osteoclastogenesis through two different pathways. Cancer Lett. 2005;223:121–8. doi: 10.1016/j.canlet.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 24.Thalmann GN, Anezinis PE, Chang SM, et al. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 1994;54:2577–81. [PubMed] [Google Scholar]
- 25.Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LW. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: role of bone stromal cells. Int J Cancer. 1994;57:406–12. doi: 10.1002/ijc.2910570319. [DOI] [PubMed] [Google Scholar]
- 26.Koeneman KS, Yeung F, Chung LW. Osteomimetic properties of prostate cancer cells: a hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. Prostate. 1999;39:246–61. doi: 10.1002/(sici)1097-0045(19990601)39:4<246::aid-pros5>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 27.Jones DH, Nakashima T, Sanchez OH, et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature. 2006;440:692–6. doi: 10.1038/nature04524. [DOI] [PubMed] [Google Scholar]
- 28.Armstrong AP, Miller RE, Jones JC, Zhang J, Keller ET, Dougall WC. RANKL acts directly on RANK-expressing prostate tumor cells and mediates migration and expression of tumor metastasis genes. Prostate. 2008;68:92–104. doi: 10.1002/pros.20678. [DOI] [PubMed] [Google Scholar]
- 29.Mori K, Le Goff B, Charrier C, Battaglia S, Heymann D, Redini F. DU145 human prostate cancer cells express functional receptor activator of NFkappaB: new insights in the prostate cancer bone metastasis process. Bone. 2007;40:981–90. doi: 10.1016/j.bone.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 30.Kaighn ME, Narayan KS, Ohnuki Y, Lechner JF, Jones LW. Establishment and characterization of a human prostatic carcinoma cell line (PC-3) Invest Urol. 1979;17:16–23. [PubMed] [Google Scholar]
- 31.Ikejiri M, Bernardo MM, Bonfil RD, et al. Potent mechanism-based inhibitors for matrix metalloproteinases. J Biol Chem. 2005;280:33992–4002. doi: 10.1074/jbc.M504303200. [DOI] [PubMed] [Google Scholar]
- 32.Wong BR, Josien R, Lee SY, Vologodskaia M, Steinman RM, Choi Y. The TRAF family of signal transducers mediates NF-kappaB activation by the TRANCE receptor. J Biol Chem. 1998;273:28355–9. doi: 10.1074/jbc.273.43.28355. [DOI] [PubMed] [Google Scholar]
- 33.Wong BR, Besser D, Kim N, et al. TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c-Src. Mol Cell. 1999;4:1041–9. doi: 10.1016/s1097-2765(00)80232-4. [DOI] [PubMed] [Google Scholar]
- 34.Wada T, Nakashima T, Hiroshi N, Penninger JM. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med. 2006;12:17–25. doi: 10.1016/j.molmed.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 35.Dalgarno D, Thilo S, Narula S, et al. Structural Basis of Src Tyrosine Kinase Inhibition with a New Class of Potent and Selective Trisubstituted Purine-based Compounds. Chemical Biology & Drug Design. 2005;67:46–57. doi: 10.1111/j.1747-0285.2005.00316.x. [DOI] [PubMed] [Google Scholar]
- 36.Pavlaki M, Zucker S. Matrix metalloproteinase inhibitors (MMPIs): the beginning of phase I or the termination of phase III clinical trials. Cancer Metastasis Rev. 2003;22:177–203. doi: 10.1023/a:1023047431869. [DOI] [PubMed] [Google Scholar]
- 37.Bonfil RD, Fridman R, Mobashery S, Cher ML. Are matrix metalloproteinases relevant therapeutic targets for prostate cancer bone metastasis? Curr Oncol. 2008;15:188–92. doi: 10.3747/co.v15i4.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Devy L, Huang L, Naa L, et al. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 2009;69:1517–26. doi: 10.1158/0008-5472.CAN-08-3255. [DOI] [PubMed] [Google Scholar]
- 39.Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 40.Boyce BF, Yoneda T, Lowe C, Soriano P, Mundy GR. Requirement of pp60c-src expression for osteoclasts to form ruffled borders and resorb bone in mice. J Clin Invest. 1992;90:1622–7. doi: 10.1172/JCI116032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thiolloy S, Halpern J, Holt GE, et al. Osteoclast-derived matrix metalloproteinase-7, but not matrix metalloproteinase-9, contributes to tumor-induced osteolysis. Cancer Res. 2009;69:6747–55. doi: 10.1158/0008-5472.CAN-08-3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene. 2008;27:6365–75. doi: 10.1038/onc.2008.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goldenberg-Furmanov M, Stein I, Pikarsky E, et al. Lyn is a target gene for prostate cancer: sequence-based inhibition induces regression of human tumor xenografts. Cancer Res. 2004;64:1058–66. doi: 10.1158/0008-5472.can-03-2420. [DOI] [PubMed] [Google Scholar]
- 44.Penno H, Nilsson O, Brandstrom H, Winqvist O, Ljunggren O. Expression of RANK-ligand in prostate cancer cell lines. Scand J Clin Lab Invest. 2009;69:151–5. doi: 10.1080/00365510802460466. [DOI] [PubMed] [Google Scholar]
- 45.Fizazi K, Bosserman L, Gao G, Skacel T, Markus R. Denosumab treatment of prostate cancer with bone metastases and increased urine N-telopeptide levels after therapy with intravenous bisphosphonates: results of a randomized phase II trial. J Urol. 2009;182:509–15. doi: 10.1016/j.juro.2009.04.023. discussion 15–6. [DOI] [PubMed] [Google Scholar]
- 46.Yu EY, Wilding G, Posadas E, et al. Phase II study of Dasatinib in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res. 2009;15:7421–8. doi: 10.1158/1078-0432.CCR-09-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Araujo J, Logothetis C. Targeting Src signaling in metastatic bone disease. Int J Cancer. 2009;124:1–6. doi: 10.1002/ijc.23998. [DOI] [PubMed] [Google Scholar]
- 48.Saad F, Lipton A. SRC kinase inhibition: Targeting bone metastases and tumor growth in prostate and breast cancer. Cancer Treat Rev. 2010;36:177–84. doi: 10.1016/j.ctrv.2009.11.005. [DOI] [PubMed] [Google Scholar]