

Abstract
Expression of tight junction proteins between brain microvascular endothelial cells (BMECs) of the blood-brain barrier (BBB) is lost during development of HIV encephalitis (HIVE). While many studies have focused on the strains of virus that induce neurological sequelae or on the macrophages/microglia that are associated with development of encephalitis, the molecular signaling pathways within the BMECs involved have yet to be resolved. We have previously shown that there is activation and disruption of an in vitro BBB model using lentivirus-infected CEMx174 cells. We and others have shown similar disruption in vivo. Therefore, it was of interest to determine if the presence of infected cells could disrupt intact cerebral microvessels immediately ex vivo, and if so, which signaling pathways were involved. Our data demonstrate that disruption of tight junctions between BMECs is mediated through activation of focal adhesion kinase (FAK) by phosphorylation at TYR-397. Inhibition of FAK activation is sufficient to prevent tight junction disruption. Thus, it may be possible to inhibit the development of HIVE by using inhibitors of FAK.
Introduction
HIV and the closely related virus SIV cause central nervous system (CNS) disease in approximately one third of infected individuals including development of encephalitis (Baskin et al, 1988; Lackner et al, 1989; Lackner et al, 1991). While breakdown of BBB occurs (Luabeya et al, 2000; MacLean et al, 2005; Mankowski et al, 1999), relatively little is known of when this begins or of the molecular mechanisms associated with the breakdown. Questions regarding molecular signaling events and viral entry can be most readily answered using ex vivo and in vitro studies, because the precise timing of viral entry varies with host and virus. (Hirsch et al, 1997; Mankowski et al, 1994; Orandle et al, 2001; Westmoreland et al, 1998).
We developed in vitro and ex vivo models of the BBB suitable for studies of SIV encephalitis (MacLean et al, 2005; MacLean et al, 2002; MacLean et al, 2004a; MacLean et al, 2004b). These studies showed that there was a requirement for SIV-infected cells to cross an in vitro BBB model to activate both the BMECs and astrocytes in a manner similar to that observed in vivo (MacLean et al, 2004a; MacLean et al, 2004b). Using immediately ex vivo microvessels obtained from encephalitic brains we demonstrated considerably lower levels of ZO-1 protein compared with microvessels obtained from control brains (MacLean et al, 2005). Here, we use our ex vivo model of the BBB to begin examining the molecular events associated with breakdown of the BBB.
Activation and translocation of focal adhesion kinase (FAK) has been reported to be a mechanism by which increased endothelial permeability occurs (Avraham et al, 2004). FAK is required for efficient angiogenesis and remodeling, with data suggesting that without FAK, there can be no reorganization of junction proteins along actin filaments to form junctions (Ilic et al, 2004; Linseman et al, 1999; Sawada and Sheetz, 2002). Therefore, it would be anticipated that FAK is involved in reorganization or disruption of tight junctions. The tyrosine at 397 is a candidate phospho-specific site of FAK activation associated with tight junction disruption events.
Phosphorylation of FAK leads to activation of the kinase and thus decreases the number of focal adhesions ‘beneath’ cells (Brown et al, 2005; Ilic et al, 2004; Orr and Murphy-Ullrich, 2004; Orr et al, 2004; Usatyuk and Natarajan, 2005). Concomitant with this, there are decreased adhesions ‘between’ BMECs (tight and adherens junctions), leading to increased permeability (Orr and Murphy-Ullrich, 2004), which is linked to development of encephalitis (Avraham et al, 2004; Dallasta et al, 1999; Kim et al, 2003; Selmaj, 1996). It has been shown in clinical SIVE cases that ZO-1 decreases following tight junction disruption in subcortical white matter and to a lesser extent in gray matter (9). ZO-1 has also been shown to decrease in vitro following transmigration of HIV positive leukocytes (Eugenin et al, 2006).
We sought to determine if activation of the FAK pathway diminishes the expression of ZO-1 and if SIV-infected monocyte/macrophages are required for development of encephalitis by disruption of tight and adherens junctions in a FAK-dependent manner. We hope this will lead to new therapeutic targets since HAART alone does not decrease the prevalence of encephalitis or peripheral neuropathies (Kusdra et al, 2002; Shiramizu et al, 2005).
Materials and Methods
Culture of virus infected cells
CEMx174 cells and primary macrophages were used for these studies. CEMx174 cells were a kind donation from Dr. Preston Marx (Tulane National Primate Research Center). Cells were maintained in RPMI (Roswell Park Memorial Institute)-1640 medium containing 10% fetal calf serum. Cultures were subdivided 1:4 once per week. Cultures infected with SIVmac251 (as previously described (MacLean et al, 2004b)) were grown exactly as the control cultures, with the exception that, upon subculturing, control CEMx174 cells were added to the cultures to a final concentration of 106 /ml, ensuring a fresh supply of cells to be infected.
Primary macrophages were isolated from Rhesus macaque (Macaca mulatta) femur bone marrow at necropsy. Briefly, 2ml bone marrow was vortexed and centrifuged at 400G at 25°C (Fisher Marathon 5000R centrifuge). Supernatant was decanted and the pellet resuspended in PBS (Gibco). This cell suspension was filtered (110 μm filter), and the filtrate consisting of enriched macrophages was plated. When nearly confluent, some plates were kept as controls, while the remainder were infected with SIVmac251 (by the same procedure as Gautam et al (Gautam et al, 2007)). Macrophages were grown in Iscove's Modified Dulbecco's Medium (Mediatech) and supplemented with 10% heat-inactivated fetal bovine serum (Gibco) and 1% penicillin/streptomycin (BioWhittaker). Macrophages were removed from culture flasks using trypsin/versene, and resuspended at a final concentration of 106 /ml. Cultures were routinely greater than 95% macrophages by immunohistochemistry with the macrophage marker HAM 56. Primary cells were determined to be productively infected by in situ hybridization for SIV RNA. Sense probe was used as a control.
Extraction of microvessels
Microvessels were extracted from frontal cortices collected from normal Rhesus macaques at scheduled necropsy, as previously described (25). In brief, meninges and contaminating vessels were removed before mincing the cortices and passing through a 320μm nylon filter. The filtrate was collected and poured through a 110μm nylon filter and rinsed until sterile PBS passed through the filter clear. Microvessels were collected from the filter by washing with M199 medium (Mediatech) into 50ml tubes. The microvessels were centrifuged at 1000 rpm for 6 minutes (Fisher Marathon 5000R centrifuge) and the supernatant decanted. Microvessels were then resuspended in M199 medium containing 10% fetal calf serum. In total, the microvessel yield from 1g of cortical tissue was resuspended in 15 mL of medium.
Incubation of microvessels with infected cells and supernatants
Slides were pre-treated with poly-L-lysine (50μg/ml in PBS) for 30 minutes to facilitate adhesion. Freshly prepared microvessels (1 g of initial cortical tissue/15 mL media) were re-suspended in medium containing SIV-infected and control CEMx174 cells, macrophages (106/mL) or their supernatants and were incubated on slides for 0, 1, 2, 4, 6, or 8 hours at 37°C. Two slides were prepared per data point. A final ratio of approximately 15:1 (infected cells:BMEC) was used for all experiments. If pre-treating with phenylarsine oxide (PAO), a fifteen minute pre-incubation occurred prior to microvessel exposure to macrophages or CEMx174 cells. Slides were fixed with 2% paraformaldehyde and stored at 4°C overnight in PBS prior to immunohistochemical staining.
Confocal microscopy
Microvessels on slides were permeabilized with PBS containing 1% bovine serum albumin and 0.1% Triton-X-100 (Sigma) for ten minutes. Following permeabilization, slides were blocked for one hour with normal goat serum (Sigma) and rinsed with PBS containing 1% BSA (Sigma). Slides were stained for confocal imaging using primary antibodies to ZO-1 and FAK at concentrations outlined in Table 1 overnight at 4°C.
TABLE 1.
Antibodies
| Antibody | Species | Supplier | Concn WB | Concn IHC |
|---|---|---|---|---|
| HAM56 | Mouse | Dako | 3.5μg/mL | |
| ZO-1 | Mouse | Zymed | 2μg/mL | 20μg/mL |
| FAK | Rabbit | Sigma | 3μg/mL | 3μg/mL |
| FAK (p397) | Mouse | BD | 0.25μg/mL | 0.25μg/mL |
| GAPDH | Rabbit | Santa Cruz | 2μg/mL | n/a |
| Rabbit IgG-HRP | Donkey | Santa Cruz | 3μg/mL | n/a |
| Mouse IgG-HRP | Bovine | Santa Cruz | 3μg/mL | n/a |
| Topro3 | n/a | Molec. Probes | 2μg/mL | 0.001 mM |
Slides were thoroughly washed and mounted using MOWIOL 4-88/ Glycerol/ DABCO (Calbiochem, La Jolla/ Sigma/ Sigma). Confocal microscopy was performed using a Leica TCS SP2 confocal microscope equipped with three lasers (Leica Microsystems, Exton, PA) to collect up to three channels simultaneously. Forty optical slices were collected at 512 × 512 pixel resolution and captured with Leica Confocal Software (Leica Microsystems, Exton, PA). Each individual slice represented a thickness of 0.4 μm.
Secondary antibodies used include: Goat anti-rabbit (heavy and light chains) conjugated to Alexa 488, appearing green (Molecular Probes, Eugene, OR); Goat anti-mouse (IgG1) Alexa conjugated to 568, appearing red (Molecular Probes, Eugene, OR). To-Pro3 iodide was used as a nuclear stain, appearing blue (Molecular Probes, Eugene, OR). Secondary antibodies were applied at a concentration of 1:1000 for 1 hour at 37°C. To-Pro3 was applied for 10 minutes.
Image analysis, quantification and statistics
Each channel of the confocal images (color) was analyzed using NIH Image (v. 1.38) to determine mean fluorescent intensity of target proteins along junctional “zippers” of microvessels. This is achieved by averaging a “stack” of images, and taking a snapshot of this mean image. Each individual image is scanned three times and background is automatically subtracted. Images were collected with a 63x objective and 2x digital zoom. From these averaged snapshots, we manually traced the microvessels with NIH Image drawing tools, and measured the pixel intensity/traced area or mean pixel intensity. The data were plotted using Microsoft Excel. Statistical significance and p-values were determined by using Student's t-test (p-values <0.05 are considered significant and are noted by an asterisk). This process allows for a standardized, reproducible and quantitative analysis of fluorescent intensity.
Inhibition of FAK phosphorylation
Phosphorylation of tyrosine residues has been shown to activate focal adhesion kinase (Linseman et al, 1999; Sawada and Sheetz, 2002). There are no specific inhibitors of FAK readily available at this time. To begin to address this issue, we used an inhibitor of tyrosine phosphorylation, phenylarsine oxide (PAO) to inhibit the activation of FAK (Linseman et al, 1999) by pre-incubating microvessels with 5μM PAO for 15 minutes before the addition of control or SIV-infected CEMx174 cells.
Determination of post-translational control
To determine if FAK is phosphorylated, and hence activated, during incubation with SIV-infected cells, relative levels of phosphorylated FAK were measured using Western blots of microvessel lysates. Microvessels were incubated with SIV-infected or control cells (either CEMx174 or primary macrophages) for the indicated times. Microvessels were then rinsed on top of cell filters (70μm pore) to remove any SIV-infected CEMx174 cells or macrophages. Samples were protected from proteases and phosphatases using Halt Inhibitor cocktail (Pierce, Rockford, IL) and were prepared on ice. Initial lysis used a Triton-X-100 detergent base containing 10mM Tris-HCl, pH 7.4, 100mM NaCl, 300mM sucrose and 0.5% Triton-X-100. The Triton insoluble pellet was gently washed twice with Tris-buffered saline (containing Halt inhibitor cocktail) and extracted using a RIPA detergent containing 10mM Tris-HCl, 140mM NaCl, 1% Triton-X-100, 1% sodium deoxycholate, 0.1% SDS, and the same protease inhibitors as above.
Immunoprecipitation
Tissue lysates were pre-cleared with 50 μl normal rabbit serum/mL of lysates and incubated 1 hour on ice. 100 μl of protein-A coupled Sepharose beads (Abcam) were then added. The lysate-bead slurry was incubated 30 minutes at 4°C with gentle agitation. Lysate-bead slurry was then centrifuged at 14,000 × g at 4°C for ten minutes. Bead pellet was discarded and supernatant was kept for immunoprecipitation.
200 μg of lysate was added with 3 μl of primary antibody and was incubated overnight at 4°C with gentle agitation. Protein-A coupled Sepharose beads (1 mL PBS-BSA 1% w/v) were prepared freshly in 400 μl lysis buffer with Halt cocktail after extensive rinsing in PBS. 70 μl of bead slurry was then added to each sample on ice and was agitated at 4°C for four hours. At the end of this incubation, the slurry was centrifuged and beads were washed in lysis buffer three times. Finally, with the supernatant removed, 25 μl of 2x loading buffer was added and beads were heat denatured at 95°C for five minutes. This was centrifuged and supernatant was retained for electrophoresis.
Electrophoresis and Western blotting
Samples were prepared in 95% Laemmli buffer with 5% β-mercaptoethanol, denatured at 95°C for 10 minutes and electrophoresed through a 7.5% SDS gel. Western blots were prepared using standard techniques (Stamatovic et al, 2003). Proteins were detected using the same primary antibodies used for confocal analysis. Secondary antibodies were conjugated to HRP and developed using SuperSignal West Fempto Luminol (Pierce). Images were captured with a Kodak Image Station 2000MM and Kodak Molecular Imaging Software v. 4.0.4.
Semi-quantitative band densitometry was calculated by measuring mean pixel with NIH Image v 1.38. To achieve this, bands were traced manually with NIH Image and pixel intensity was measured within the selected area. Data were then corrected for GAPDH loading controls (GAPDH discrepancy across conditions: <5%).
Results
Preparation of Microvessels
Isolated control microvessels typically appear as shown in Figure 1 in lengths or small networks of up to several hundred microns. Nuclei of endothelial cells are evident along the lengths of these control microvessels with hematoxylin staining at low magnification (Figure 1A) and at higher magnification (Figure 1B).
Figure 1. Isolated microvessel preparation and infectivity of primary macrophages.

Isolated microvessels frequently appear as individual lengths or small networks as shown here in light microscopy using hematoxylin staining. The elongated nuclei of BMECs are evident along the lengths of these microvessels. Figure 1A shows an example of an entire network and (B) shows a 100x image of the same network. Primary cultures of bone marrow-derived macrophages (HAM56, in green) were infected with SIVmac251 (in situ hybridization, red). Control macrophage cultures were positive only for HAM56 (C). Productively infected cultured macrophages (D) were double positive. Cytospins of CEMx174 cells were shown to be infected with SIVmac251 (in situ hybridization, red) (E).
In order to determine that our stimulus cells (CEMx174 or primary macrophages) were productively infected with SIVmac251, we combined in situ hybridization and immunohistochemistry. We stained macrophages infected with SIV (in red) and control macrophages (stained with HAM56, green) to verify the level of infectivity. No detectable SIV RNA was found in control cultures (Figure 1C). Effectively all (>90%) bone marrow-derived macrophages were productively-infected with SIVmac251 as shown with in situ hybridization 72 hours after infection (Figure 1D). As with other studies, generally 10% of CEMx174 cells were productively infected (red) with SIVmac251 (Sabine Lange, personal communication, in situ hybridization of cytospins, Figure 1E).
Lentiviral-induced disruption of ZO-1 patterning
To investigate BBB disruption following exposure to SIV-infected cells, we examined changes in the expression of tight junction protein ZO-1 in microvessels incubated with control CEMx174 cells and SIVmac251-infected CEMx174 cells (n=20 each data point, both groups). Normal microvessels incubated in the presence of uninfected CEMx174 cells, demonstrate a linear “zipper” pattern of ZO-1 expression, with rings of protein expression between BMECs (Figure 2A). When brain microvessels were incubated in the presence of SIVmac251-infected CEMx174 cells, a loss of ZO-1 expression was observed along the length of microvessels during the first eight hours of incubation. At four hours, these changes can be observed (Figure 2B). Freshly isolated microvessels express a uniform “zipper” expression of ZO-1 protein (Figure 2C).
Figure 2. Loss of ZO-1 expression on microvessels following incubation with SIV-infected CEMx174 cells.
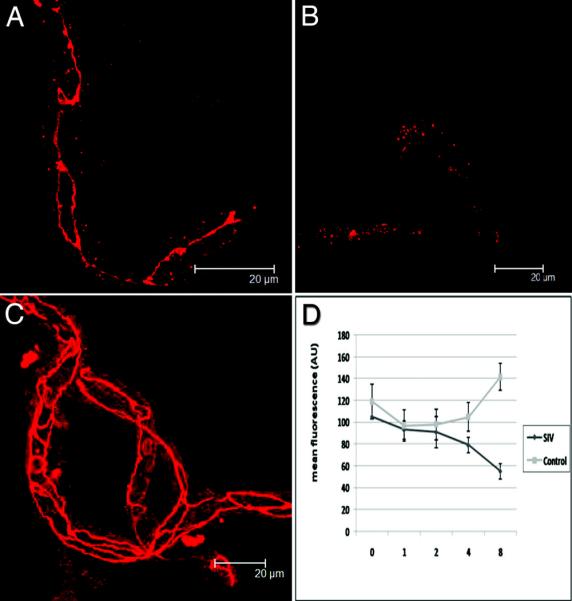
At top left (A), microvessels incubated with control CEMx174 cells for 4 hours, showing the distinctive linear expression of ZO-1 with ring-like patterns at cell junctions. At top right (B), a microvessel that has been incubated with SIVmac251-infected CEMx174 cells for 4 hours showing diffuse, punctate expression of ZO-1. Freshly isolated microvessels without treatment (C) show complex and continuous linear patterns of ZO-1. Note the loss of linear staining seen in B. Using NIH Image v1.62 we determined mean fluorescence intensities of 10μm lengths of microvessels. We measured along five non-overlapping sections of each of at least four vessels in areas anticipated to be rich for ZO-1 expression. The graph (D) demonstrates that although there may be an initial loss of expression of ZO-1 in control vessels, there is a steady loss of ZO-1 fluorescent intensity after 2 hours incubation with SIV-infected CEMx174 cells. Error bars represent standard errors of the means.
Statistical evaluation of decreased ZO-1 intensity
When fluorescence was quantified in these microvessels over time (Figure 2D), A significant decrease in ZO-1 intensity in SIV-exposed microvessels is evident at the 8-hour time point versus controls (p<0.01). While there is a trend toward increased ZO-1 fluorescence in the control group, this effect is not statistically significant over 8 hours (p=0.363). Since maintenance of tight junctions is dependent on the proper organization of junctional scaffolding proteins such as ZO-1, it would be anticipated that there would be disruption of the BBB resulting from this altered ZO-1 patterning.
Focal adhesion kinase expression precedes reduction of ZO-1 expression
To establish if a change in ZO-1 expression is linked to expression of focal adhesion kinase (FAK), microvessels were incubated and stained as above (n=20 each data point, each group). Freshly extracted microvessels from control macaques expressed undetectable (or barely detectable) levels of FAK after incubation with CEMx174 cells for 1, 2, 4, or 6 hours (Figure 3A-D, respectively). This suggests that the presence of CEMx174 cells in proximity to the BBB is not sufficient in and of itself for activation of FAK.
Figure 3. Expression of focal adhesion kinase (FAK) in freshly isolated microvessels.
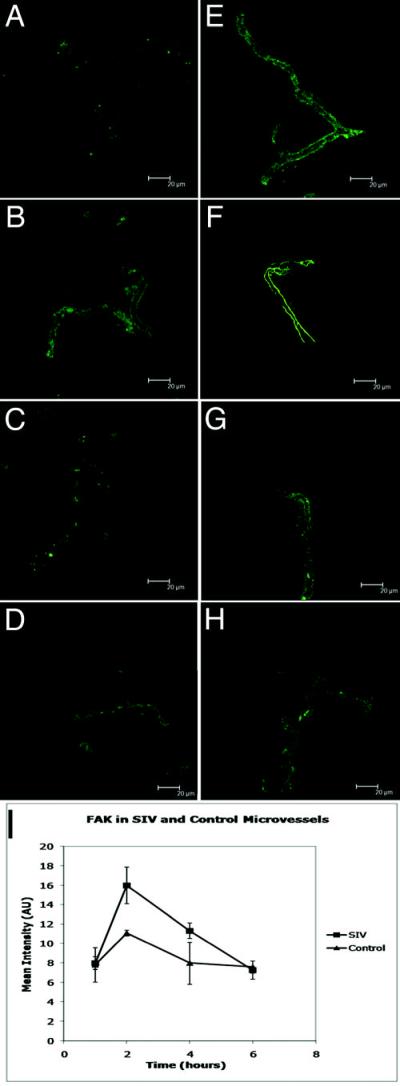
Microvessels incubated with control CEMx174 cells expressed low levels of FAK (green) at time points 1, 2, 4 and 6 hours (A-D, respectively). Following one hour incubation with SIVmac251-infected CEMx174 cells, there was minimally (not significant) increased expression of FAK (E). At two hours, the expression of FAK was similar to the normal expression of ZO-1 (F). The levels of expression of FAK then begins to wane over the next four hours (G), returning to basal levels by six hours (H). As the graph represents (I), FAK expression significantly increases at the 2- hour time point when vessels are exposed to SIV (p=0.004), and then subsides to control levels.
We then investigated the effects of incubation of microvessels with SIVmac251-infected CEMx174 cells as above (Figures 3E-H, respectively). Following incubation of these microvessels with SIV-infected CEMx174 cells, FAK expression patterns changed. After a one-hour incubation, SIV treated microvessels began to express FAK in a similar pattern to ZO-1 expression (Figure 3E). Following incubation of the microvessels for two hours with SIVmac251-infected CEMx174 cells, the expression of FAK increased further and appeared to be continuous along the length of the microvessels (Figure 3F). At 4- and 6- hour time points, the level of FAK expression decreases, with the pattern of FAK immunostaining most closely resembling the staining seen at one hour points (Figures 3G and H, respectively).
Analysis of mean fluorescent intensity in the green channel (FAK immunostaining) indicated transient increases of FAK expression at the two- and four- hour time points (Figure 3I). At the two-hour time point, there is significant 1.44 fold increase in FAK expression (p=<0.005, n=20).
The changes in FAK and ZO-1 expression can be viewed concurrently as illustrated in the three-hour incubations shown in Figure 4 with FAK in green, ZO-1 in red and nuclei in blue. Figure 4A is a three-hour incubation of control microvessels. Figure 4B is a three-hour incubation of microvessels with non-infected CEMx174 cells and shows little difference from the control. However, the addition of SIV-infected CEMx174 cells for 3 hours results in increased FAK, and decreased ZO-1 (Figure 4C).
Figure 4. Concurrent views of FAK and ZO-1 in microvessels with CEMx174 cells.
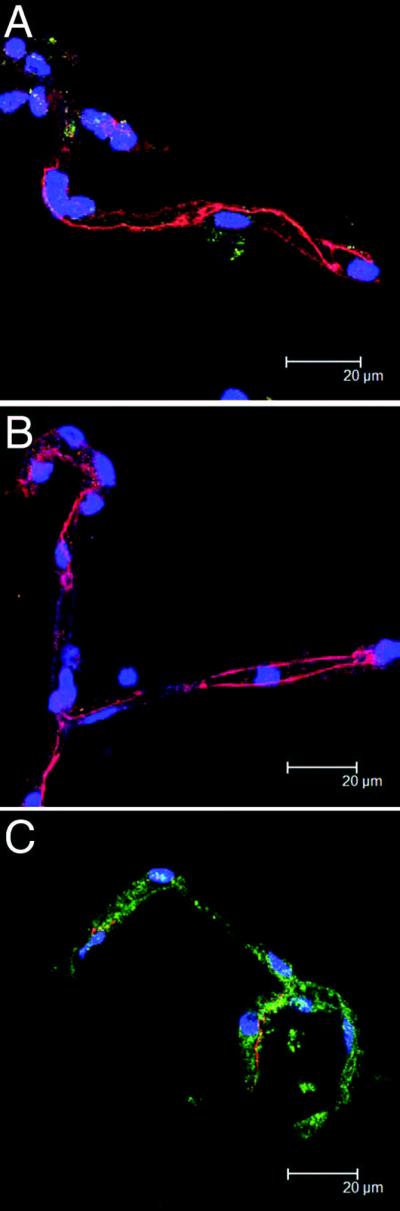
Microvessels are shown with no CEMx174 cells after an incubation of 3 hours (A). Over the same 3 hour time period, microvessels are incubated with uninfected CEMx174 cells and show little difference in FAK (green) and ZO-1 (red) compared with controls (B). Isolated microvessels incubated with SIV-infected CEMx174 cells for 3 hours (C), however, show markedly decreased ZO-1 patterning and more robust FAK fluorescence.
Macrophage-induced activation of FAK and disruption of ZO-1
These effects, seen with the CEMx174 cell line were also observed when microvessels were exposed to SIVmac251-infected primary bone marrow-derived macrophages (Figure 5). Figure 5A shows microvessels incubated with uninfected primary bone marrow-derived macrophages for three hours. In figure 5B, freshly isolated microvessels were incubated with SIVmac251-infected primary macrophages for three hours. FAK fluorescence (red) is brighter in the SIVmac251 group than in the matched control, and ZO-1 (green) shows the opposite trend, appearing degraded in the SIVmac251 group.
Figure 5. FAK and ZO-1 altered by primary SIV-infected macrophages and macrophage supernatants.

Isolated microvessels were incubated for three hours with bone marrow derived macrophages that were either uninfected (A) or infected with SIVmac251 (B). At three hours, the control microvessels show strong ZO-1 staining (green) and very little FAK (red). In the SIV exposed microvessels, FAK is more robust and ZO-1 diminished, with gaps along the length of the vessel.
Determination of specificity of signal transduction pathways
To show that the FAK pathway is involved in BBB tight junction regulation in neuroAIDS, we used PAO, a known inhibitor of FAK (Linseman et al, 1999; Retta et al, 1996; Sawada and Sheetz, 2002; Yuan et al, 1998). Microvessels were incubated in 5μM PAO for 15 minutes before incubating with SIVmac251-infected (and control) CEMx174 cells (Figure 6). In the presence of PAO, SIV-infected CEMx174 cells not only did not induce any FAK expression (Figure 6A), but there was no loss of ZO-1 expression, indicating a specificity in the signal transduction pathway induced by SIV-infected CEMx174 cells. Similarly, incubation of PAO-treated microvessels with control CEMx174 cells showed no change in ZO-1 expression (Figure 6B).
Figure 6. Inhibition of BBB disruption by PAO.

Microvessels were incubated for fifteen minutes in the presence of 5μm PAO. Control (A) or SIV-infected CEMx174 cells (B) were then added to the microvessels and incubated for an additional 2 hours before fixation and staining for ZO-1 (red) and FAK (green). It was clear that PAO pretreatment prevented the increased expression of FAK, and the decreased expression of ZO-1 following incubation with SIVmac251-infected CEMx174 cells (B). Both images are two-color, red-green images.
Expression of FAK protein
Total FAK protein in microvessel lysates rapidly increases upon exposure to SIVmac251 infected macrophage supernatant (Figure 7A). Additionally, a similar analysis of total FAK protein over time showed an increase following exposure to SIV-infected macrophage supernatant, with up to a two-fold increase at two hours incubation (Figure 7B). In order to more accurately assess whether our confocal results indicated an actual change in the quantity of activated tight-junction associated FAK, we also probed microvessel Western blots for phospho-specific FAK Tyr-397 protein. When probed for FAK Tyr-397 by Western blotting, vessel lysates that had been incubated with SIV-infected macrophages for two hours showed increased expression in lane 2 (Figure 7C) compared to incubation with uninfected macrophages in lane 1 compared to loading control. Band densitometry at this 2 hour time point indicated that FAK Tyr-397 increased by 1.57 fold.
Figure 7. Western blot of FAK expression.

FAK protein increases transiently when incubated with SIVmac251-infected macrophages. At 60 and 75 minutes post-incubation, FAK increases over control lysates (A). Band densitometry was performed of total FAK/area in a time course of SIVmac251 exposed or non-exposed microvessels subjected to a 4 hour time course (B). Total FAK protein in these samples increased at up to 2-fold during the course of the experiment. Expression of FAK397 is increased in microvessels incubated with SIV-infected versus control macrophages (C).
Discussion
Our experiments indicate activation of immediately ex vivo cerebral microvessels following incubation with SIVmac251-infected mononuclear cells that is mediated via phosphorylation of FAK and subsequent alteration in the expression of the tight junction anchorage protein ZO-1. Activation of FAK is rapid, occurring within two hours, with phosphorylation of tyrosine residue 397.
Encephalitis associated with SIV infection of macaques and HIV infection of humans is characterized by astrocyte and microglia activation paired with productive infection of macrophages and multinucleated giant cells (Persidsky and Gendelman, 1997; Persidsky et al, 1995). This is accompanied by activation of the cerebral microvasculature (Persidsky et al, 1997; Sasseville et al, 1994; Sasseville et al, 1992). We and others have shown that tight junctions of microvessels are disrupted during terminal AIDS (Andras et al, 2003; MacLean et al, 2005; Persidsky et al, 1997). By examining ex vivo microvessels, we found that disruption of the BBB, manifested by changes in ZO-1 expression did not occur at discrete spots (MacLean et al, 2005). Rather, microvessels obtained from SIVE brains have diminished ZO-1 expression over distances of several dozen microns and multiple cell lengths.
ZO proteins are thought to link occludin/claudins to F-actin filaments via ZO-associated protein. Decreases in ZO-1 have been correlated with increased permeability of the tight junction, and BBB disruption (Anathbandhu et al, 2007; Kebir et al, 2007). In macaques with SIV encephalitis, ZO-1 staining in medium sized vessels becomes patchy or decreased, especially in areas associated with perivascular macrophages or multi-nucleated giant cells (Luabeya et al, 2000). Similar results have been produced by evaluating the significant ZO-1 repatterning in small and medium-sized vessels that occurs in HIV encephalitis patients during a postmortem study (Dallasta et al, 1999). We chose to examine ZO-1 because of its relevance in other studies looking at BBB disruption, and also because it is thought to be the primary component of anchorage sites in common for transmembrane tight junction proteins like JAMs, Claudins, and Occludin. It is therefore likely that the degradation or reorganization of ZO-1 results in tight junction integrity dysregulation.
Recently, Avraham and colleagues have shown that the HIV protein Tat can induce increased phosphorylation of FAK leading to increased endothelial permeability (Avraham et al, 2004). In addition to Tat, gp120 from HIV has been shown to cause disruption of tight junctions (Avraham et al, 2004; Kanmogne et al, 2005), leading to vascular dysfunction (Lohmann et al, 2004). Whether or not Tat is free in the extracellular space is still a matter of speculation. Further complicating this issue, purified Tat has been shown to have no effect on ZO-1 or occludin in an in vitro model (Andras et al, 2003), although ZO-2 and claudins 1&5 were diminished. Our studies show that primary bone marrow-derived macrophages infected with pathogenic SIV can activate FAK in a time course similar to that observed with purified Tat, an event followed by the reorganization of ZO-1 in freshly ex vivo cortical microvessels.
FAK is activated by intracellular tyrosine phosphatases (Leu et al, 2003; Lohmann et al, 2004). Increased FAK expression was blocked by the addition of the tyrosine phosphatase inhibitor PAO. This would have been expected to maintain the expression of the tight junction protein ZO-1 (Wachtel et al, 1999), and thus inhibit any increase in vascular permeability (Staddon et al, 1995). Tyrosine 397 (Tyr-397) is thought to be the primary site of phosphorylation on the n-terminal domain of FAK. Activation of FAK at Tyr-397 is thought to induce further phosphorylation of the protein at Tyr-576 and Tyr-577 by way of positive feedback (Ruest et al, 2000).
It will be of interest to determine if other tight junction proteins are also affected by FAK activation, or if other tyrosine phosphatase inhibitors are as effective at blocking FAK activation by HIV-infected cells. These data suggest that by inhibiting tyrosine phosphorylation of FAK we can inhibit mononuclear cell-mediated degradation of BBB junctional components. This could be of importance not only in lentiviral-associated mononuclear infiltration (Potula et al, 2005; Stins et al, 2003), but also in other neurovascular infections such as those associated with Cryptococcus (Chen et al, 2003) and Borrelia (Ramesh and Philipp, 2005) infections.
Combined, these studies show that HIV-infected mononuclear cells induce activation of the FAK pathway within intact cerebral microvessels and that this activation is dependent on tyrosine phosphatase activity. Selectively targeting FAK activation may lead to new strategies for inhibiting development of AIDS dementia by preventing breakdown of BBB.
Acknowledgements
This work was supported by PHS grants: AA13828, MH077544, MH61192, RR00164, AA9803, and RR20159. N. Scott Ivey is supported by a teaching assistantship from the School of Science and Engineering, Tulane University. Nicole Renner is supported by a fellowship from The State of Louisiana Board of Regents (LEQSF(2007-12)-GF-15).
We thank Dr. Preston Marx for supplying CEMx174 cells and SIVmac251 viral stocks. We also thank Robin Rodriguez for excellent assistance with preparation of the figures.
References
- Anathbandhu C, Yang B, Gendelman HE, Persidsky Y, Kanmogne GD. STAT1 signaling modulates HIV-1-induced inflammatory responses and leukocyte transmigration across the blood-brain barrier. Blood. 2007 doi: 10.1182/blood-2007-05-091207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andras IE, Pu H, Deli MA, Nath A, Hennig B, Toborek M. HIV-1 Tat protein alters tight junction protein expression and distribution in cultured brain endothelial cells. J Neurosci Res. 2003;74:255–65. doi: 10.1002/jnr.10762. [DOI] [PubMed] [Google Scholar]
- Avraham HK, Jiang S, Lee TH, Prakash O, Avraham S. HIV-1 Tat-mediated effects on focal adhesion assembly and permeability in brain microvascular endothelial cells. J Immunol. 2004;173:6228–33. doi: 10.4049/jimmunol.173.10.6228. [DOI] [PubMed] [Google Scholar]
- Baskin GB, Murphey-Corb M, Watson EA, Martin LN. Necropsy findings in rhesus monkeys experimentally infected with cultured simian immunodeficiency virus (SIV)/delta. Vet Pathol. 1988;25:456–67. doi: 10.1177/030098588802500609. [DOI] [PubMed] [Google Scholar]
- Brown MC, Cary LA, Jamieson JS, Cooper JA, Turner CE. Src and FAK Kinases Cooperate to Phosphorylate PKL, Stimulate Its Focal Adhesion Localization, and Regulate Cell Spreading and Protrusiveness. Mol Biol Cell. 2005 doi: 10.1091/mbc.E05-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Stins MF, Huang SH, Chen YH, Kwon-Chung KJ, Chang Y, Kim KS, Suzuki K, Jong AY. Cryptococcus neoformans induces alterations in the cytoskeleton of human brain microvascular endothelial cells. J Med Microbiol. 2003;52:961–70. doi: 10.1099/jmm.0.05230-0. [DOI] [PubMed] [Google Scholar]
- Dallasta LM, Pisarov LA, Esplen JE, Werley JV, Moses AV, Nelson JA, Achim CL. Blood-brain barrier tight junction disruption in human immunodeficiency virus-1 encephalitis. Am J Pathol. 1999;155:1915–27. doi: 10.1016/S0002-9440(10)65511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci. 2006;26:1098–106. doi: 10.1523/JNEUROSCI.3863-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam R, Carter AC, Katz N, Butler IF, Barnes M, Hasegawa A, Ratterree M, Silvestri G, Marx PA, Hirsch VM, Pandrea I, Apetrei C. In vitro characterization of primary SIVsmm isolates belonging to different lineages. In vitro growth on rhesus macaque cells is not predictive for in vivo replication in rhesus macaques. Virology. 2007;362:257–70. doi: 10.1016/j.virol.2006.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch V, Adger-Johnson D, Campbell B, Goldstein S, Brown C, Elkins WR, Montefiori DC. A molecularly cloned, pathogenic, neutralization-resistant simian immunodeficiency virus, SIVsmE543-3. J Virol. 1997;71:1608–20. doi: 10.1128/jvi.71.2.1608-1620.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic D, Kovacic B, Johkura K, Schlaepfer DD, Tomasevic N, Han Q, Kim JB, Howerton K, Baumbusch C, Ogiwara N, Streblow DN, Nelson JA, Dazin P, Shino Y, Sasaki K, Damsky CH. FAK promotes organization of fibronectin matrix and fibrillar adhesions. J Cell Sci. 2004;117:177–87. doi: 10.1242/jcs.00845. [DOI] [PubMed] [Google Scholar]
- Kanmogne GD, Primeaux C, Grammas P. HIV-1 gp120 proteins alter tight junction protein expression and brain endothelial cell permeability: implications for the pathogenesis of HIV-associated dementia. J Neuropathol Exp Neurol. 2005;64:498–505. doi: 10.1093/jnen/64.6.498. [DOI] [PubMed] [Google Scholar]
- Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–5. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TA, Avraham HK, Koh YH, Jiang S, Park IW, Avraham S. HIV-1 Tat-mediated apoptosis in human brain microvascular endothelial cells. J Immunol. 2003;170:2629–37. doi: 10.4049/jimmunol.170.5.2629. [DOI] [PubMed] [Google Scholar]
- Kusdra L, McGuire D, Pulliam L. Changes in monocyte/macrophage neurotoxicity in the era of HAART: implications for HIV-associated dementia. Aids. 2002;16:31–8. doi: 10.1097/00002030-200201040-00005. [DOI] [PubMed] [Google Scholar]
- Lackner AA, Marx PA, Lerche NW, Gardner MB, Kluge JD, Spinner A, Kwang HS, Lowenstine LJ. Asymptomatic infection of the central nervous system by the macaque immunosuppressive type D retrovirus, SRV-1. J Gen Virol. 1989;70:1641–51. doi: 10.1099/0022-1317-70-7-1641. [DOI] [PubMed] [Google Scholar]
- Lackner AA, Smith MO, Munn RJ, Martfeld DJ, Gardner MB, Marx PA, Dandekar S. Localization of simian immunodeficiency virus in the central nervous system of rhesus monkeys. Am J Pathol. 1991;139:609–21. [PMC free article] [PubMed] [Google Scholar]
- Leu TH, Su SL, Chuang YC, Maa MC. Direct inhibitory effect of curcumin on Src and focal adhesion kinase activity. Biochem Pharmacol. 2003;66:2323–31. doi: 10.1016/j.bcp.2003.08.017. [DOI] [PubMed] [Google Scholar]
- Linseman DA, Sorensen SD, Fisher SK. Attenuation of focal adhesion kinase signaling following depletion of agonist-sensitive pools of phosphatidylinositol 4,5-bisphosphate. J Neurochem. 1999;73:1933–44. [PubMed] [Google Scholar]
- Lohmann C, Krischke M, Wegener J, Galla HJ. Tyrosine phosphatase inhibition induces loss of blood-brain barrier integrity by matrix metalloproteinase-dependent and -independent pathways. Brain Res. 2004;995:184–96. doi: 10.1016/j.brainres.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Luabeya MK, Dallasta LM, Achim CL, Pauza CD, Hamilton RL. Blood-brain barrier disruption in simian immunodeficiency virus encephalitis. Neuropathol Appl Neurobiol. 2000;26:454–62. doi: 10.1046/j.1365-2990.2000.00275.x. [DOI] [PubMed] [Google Scholar]
- MacLean AG, Belenchia GE, Bieniemy DN, Moroney-Rasmussen TA, Lackner AA. Simian immunodeficiency virus disrupts extended lengths of the blood--brain barrier. J Med Primatol. 2005;34:237–42. doi: 10.1111/j.1600-0684.2005.00121.x. [DOI] [PubMed] [Google Scholar]
- MacLean AG, Orandle MS, MacKey J, Williams KC, Alvarez X, Lackner AA. Characterization of an in vitro rhesus macaque blood-brain barrier. J Neuroimmunol. 2002;131:98–103. doi: 10.1016/s0165-5728(02)00256-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean AG, Rasmussen TA, Bieniemy D, Lackner AA. Activation of the blood-brain barrier by SIV (simian immunodeficiency virus) requires cell-associated virus and is not restricted to endothelial cell activation. Biochem Soc Trans. 2004a;32:750–2. doi: 10.1042/BST0320750. [DOI] [PubMed] [Google Scholar]
- MacLean AG, Rasmussen TA, Bieniemy DN, Alvarez X, Lackner AA. SIV-induced activation of the blood-brain barrier requires cell-associated virus and is not restricted to endothelial cell activation. J Med Primatol. 2004b;33:236–42. doi: 10.1111/j.1600-0684.2004.00077.x. [DOI] [PubMed] [Google Scholar]
- Mankowski JL, Queen SE, Kirstein LM, Spelman JP, Laterra J, Simpson IA, Adams RJ, Clements JE, Zink MC. Alterations in blood-brain barrier glucose transport in SIV-infected macaques. J Neurovirol. 1999;5:695–702. doi: 10.3109/13550289909021298. [DOI] [PubMed] [Google Scholar]
- Mankowski JL, Spelman JP, Ressetar HG, Strandberg JD, Laterra J, Carter DL, Clements JE, Zink MC. Neurovirulent simian immunodeficiency virus replicates productively in endothelial cells of the central nervous system in vivo and in vitro. J Virol. 1994;68:8202–8. doi: 10.1128/jvi.68.12.8202-8208.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orandle MS, Williams KC, MacLean AG, Westmoreland SV, Lackner AA. Macaques with rapid disease progression and simian immunodeficiency virus encephalitis have a unique cytokine profile in peripheral lymphoid tissues. J Virol. 2001;75:4448–52. doi: 10.1128/JVI.75.9.4448-4452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr AW, Murphy-Ullrich JE. Regulation of endothelial cell function BY FAK and PYK2. Front Biosci. 2004;9:1254–66. doi: 10.2741/1239. [DOI] [PubMed] [Google Scholar]
- Orr AW, Pallero MA, Xiong WC, Murphy-Ullrich JE. Thrombospondin induces RhoA inactivation through FAK-dependent signaling to stimulate focal adhesion disassembly. J Biol Chem. 2004;279:48983–92. doi: 10.1074/jbc.M404881200. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Gendelman HE. Development of laboratory and animal model systems for HIV-1 encephalitis and its associated dementia. J Leukoc Biol. 1997;62:100–6. doi: 10.1002/jlb.62.1.100. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Nottet HS, Sasseville VG, Epstein LG, Gendelman HE. The development of animal model systems for HIV-1 encephalitis and its associated dementia. J Neurovirol. 1995;1:229–43. doi: 10.3109/13550289509114019. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Stins M, Way D, Witte MH, Weinand M, Kim KS, Bock P, Gendelman HE, Fiala M. A model for monocyte migration through the blood-brain barrier during HIV-1 encephalitis. J Immunol. 1997;158:3499–510. [PubMed] [Google Scholar]
- Potula R, Poluektova L, Knipe B, Chrastil J, Heilman D, Dou H, Takikawa O, Munn DH, Gendelman HE, Persidsky Y. Inhibition of indoleamine 2,3-dioxygenase (IDO) enhances elimination of virus-infected macrophages in an animal model of HIV-1 encephalitis. Blood. 2005;106:2382–90. doi: 10.1182/blood-2005-04-1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh G, Philipp MT. Pathogenesis of Lyme neuroborreliosis: mitogen-activated protein kinases Erk1, Erk2, and p38 in the response of astrocytes to Borrelia burgdorferi lipoproteins. Neurosci Lett. 2005;384:112–6. doi: 10.1016/j.neulet.2005.04.069. [DOI] [PubMed] [Google Scholar]
- Retta SF, Barry ST, Critchley DR, Defilippi P, Silengo L, Tarone G. Focal adhesion and stress fiber formation is regulated by tyrosine phosphatase activity. Exp Cell Res. 1996;229:307–17. doi: 10.1006/excr.1996.0376. [DOI] [PubMed] [Google Scholar]
- Ruest PJ, Roy S, Shi E, Mernaugh RL, Hanks SK. Phosphospecific antibodies reveal focal adhesion kinase activation loop phosphorylation in nascent and mature focal adhesions and requirement for the autophosphorylation site. Cell Growth Differ. 2000;11:41–8. [PubMed] [Google Scholar]
- Sasseville VG, Newman W, Brodie SJ, Hesterberg P, Pauley D, Ringler DJ. Monocyte adhesion to endothelium in simian immunodeficiency virus- induced AIDS encephalitis is mediated by vascular cell adhesion molecule-1/alpha 4 beta 1 integrin interactions. Am J Pathol. 1994;144:27–40. [PMC free article] [PubMed] [Google Scholar]
- Sasseville VG, Newman WA, Lackner AA, Smith MO, Lausen NC, Beall D, Ringler DJ. Elevated vascular cell adhesion molecule-1 in AIDS encephalitis induced by simian immunodeficiency virus. Am J Pathol. 1992;141:1021–30. [PMC free article] [PubMed] [Google Scholar]
- Sawada Y, Sheetz MP. Force transduction by Triton cytoskeletons. J Cell Biol. 2002;156:609–15. doi: 10.1083/jcb.200110068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmaj K. Pathophysiology of the blood-brain barrier. Springer Semin Immunopathol. 1996;18:57–73. doi: 10.1007/BF00792609. [DOI] [PubMed] [Google Scholar]
- Shiramizu B, Gartner S, Williams A, Shikuma C, Ratto-Kim S, Watters M, Aguon J, Valcour V. Circulating proviral HIV DNA and HIV-associated dementia. Aids. 2005;19:45–52. doi: 10.1097/00002030-200501030-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staddon JM, Herrenknecht K, Smales C, Rubin LL. Evidence that tyrosine phosphorylation may increase tight junction permeability. J Cell Sci. 1995;108(Pt 2):609–19. doi: 10.1242/jcs.108.2.609. [DOI] [PubMed] [Google Scholar]
- Stamatovic SM, Keep RF, Kunkel SL, Andjelkovic AV. Potential role of MCP-1 in endothelial cell tight junction ‘opening’: signaling via Rho and Rho kinase. J Cell Sci. 2003;116:4615–28. doi: 10.1242/jcs.00755. [DOI] [PubMed] [Google Scholar]
- Stins MF, Pearce D, Di Cello F, Erdreich-Epstein A, Pardo CA, Sik Kim K. Induction of intercellular adhesion molecule-1 on human brain endothelial cells by HIV-1 gp120: role of CD4 and chemokine coreceptors. Lab Invest. 2003;83:1787–98. doi: 10.1097/01.lab.0000107008.13321.c8. [DOI] [PubMed] [Google Scholar]
- Usatyuk PV, Natarajan V. Regulation of Reactive Oxygen Species-Induced Endothelial Cell-Cell and Cell-Matrix Contacts by Focal Adhesion Kinase and Adherens Junction Proteins. Am J Physiol Lung Cell Mol Physiol. 2005 doi: 10.1152/ajplung.00211.2005. [DOI] [PubMed] [Google Scholar]
- Wachtel M, Frei K, Ehler E, Fontana A, Winterhalter K, Gloor SM. Occludin proteolysis and increased permeability in endothelial cells through tyrosine phosphatase inhibition. J Cell Sci. 1999;112(Pt 23):4347–56. doi: 10.1242/jcs.112.23.4347. [DOI] [PubMed] [Google Scholar]
- Westmoreland SV, Halpern E, Lackner AA. Simian immunodeficiency virus encephalitis in rhesus macaques is associated with rapid disease progression. J Neurovirol. 1998;4:260–8. doi: 10.3109/13550289809114527. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Meng FY, Huang Q, Hawker J, Wu HM. Tyrosine phosphorylation of paxillin/pp125FAK and microvascular endothelial barrier function. Am J Physiol. 1998;275:H84–93. doi: 10.1152/ajpheart.1998.275.1.H84. [DOI] [PubMed] [Google Scholar]