

Abstract
The mechanisms by which alcohol consumption accelerates liver disease in patients with chronic hepatitis C virus (HCV) are not well understood. To identify the characteristics of molecular pathways affected by alcohol in HCV patients, we fit probe-set level linear models that included the additive effects as well as the interaction between alcohol and HCV. The study included liver tissue samples from 78 patients, 23 (29.5%) with HCV-cirrhosis, 13 (16.7%) with alcohol-cirrhosis, 23 (29.5%) with HCV/alcohol cirrhosis and 19 (24.4%) with no liver disease (no HCV/no alcohol group). We performed gene-expression profiling by using microarrays. Probe-set expression summaries were calculated by using the robust multiarray average. Probe-set level linear models were fit where probe-set expression was modeled by HCV status, alcohol status, and the interaction between HCV and alcohol. We found that 2172 probe sets (1895 genes) were differentially expressed between HCV cirrhosis versus alcoholic cirrhosis groups. Genes involved in the virus response and the immune response were the more important upregulated genes in HCV cirrhosis. Genes involved in apoptosis regulation were also overexpressed in HCV cirrhosis. Genes of the cytochrome P450 superfamily of enzymes were upregulated in alcoholic cirrhosis, and 1230 probe sets (1051 genes) had a significant interaction estimate. Cell death and cellular growth and proliferation were affected by the interaction between HCV and alcohol. Immune response and response to the virus genes were downregulated in HCV-alcohol interaction (interaction term alcohol*HCV). Alcohol*HCV in the cirrhotic tissues resulted in a strong negative regulation of the apoptosis pattern with concomitant positive regulation of cellular division and proliferation.
INTRODUCTION
Alcoholic liver disease (ALD) and chronic hepatitis C virus (HCV) infection are the most frequent chronic liver diseases in the Western world. In addition, ALD and HCV frequently coexist in the same individual. Although both diseases alone have a similar progression sequence leading to cirrhosis in approximately 15% of patients within 10–20 years, their coexistence dramatically accelerates disease progression in a synergistic manner (1). This synergism affects both fibrosis progression and the development of hepatocellular carcinoma. The relationship between ALD and HCV was first described in populations of alcoholic individuals with liver disease (2) and confirmed later by investigators who compared the prevalence of HCV markers in alcoholic individuals with and without liver disease and found a significantly higher prevalence in those with liver disease (2–4).
Even though the coexistence of HCV and alcoholism has been associated with accelerated hepatic injury, the pathogenesis is not fully understood but is suspected to be multifactorial (1–4). Liver biopsy samples in HCV-infected patients with reported alcohol consumption characteristically show a pattern of hepatic injury consistent with chronic viral hepatitis, indicating that the alcohol plays a role in potentiating the effects of HCV, rather than causing traditional alcohol-related liver injury (5,6). The use of alcohol has been found to be associated with immune control of HCV and to affect HCV replication and/or viral clearance. This final effect may have an impact on the evolution of HCV quasispecies, presumably through its effects on the immune system (7,8).
DNA microarray studies offer a robust method for unbiased analysis of whole-genome messenger RNA (mRNA) expression patterns. Expression profiling with DNA microarrays has been used to identify molecular network responses to ethanol in cell culture (10,11), animal models (9–12), and humans (13,14). Expression profiling has led to the identification of novel gene networks that respond to ethanol or differ across animal strains with differing responses to ethanol. In similar research, many studies have been performed to evaluate the gene expression patterns associated with chronic HCV infection (15–17).
In the present study, to identify molecular pathways affected by the addition of alcohol in HCV, we modeled gene expression, including alcoholic cirrhosis and HCV cirrhosis as separate conditions and their interaction term (alcohol*HCV). A better understanding of the underlying molecular mechanisms of alcohol-HCV interaction could help investigators to develop novel targeted treatment options.
PATIENTS AND METHODS
Patients and Samples
In this study we evaluated 78 liver samples, including 23 (29.5%) from patients with cirrhosis due to HCV infection, 13 (16.7%) from patients with cirrhosis due to alcohol, and 23 (29.5%) from patients with cirrhosis due to both HCV and alcohol. In addition, 19 (24.4%) of the liver samples were from donors with normal livers and were included in the study as a group with neither HCV-nor alcohol-associated liver damage. Liver function and histopathology for the samples from the healthy liver donors were shown to be normal. All 19 of these donors were seronegative for HCV antibodies. Characteristics of the patients who donated liver tissue are shown in Supplemental Table 1.
Table 1.
Summary of the more important findings for each analysis.a
| Alcohol cirrhotic tissues versus normal livers | HCV cirrhotic tissues versus normal livers | HCV versus alcohol cirrhotic tissues | Significant interaction analysis | |
|---|---|---|---|---|
| Number of differentially expressed probe sets/genes | 1406/1272 | 2222/1841 | 2172/1896 | 1230/1051 |
| Associated Network Functions (five top-scored networks) |
|
|
|
|
| Top molecular and cellular functions | Cellular growth and proliferation (P 1.47E-04 to 4.24E-02), 314 genes; Gene expression (P 1.92E-04 to 3.81E-02), 236 genes; Cell Cycle (P 4.81E-04 to 4.42E-02) 148 genes; Amino acid metabolism (P 7.68E-04 to 4.51E-02), 54 genes; Posttranslational modification (P 7.68E-04 to 4.38E-02), 135 genes |
Cellular growth and proliferation (P 1.12E-13 to 4.03E-03), 402 genes; Cell death (P 2.80E-13 to 3.90E-03), 361 genes; Cellular movement (P 2.88E-12 to 3.83E-03), 211 genes; Cellular development (P 1.76E-09 to 3.76E-03), 260 genes; Cell-to-cell signaling and interaction (P 6.38E-08 to 4.26E-03), 248 genes; |
Gene expression (P 1.49E-09 to 1.21E-02), 285 genes; RNA posttranscriptional modification; (P 2.50E-09 to 7.19E-05), 51 genes; Cell death (P 1.35E-07 to 1.21E-02), 326 genes; Cellular growth and proliferation (P 2.20E-07 to 1.21E-02), 354 genes; Protein synthesis (P 2.47E-06 to 5.74E-03), 107 genes |
Cell death (P 1.00E-08 to 2.19E-02), 228 genes; Cellular growth and proliferation (P 8.57E-08 to 2.12E-02), 267 genes; Cellular compromise (P 2.88E-05 to 1.17E-02), 14 genes; Cell morphology (P 3.36E-05 to 2.21E-02), 144 genes; Cell-to-cell signaling and interaction (P 6.79E-05 to 1.63E-02) 96 genes |
| Top canonical pathways | JAK/Stat signaling (P = 5.39E-04), Protein ubiquitination pathway (P = 8.22E-04), VEGF signaling (P = 1.06E-03) |
Hepatic fibrosis/hepatic stellate cell activation (P = 9.17E-06), Phospholipase C signaling (P = 1.62E-05), G-protein–coupled receptor signaling (P = 3.46E-05), Thrombin signaling (P = 4.21E-05) |
Death-receptor signaling (P = 1.12E-04), ILK signaling (P = 1.82E-04), B-cell–receptor signaling (P = 6.3E-04) |
Antigen-presentation pathway (P = 7.92E-07), IL-4 signaling (P = 6.92E-03), Riboflavin metabolism (P = 1.27E-02), Hepatic fibrosis/hepatic stellate cell activation (P = 1.32E-02 ) coagulation system (P = 2.19E-02) |
| Top overexpressed genes (fold changes) |
NTN3 (19.62) IGKC (11.95) IGL@ (10.56) HLA-C (7.08) IGHM (6.8) HBB (includes EG:3043) (6.65) ZCWPW2 (includes EG:152098) (5.73) MAFF (5.51) IL8 (5.4) HBA1 (5.26) |
IFI27 (21.72) NTN3 (19.62) IGKC (16.04) CXCL10 (10.89) IGL@ (10.56) HLA-DQB1 (9.98) IFIT1 (9.07) AKR1B10 (8.09) HBB (includes EG:3043) (8.01) IGHM (7.68) |
IFI27 (11.7) ISG15 (5.73) CXCL10 (5.68) IFIT1 (5.13) MX1 (4.09) HSP90B1 (4.06) ID1 (3.73) ISG20 (3.66) IFI44 (3.33) MCL1 (3.27) |
Not applicable |
| Top underexpressed genes (fold changes) |
S100A8 (−5.57) FCN2 (−3.58) RFC5 (−3.54) SAA2 (−3.47) TFPI (−3.30) ST6GAL1 (−3.1) KCNN2 (−3.1) ABCA1 (−2.99) TGM2 (−2.97) LMAN1 (−2.95) |
LPA (−4.5) ATF5 (−4.49) STEAP1 (−3.8) PZP (−3.63) SAA2 (−3.47) MT1M (−3.45) TFPI (−3.3) DHODH (−3.3) SLCO4C1 (−3.2) KCNN2 (−3.1) |
BBOX1 (−4.75) ATP8B1 (−4.0) SLC35E1 (−2.55) PRG2 (includes EG:79948) (−2.46) RBM25 (includes EG:58517) (−2.41) DBP (−2.39) ZC3H7Ba (−2.37) POLR1B (−2.26) UBB (−2.21) |
Not applicable |
Analysis performed by using Ingenuity Pathways Analysis tools 7.0 (http://www.ingenuity.com).
None of the HCV-infected patients in this study were receiving antiviral treatment at the time the liver sample was obtained. Pretransplantation alcohol intake was assessed by the attending physician at the time of evaluation for liver transplantation. Significant alcohol consumption was defined as >50 g/d for a period of >5 years or >80 g/d for a shorter period. This information was obtained from chart review and patient questionnaire.
The research protocol was approved by the institutional review board, and informed consent was obtained from all patients. Normal livers and cirrhotic liver samples were obtained through the Liver Tissue Cell Distribution System, Richmond, VA, USA, which was funded by NIH Contract #N01-DK-7-0004/HHSN267200700004C.
Sample Collection and Pathological Data
At the time of collection, liver tissue samples were placed in liquid nitrogen or RNA-later solution (Ambion, Austin, TX, USA) and stored at − 80°C until use. The cirrhotic tissues from explanted livers were classified by using Knodell score and Ishak grade (18).
RNA Isolation, cDNA Synthesis and in vitro Transcription for Labeled cRNA Probe
The sample preparation protocol was performed according to the Affymetrix GeneChip® Expression Analysis Manual (Santa Clara, CA, USA). Briefly, total RNA was reverse transcribed by using T7-polydT primer and converted into double-stranded cDNA (One-Cycle Target Labeling and Control Reagents, Affymetrix, Santa Clara, CA, USA), with templates being used for an in vitro transcription reaction to yield biotin-labeled antisense cRNA. According to the Affymetrix GeneChip protocol, the labeled cRNA was chemically fragmented and made into the hybridization cocktail, which was then hybridized to HG-U133A 2.0 GeneChips. The array image was generated by the high-resolution GeneChip® Scanner 3000 by Affymetrix. The data were analyzed by using Affymetrix Microarray Suite Software, Version 5.0, and Bioconductor packages (19) available in the R programming environment (20).
Microarray Data Analysis
After image analysis, probe-set expression summaries were calculated by using the robust multiarray average.
We conducted an unsupervised analysis by first filtering the data set by retaining only the most variable probe sets, for which the range of each probe set was calculated and probe sets having a range among the top 1% were retained. Thereafter, using the Ward method we applied agglomerative hierarchical clustering to the filtered data set using the 1-Pearson correlation as the dissimilarity measure.
For each probe set, we compared HCV cirrhotic liver tissue versus normal liver tissue and alcohol-cirrhotic tissue versus normal liver tissue using a two-sample t test with the Satterwaite approximation to the degrees of freedom to account for unequal variances. Thereafter, probe-set level linear models were fit such that probe-set expression was modeled by HCV status, alcohol (EtOH) status and the interaction between HCV and alcohol. Specifically, the model was taken to be:
where yij represents the log2 intensity of probe set j for subject i, μj represents the baseline log2 intensity for probe set j, HCVi is a dichotomous variable representing the HCV status of subject i and β1j represents the effect due to HCV status on probe set j. EtOHi is a dichotomous variable representing the alcohol status of subject i and β2j represents the effect due to alcohol status on probe set j;β3j represents the effect of the combination of HCV and alcohol on probe set j, and eij represents the random error term. Of particular interest were those probe sets for which the β3j term was significant, indicating that the effect of the combination of HCV and alcohol on gene expression was not simply additive, but either synergistic or antagonistic. To control for multiple hypothesis tests, a Bonferroni correction was used for all comparisons.
Interaction Networks and Functional Analysis
Gene ontology and gene interaction analyses were executed by using Ingenuity Pathways Analysis tools 7.0 (http://www.ingenuity.com) (Ingenuity Systems, Redwood City, CA, USA).
Validation of Microarray Results
We carried out a quantitative reverse-transcriptase–real-time PCR (QPCR) for caspase 1, apoptosis-related cysteine peptidase (CASP1), interleukin 8 (IL-8), IL-15, platelet derived growth factor C (PDGFC), myxovirus (influenza virus) resistance 1 (Mx1), and (transforming growth factor [TGF]-β1) mRNAs from the same RNA samples that were subjected to microarray study. We performed reverse transcription of total RNA using TaqMan® Reverse Transcription Reagents (Applied Biosystems, Foster City, CA) according to the manufacturer’s protocol. Real-time PCR reactions then were carried out in an ABI Prism 7700 Sequence Detection System, using TaqMan® Gene Expression Assays (Applied Biosystems). Data was analyzed according to the comparative cycle threshold method and was normalized with a housekeeping gene (β2-microglobulin). The Pearson correlation coefficient (r) was calculated to examine the relation between microarray and real-time PCR results. P values < 0.05 were considered significant.
All supplementary materials are available online at www.molmed.org.
RESULTS
Normal Liver Selection and Analysis
The selection of the normal liver group was critical for the analysis. We studied gene expression profiles using high-density oligonucleotide arrays in 19 donor liver samples. To identify probe sets significantly correlated with donor age, we fit probe-set–specific linear models that included age as the independent variable and obtained the significance of the estimated slope parameter. Probe sets with a raw P value < 0.001 were considered significant. The false-discovery rate (FDR) method of Benjamini and Hochberg was used to adjust for multiple comparisons. Donor liver groups were classified by age as <25, 25–50 and >50 years, and a Jonckheere-Terpstra test for trend was applied to identify probe sets with a significant monotonic trend with age. For the trend tests, the FDR was estimated by using the q-value method (Storey and Tibshirani). For each probe set a two-sample t test assuming unequal variances was used to compare normal livers with respect to race (white versus African American), sex and cause of death (stroke/cerebrovascular accident [CVA] versus trauma). The FDR was estimated by using the q-value method (Storey and Tibshirani). Donor characteristics included median age 41 (range 9–66) years, 68.4% male and 73% white. Causes of donor death were stroke/CVA (n = 7), trauma (n = 9) and other (n = 3). With the use of a raw P value of < 0.001, 21 probe sets were significantly associated with age. When we examined probe-set expression values with a significant monotonic increasing and decreasing trend with age (P < 0.001), 11 probe sets were significantly increasing and 11 probe sets were significantly decreasing. For the tests of significance that compared cause of death due to stroke/CVA to trauma, only 10 probe sets were significant at the 0.001 level. Fifty-six probe sets were significantly different between males and females, with the probe sets with very small P values and FDR being located on the x or y chromosome as expected. However, 288 probe sets were significantly different for race comparison (white versus African American) (P < 0.001). After performing these analyses and recognizing a high FDR was associated with the probe sets identified in most of these analyses, we concluded that race was the more critical variable for which to control, with respect to normal tissues. Therefore we matched the race distribution between our control and study groups.
Cluster Analysis
For the unsupervised analysis, after we applied our variance-based filter the data set consisted of 223 probe sets. The heatmap resulting from the agglomerative hierarchical clustering procedure is shown in Figure 1. The unsupervised analysis showed that all the normal liver samples clustered together. Moreover, we also observed that the samples with alcoholic cirrhosis tended to cluster together in the same group whereas the tissue samples with HCV-cirrhosis and those with the combination of HCV and alcohol clustered together in a different group. These findings might indicate that the dominance of the HCV-cirrhotic gene expression pattern in those cirrhotic tissues was associated with the interaction of HCV and alcohol.
Figure 1.

Unsupervised cluster analysis. An unsupervised analysis was conducted by first filtering the data set by retaining only the most variable probe sets, for which the range of each probe set was calculated and probe sets having a range among the top 1% were retained. Thereafter, agglomerative hierarchical clustering performed using the Ward method was applied to the filtered data set with the 1-Pearson correlation as the dissimilarity measure. Blue light boxes, normal liver tissues; black boxes, alcohol-cirrhotic liver tissues; pink boxes, HCV-cirrhotic liver tissues; green boxes, alcohol-HCV cirrhotic liver tissues.
Molecular Pathways Involved in Alcohol-Cirrhosis and HCV-Cirrhosis
By comparing signatures of normal liver tissue and tissue affected by other liver diseases, we were able to exploit the molecular profiles of particular disease processes. Specifically, 2565 probe sets were significantly differentially expressed when we compared alcoholic cirrhotic versus normal liver tissues. Furthermore, 3370 probe sets were significantly differentially expressed when we compared HCV versus normal liver, with 916 probe sets being significant in both comparisons (Figure 2). Because main effects cannot be directly interpreted when interaction is present, only the 1406 alcohol-specific probe sets (corresponding to 1272 unique genes) and the 2222 HCV-specific probe sets (corresponding to 1,841 unique genes) were further examined by using Ingenuity Pathway Analysis.
Figure 2.
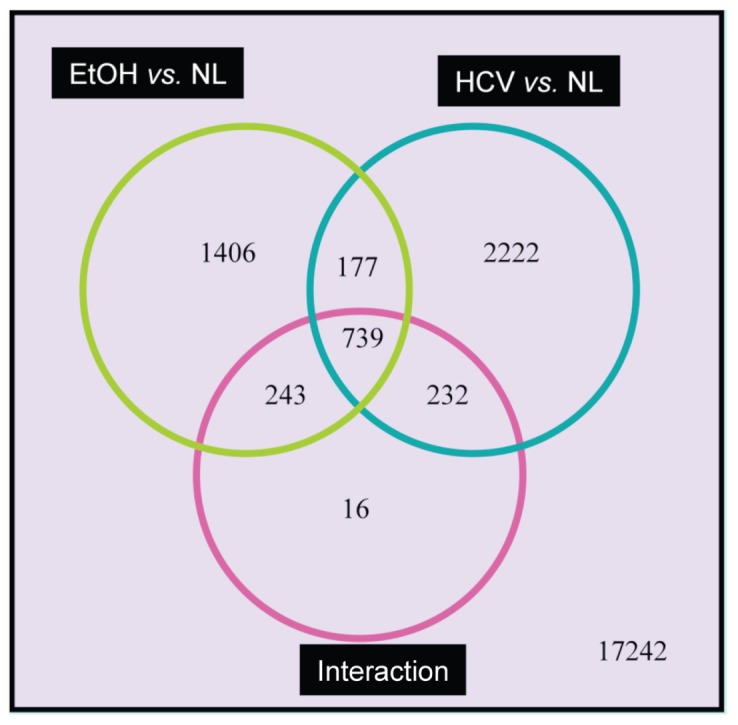
Venn diagram. For each sample, RNA was extracted and after being converted to biotin-labeled cRNA was hybridized to an Affymetrix HG-U133Av2 GeneChip. After image analysis, probe-set expression summaries were calculated by using the robust multiarray average. Thereafter, probe-set–level linear models were fit such that probe-set expression was modeled by HCV status, alcohol status, and the interaction between HCV and alcohol. Of particular interest were those probe sets for which all three terms (HCV, Alcohol, and HCV*Alcohol) were significant. To control for multiple hypothesis tests, probe sets were significant at an α level of 0.0001.
Core analysis was performed to interpret the data set in the context of biological processes, pathways and molecular networks. From the analysis of the differentially expressed genes in alcoholic cirrhotic samples compared with normal livers, 100 networks were identified (46 networks with a score higher than 15). The top canonical pathways associated with these genes included pyrimidine metabolism, NF-kB signaling, CD28 signaling in T-helper cells, and glycolisis/gluconeogenesis. The most overexpressed genes included genes involved in antigen presentation (IGKC, HLA-C and IGHM) and inflammatory response (IL8). IL-8, a cytokine produced by a host of cells, including monocytes, macrophages, Kupffer cells and hepatocytes, can activate neutrophils. Peripheral neutrophilia and liver neutrophil infiltration are frequently noted in patients with alcoholic liver disease. However, the relationship between IL-8 and different stages of alcoholic liver disease is uncertain. Genes involved in the immune response were downregulated in alcoholic cirrhosis (IRAK1, IL17RA, IL13RA1, LRIG1 and IKBKG, among others). In a recent study, Lemmers et al. (22) studied the IL-17 pathway in alcoholic cirrhosis and alcoholic hepatitis. In the present study, IL-17B gene expression was upregulated in alcoholic cirrhotic tissues. The protein encoded by this gene is a T-cell–derived cytokine that shares sequence similarity with IL-17. This cytokine was reported to stimulate the release of tumor necrosis factor-α (TNF-α) and IL-1 β from a monocytic cell line.
Growth factors were overexpressed in alcoholic cirrhotic tissues compared with normal liver tissues. Also overexpressed in alcoholic cirrhosis were CD96 (the a type I membrane protein encoded by this gene belongs to the immunoglobulin superfamily and may play a role in the adhesive interactions of activated T and NK cells during the late phase of the immune response) and CD7 (the protein encoded by this gene is found on thymocytes and mature T cells).
Moreover, ALDH3B1 (aldehyde dehydrogenase 3 family, member B1), was also overexpressed in alcoholic cirrhrotic tissues. The aldehyde dehydrogenases are a family of isozymes that may play a major role in the detoxification of aldehydes generated by alcohol metabolism and lipid peroxidation.
From the analysis of the differentially expressed genes in HCV cirrhotic tissues compared with normal livers, we identified 94 networks (45 with a score higher than 15). The top-scoring network is shown in Figure 3. Cell-to-cell signaling and interaction and cellular development were identified as the principal molecular and cellular functions associated with these genes.
Figure 3.

The top-scoring network of interactions among the 3370 probe sets identified as significantly differentially expressed when we compared HCV with normal samples. The probe sets were subsequently analyzed by using the Ingenuity pathway analysis software (https://analysis.ingenuity.com). This software is designed to identify dynamically generated biological networks, global canonical pathways and global functions. At interconnections of significant functional networks, protein nodes appeared in different shades of red and green or white depending on being upregulated and downregulated or no-change, respectively, in HCV-cirrhotic samples.
A characteristic pattern was observed in HCV cirrhotic tissues showing overexpression of interferon-inducible genes (IFITM1, IFI27 and ISG20, among others) and genes involved in viral response (MX1, PRKRA and IL18, among other). Also, genes related to immune response emerged as important in HCV cirrhotic tissues (IL15, IL8, IL10RA, IL18, FCGR2A and CD48, among others). Specifically, genes related to natural killer cells were upregulated, including IL15, CCL5, KLRC4, KLRB1, KIR2DL3 and CD44. The protein encoded by the IL15 gene is a cytokine that regulates activation and proliferation of T cells and natural killer cells. Moreover, IL-15 signaling was identified as one of the top canonical pathways (P = 0.027).
We also observed overexpression of the gene that encodes IL-18, an interferon γ–inducing factor that plays a critical role in the T-helper type 1 response required for host defense against viruses. Antibodies to IL-18 have been found to prevent liver damage in a murine model (23).
Another important finding was that chemokine genes were upregulated in this group (CCL21, CCL4, CCL5, CXCL9 and CXCL10). Moreover, genes involved in the cell-mediated immune response as granzyme A and granzyme B were up-regulated. The proteins encoded by these genes are crucial for the rapid induction of target-cell apoptosis by cytolytic T lymphocytes (CTL) in cell-mediated immune responses.
Genes Differentially Expressed in Alcoholic Cirrhosis versus HCV Cirrhosis
We found 2172 probe sets (corresponding to 1896 unique genes) that were differentially expressed. This finding was revealed by the comparison analysis of 23 tissues with HCV cirrhosis versus 13 tissues with alcoholic cirrhosis performed by using a two-sample t test with a Bonferroni correction (P < 0.05/22,277 considered significant). From this analysis, 89 networks were identified (39 with a score higher than 15). RNA posttranscriptional modifications (P = 6.7E-08 to 3.9E-03, 42 genes); cell death (P = 1.2E-06 to 5.2E-03, 348 genes); cellular development (P = 8.5E-06 to 5.5E-03), gene expression, and cellular growth and proliferation were the principally associated molecular and cellular functions.
Interferon and interferon-inducible genes were upregulated in HCV cirrhotic tissues compared with alcoholic cirrhotic tissues (MX1, IFI27, PRKRA, IFI 44, ISG20 and IFI16, among others). The gene expression levels of the growth factors TGF-β1, VEGFA and PDGFC were also upregulated in HCV cirrhotic tissues. Moreover, the expression of IL-8 and IL-15 was increased in these tissues.
Gene symbols for all Affymetrix probe sets were obtained using the annotate Bioconductor package. Thereafter, all probe sets annotated to interrogate specific genes in the pathways of interest (response to virus, immune and inflammatory response, synthesis of cholesterol steroids and other lipids, solute transporters, growth factors and growth factor receptors and apoptosis) were retained, and agglomerative hierarchical clustering using the Ward method with 1-Pearson (1 minus Pearson correlation) as the dissimilarity measure was applied to each pathway. The resulting heatmap and dendrograms for the specific pathway related with response to the virus are shown in Figure 4. Once again, the gene expression pattern of HCV cirrhotic tissues is more similar to the profiles of tissues with cirrhosis due to both HCV and alcohol.
Figure 4.

Supervised cluster analysis for response to virus genes. Gene symbols for all Affymetrix probe sets were obtained by using the Bioconductor annotation package. Thereafter, all probe sets annotated to interrogate specific genes in the pathways of interest (response to virus) were retained, and agglomerative hierarchical clustering was performed by using the Ward method with 1-Pearson as the dissimilarity measure was applied to the pathway. Blue light boxes, normal liver tissues; black boxes, alcohol-cirrhotic liver tissues; pink boxes, HCV-cirrhotic liver tissues; green boxes, alcohol-HCV cirrhotic liver tissues.
Interaction between HCV and Ethanol
It was of great interest to study the interaction between ethanol and HCV in the cirrhotic tissues. Probe-set–level linear models were fit such that probe-set expression was modeled by HCV status, alcohol status, and the interaction between HCV and alcohol. Among the 22,277 probe-set–specific linear models, 1230 probe sets (corresponding to 1051 unique genes) had a significant interaction. The estimates of the interaction term, which provides information on the expected change in gene expression when both HCV and alcohol are causative factors of cirrhosis, are reported in Supplemental Table 2.
The top molecular and cellular functions affected by the interaction between HCV and alcohol were cell death and cellular growth and proliferation. The top canonical pathways for those genes were the antigen-presentation pathway (P = 0.0079), IL-4 signaling (P = 0.0069), hepatic fibrosis/hepatic stellate cell activation (P = 0.013) and the coagulation system (P = 0.02).
A substantial number of genes involved in apoptosis were present in the interaction analysis (TRADD, CASP1, AVEN, BAD, TNFRSF21, BCL2 and BAK1, among others). For some of the genes involved in apoptosis that were affected by the interaction between HCV and alcohol, interaction plots were constructed to enable visualization of the expression changes based on the HCV status (HCV positive versus negative) or alcohol status (yes versus no) (Figure 5).
Figure 5.

Significant interactions. For CASP1, BCL2, TRADD and BAD genes, interaction plots were constructed to enable visualization of the expression changes based on the HCV status (HCV positive versus negative) or alcohol status (yes versus no). In each plot one line represents alcohol = yes (solid line) and the other line represents alcohol = no (dashed line). Each line is formed by connecting two points: the group mean expression for HCV-negative patients to the group mean expression for HCV-positive patients.
Among the genes having a significant interaction, genes involved in immune response had a negative estimate for the interaction term. These genes included genes associated with immunoglobulins (IGBP1, IGL, IGKC), chemokines (CXCL12, CXCL6, CXCR4, CXCR6), integrins (ITGBL1) and immune cell receptors (TRA@, TRBC1, TICAM2). Also, a negative estimate for the interaction term was associated with matrix metal-loproteinase (MMP) genes (MMP7, MMP2), genes involved in cell-to-cell and cell-to-matrix interactions (THBS1, TIMP3, BGN, DCN, COL6A3, COL1A2, among others).
Interestingly, IFNAR1 and IRF2 genes had a significant positive interaction. The protein encoded by this gene is a type I membrane protein that forms one of the two chains of a receptor for interferons α and β. Binding and activation of the receptor stimulates Janus protein kinases, which in turn phosphorylate several proteins, including STAT1 and STAT2. The encoded protein also functions as an antiviral factor. IRF2 encodes interferon regulatory factor 2, a member of the interferon regulatory transcription factor (IRF) family. IRF2 competitively inhibits the IRF1-mediated transcriptional activation of interferons (α and β), and presumably other genes that employ IRF1 for transcription activation. Table 1 summarizes the more important findings for each of the described tissue comparison analysis.
Gene Expression Quantitation by Using QPCR
We used two criteria to select the genes for the QPCR microarray assay: the genes were in the top of the differentially expressed genes and/or the genes were among the top scoring networks and pathways when we used interaction networks and functional analysis for the different comparison analyses.
Expression levels of the CASP1, IL-8, IL-15, PDGFC, MX1, and TGF-β1 mRNAs were further confirmed by using QPCR. These genes were identified as significantly differentially expressed between different tissues types. The results from the microarray were reproduced by QPCR (r = 0.82, P < 0.001; r = 0.71, P = 0.002; r = 0.65, P < 0.001; r = 0.7, P < 0.001; r = 0.73, P < 0.001; and r = 0.61 P < 0.001 respectively).
DISCUSSION
The underlying molecular mechanisms of alcohol and HCV-mediated liver disease are complex and they are still incompletely understood despite decades of intensive investigative efforts. Both alcohol and HCV can reproduce the four sequential hallmarks of liver disease: steatosis, steatohepatitis, fibrosis and hepatocellular carcinoma (1).
In the present study, characteristic gene expression patterns were observed in HCV cirrhosis and alcoholic cirrhosis. Comparisons between normal liver tissues and HCV-cirrhotic and alcohol- cirrhotic liver tissues showed the principal pathways and molecular and cellular functions affected in each condition. Also, a direct comparison analysis was performed between the cirrhotic tissues with the two different etiologies. Moreover, the direct effect of the interaction between alcohol and HCV in the liver tissue permitted the identification of genes synergistically and antagonistically affected by the combination of alcohol and HCV.
The liver is the primary site of alcohol metabolism, and therefore is vulnerable to severe, chronic injury. Chronic heavy alcohol consumption damages the liver (as manifested by fatty liver, steatosis, fibrosis, cirrhosis and hepatic cancer) and impairs the liver’s ability to regenerate (1–4). Fibrosis of the liver is a wound-healing process that occurs in response to persistent hepatocellular injury. Although the major mechanisms of fibrogenesis are independent of the origin of liver injury, alcoholic liver fibrosis features several special characteristics. An important aspect in alcoholic liver disease is the pronounced inflammatory response of Kupffer cells and other types of leukocytes (macrophages, neutrophils, lymphocytes).
All mature liver cells, including hepatocytes, Kupffer cells, hepatic stellate cells and endothelial cells, as well as oval stem cells, have been implicated in alcohol-induced liver damage and impaired regeneration. Acetaldehyde, an immediate metabolite of ethanol, is primarily produced in hepatocytes and can activate hepatic stellate cells (HSC) in a paracrine manner. Activation of HSC is the primary event that triggers the process of fibrogenesis. Kupffer cells have been implicated as mediators of alcoholic liver injury through the release of free radicals as well as generation of inflammatory and fibrogenic mediators in response to alcohol and lipopolysaccharide (1–4). In the present study, the top overexpressed genes that were identified as associated with alcoholic cirrhosis compared with normal livers included genes involved in antigen presentation, inflammatory response and growth factors.
Chronic ethanol intake is also associated with accelerated liver damage when it coexists with conditions such as HCV infection. Reported data suggest that HCV is not directly cytopathic to the hepatocyte, and the main focus of pathology of HCV infection has been on inflammation from the immune response as the main contributor of reactive oxygen species from macrophages and neutrophils (24). Alcohol is considered an accelerant of illness in individuals infected with HCV (25,26). There is a higher rate of HCV infection among alcoholics than the general population and there is a 40% increased risk of morality in alcohol abusers with HCV (27).
Global gene expression profiling of liver tissue revealed characteristic portraits in HCV and ethanol-induced cirrhosis. In general, gene expression changes in cirrhotic livers were more relevant in livers with HCV infection than in those with alcohol-induced disease when both were compared with normal livers. Lederer et al. (28), in a recent study, performed global transcriptional profiling using oligonucleotide microarrays on liver biopsy samples from patients with cirrhosis caused by either chronic alcohol consumption or HCV infection. As the authors discussed in the article, this was a small cross-sectional study, but the gene expression profiles in cirrhotic livers from alcoholic liver disease were distinct from the gene expression profiles of HCV-induced cirrhosis. These results are in concordance with our findings. Moreover, in HCV-cirrhotic tissues, we observed upregulation of genes that might reflect the antiviral response of liver hepatocytes induced by chronic HCV infection. Previous studies (15,17) showed overexpression of several of these genes, including IFI27, ISG15, MX1, IL8 and IFIT1.
In the present study we included an important number of samples per group and moreover, we studied the interaction of HCV and alcohol in cirrhosis induction. Little is known about the combined effects of ethanol and HCV on the pathogenesis of liver disease. Proposed mechanisms of interaction include enhancement of viral replication and increased oxidative stress and cytotoxicity, as well as impairment of immune response (29).
Both HCV and alcohol are known stimuli of hepatic oxidative stress and lipid peroxidation, suggesting that coexistence of these factors might enhance these pathways (30), thereby leading to increased activation of liver fibrogenic cells and subsequent acceleration of fibrogenesis. Thus, chronic administration of alcohol to HCV-core transgenic mice resulted in additive hepatic lipid peroxidation, synergistic induction of the profibrogenic cytokine TGF-β1 and activation of HSC (31). Experimental data suggest that alcohol-induced impairment of immunity may account for the high rates of persistent viral infection reported in excessive drinkers (32).
In the present study, we observed that the interaction of HCV and alcohol was characterized by downregulation of many genes involved in the immune response. Acute alcohol ingestion results in generalized suppression of the immune system (33). The antigen-specific CTL that are produced in response to HCV infection migrate to the liver, where they cause apoptosis of infected hepatocytes via interaction of Fas ligand expressed on activated CTL and Fas on hepatocytes (34,35), and this action may be enhanced by alcohol (36).
According to our analysis, the top canonical pathways affected for interaction of HCV and ethanol were antigen presentation, IL-4 signaling, hepatic fibrosis/HSC activation, and coagulation. Evidently, fibrosis is exacerbated from the interaction of both etiologies. Specific analysis of the hepatic fibrosis/HSC activation canonical pathway revealed that the principal genes were BAX, BCL2, COL1A2, CTGF, FGFR1, FGR2, MMP2 and PDGFRB, among others.
Connective tissue growth factor (CTGF = CCN2), has been recognized as an important player in fibrogenic pathways on the basis of findings in non-hepatic tissues and emerging results from liver fibrosis. By changing the activity ratio of TGF-β to its antagonist, bone-morphogenetic protein 7, CTGF has been proposed as a fibrogenic master switch for epithelial-mesenchymal transition (37).
HSC are known to synthesize the excess matrix that characterizes liver fibrosis and cirrhosis. Activated HSC express the matrix-degrading MMP enzymes and the tissue inhibitors of metalloproteinases (TIMPs). During spontaneous recovery from experimental liver fibrosis, the expression of TIMP-1 declines and hepatic collagenolytic activity increases. This process is accompanied by HSC apoptosis. In a recent publication, Hartland et al. (38) reported a role for both N-cadherin and MMP-2 in mediating HSC apoptosis, in which N-cadherin works to provide cell-survival stimulus and MMP-2 promotes HSC apoptosis concomitant with N-cadherin degradation. In the present study, MMP2 and CDH1 were negatively affected by the interaction of HCV and alcohol as well as macrophage-derived MMP-7 and TIMP3.
In a recent publication, Kendall et al. (39) suggested that cleavage of proNGF by MMP7 during the early phase of recovery from liver fibrosis alters the pro/mature NGF balance to facilitate hepatic myofibroblast loss. Whereas fibrosis develops in the absence of p75(NTR) signaling, the dominant effects of loss of p75(NTR) ligand–mediated events are the retardation of liver fibrosis resolution via regulation of hepatic myofibroblast proliferation and apoptosis, and the reduction of hepatocyte- and oval-cell proliferation.
Apoptosis was also affected by the interaction, because it was well reflected for the negative effect in CASP1 by the interaction and it was overexpressed in HCV cirrhotic tissues compared with both normal and alcoholic cirrhotic liver tissue. Similarly, TNFRSF21 and BCL2 were negatively affected for the interaction.
We are aware of the limitation that cellular heterogeneity of tissue samples introduces to the analysis. Liver tissue samples used for microarray analysis contained a mixture of different cells and cell infiltrates. Thus, with the exception of cell-specific genes (for example, E-selectin), the source of mRNA was unknown, and this limited our ability to interpret the cellular signatures. However, in this study we aimed to evaluate the global differences that characterize cirrhosis related to alcohol versus HCV, and moreover, how the interaction of both etiologies affects the overall molecular pathways.
In conclusion, according to our results, the characteristic profiles of the tissues under the additive/synergic effect of HCV and alcohol were characterized by an increase in fibrosis development and apoptosis inhibition and a decrease in immune response. These initial findings will contribute to further studies to distinguish which pathways would be better targets for new therapeutic alternatives for end-stage liver disease.
Supplemental Data
ACKNOWLEDGMENTS
This project was partially supported by the Commonwealth Foundation for Cancer Research as a pilot project of the Massey Cancer Center.
Footnotes
This work has been presented in part at the American Transplant Congress meeting, Boston, Massachusetts, 2009.
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Online address: http://www.molmed.org
REFERENCES
- 1.Mueller S, Millonig G, Seitz HK. Alcoholic liver disease and hepatitis C: a frequently underestimated combination. World J. Gastroenterol. 2009;15:3462–71. doi: 10.3748/wjg.15.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singal AK, Anand BS. Mechanisms of synergy between alcohol and hepatitis C virus. J Clin. Gastroenterol. 2007;41:761–72. doi: 10.1097/MCG.0b013e3180381584. [DOI] [PubMed] [Google Scholar]
- 3.Bhattacharya R, Shuhart MC. Hepatitis C and alcohol: interactions, outcomes, and implications. J. Clin. Gastroenterol. 2003;36:242–52. doi: 10.1097/00004836-200303000-00012. [DOI] [PubMed] [Google Scholar]
- 4.Gitto S, Micco L, Conti F, Andreone P, Bernardi M. Alcohol and viral hepatitis: a mini-review. Dig. Liver Dis. 2009;41:67–70. doi: 10.1016/j.dld.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 5.Anderson S, Nevins CL, Green LK, El-Zimaity H, Anand BS. Assessment of liver histology in chronic alcoholics with and without hepatitis C virus infection. Dig. Dis. Sci. 2001;46:1393–98. doi: 10.1023/a:1010671300507. [DOI] [PubMed] [Google Scholar]
- 6.Tamai T, et al. Effects of alcohol consumption on histological changes in chronic hepatitis C: a clinicopathological study. Alcohol Clin. Exp. Res. 2000;24:106S–11S. [PubMed] [Google Scholar]
- 7.Tanaka T, et al. Contribution of hepatitis C virus to the progression of alcoholic liver disease. Alcohol Clin. Exp. Res. 2. 2000;4:112S–116S. [PubMed] [Google Scholar]
- 8.Anand BS, Velez M. Influence of chronic alcohol abuse on hepatitis C virus replication. Dig. Dis. 2000;18:168–71. doi: 10.1159/000051390. [DOI] [PubMed] [Google Scholar]
- 9.Thibault C, et al. Expression profiling of neural cells reveals specific patterns of ethanol-responsive gene expression. Mol. Pharmacol. 2000;58:1593–600. doi: 10.1124/mol.58.6.1593. [DOI] [PubMed] [Google Scholar]
- 10.Hassan S, Duong B, Kim KS, Miles MF. Pharmacogenomic analysis of mechanisms mediating ethanol regulation of dopamine beta-hydroxylase. J. Biol. Chem. 2003;278:29960–9. doi: 10.1074/jbc.M305040200. [DOI] [PubMed] [Google Scholar]
- 11.Rimondini R, Arlinde C, Sommer W, Heilig M. Long-lasting increase in voluntary ethanol consumption and transcriptional regulation in the rat brain after intermittent exposure to alcohol. FASEB J. 2002;16:27–35. doi: 10.1096/fj.01-0593com. [DOI] [PubMed] [Google Scholar]
- 12.Saito M, Smiley J, Toth R, Vadas C. Microarray analysis of gene expression in rat hippocampus after chronic ethanol treatment. Neurochem. Res. 2002;27:1221–9. doi: 10.1023/a:1020937728506. [DOI] [PubMed] [Google Scholar]
- 13.Kerns RT, et al. Ethanol-responsive brain region expression networks: implications for behavioral responses to acute ethanol in DBA/2J versus C57BL/6J mice. J. Neurosci. 2005;25:2255–66. doi: 10.1523/JNEUROSCI.4372-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewohl JM, et al. Gene expression in human alcoholism: microarray analysis of frontal cortex. Alcohol Clin. Exp. Res. 2000;24:1873–82. [PubMed] [Google Scholar]
- 15.Smith MW, et al. Hepatitis C virus and liver disease: global transcriptional profiling and identification of potential markers. Hepatology. 2003;38:1458–67. doi: 10.1016/j.hep.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 16.Shackel NA, McGuinness PH, Abbott CA, Gorrell MD, McCaughan GW. Insights into the pathobiology of hepatitis C virus-associated cirrhosis: analysis of intrahepatic differential gene expression. Am. J. Pathol. 2002;160:641–54. doi: 10.1016/S0002-9440(10)64884-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bièche I, et al. Molecular profiling of early stage liver fibrosis in patients with chronic hepatitis C virus infection. Virology. 2005;332:130–44. doi: 10.1016/j.virol.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 18.Knodell RG, et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology. 1981;1:431–5. doi: 10.1002/hep.1840010511. [DOI] [PubMed] [Google Scholar]
- 19.Gentleman RC, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R30. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.R Development Core Team. R: a language and environment for statistical computing [computer program] Vienna (Austria): R Foundation for Statistical Computing; 2007. [Google Scholar]
- 21.Swiatkowska-Stodulska R, Bakowska A, Drobińska-Jurowiecka A. Interleukin-8 in the blood serum of patients with alcoholic liver disease. Med. Sci. Monit. 2006;12:CR215–20. [PubMed] [Google Scholar]
- 22.Lemmers A, et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology. 2009;49:646–57. doi: 10.1002/hep.22680. [DOI] [PubMed] [Google Scholar]
- 23.Sharma A, Chakraborti A, Das A, Dhiman RK, Chawla Y. Elevation of interleukin-18 in chronic hepatitis C: implications for hepatitis C virus pathogenesis. Immunology. 2009;128:e514–22. doi: 10.1111/j.1365-2567.2008.03021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loguercio C, Federico A. Oxidative stress in viral and alcoholic hepatitis. Free Radic. Biol. Med. 2003;34:1–10. doi: 10.1016/s0891-5849(02)01167-x. [DOI] [PubMed] [Google Scholar]
- 25.Safdar K, Schiff ER. Alcohol and hepatitis C. Semin. Liver Dis. 2008;24:305–15. doi: 10.1055/s-2004-832942. [DOI] [PubMed] [Google Scholar]
- 26.Mallat A, Hezode C, Lotersztajn S. Environmental factors as disease accelerators during chronic hepatitis C. J. Hepatol. 2008;48:657–65. doi: 10.1016/j.jhep.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 27.Lederer SL, et al. Distinct cellular responses differentiating alcohol- and hepatitis C virus-induced liver cirrhosis. Virol. J. 2006;3:98. doi: 10.1186/1743-422X-3-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jamal MM, Morgan TR. Liver disease in alcohol and hepatitis C. Best Pract. Res. Clin. Gastroenterol. 2003;17:649–62. doi: 10.1016/s1521-6918(03)00018-0. [DOI] [PubMed] [Google Scholar]
- 29.Otani K, et al. Hepatitis C virus core protein, cytochrome P450 2E1, and alcohol produce combined mitochondrial injury and cytotoxicity in hepatoma cells. Gastroenterology. 2005;128:96–107. doi: 10.1053/j.gastro.2004.10.045. [DOI] [PubMed] [Google Scholar]
- 30.Perlemuter G, et al. Alcohol and hepatitis C virus core protein additively increase lipid peroxidation and synergistically trigger hepatic cytokine expression in a transgenic mouse model. J. Hepatol. 2003;39:1020–27. doi: 10.1016/s0168-8278(03)00414-8. [DOI] [PubMed] [Google Scholar]
- 31.Piasecki BA, et al. Influence of alcohol use, race, and viral coinfections on spontaneous HCV clearance in a US veteran population. Hepatology. 2004;40:892–99. doi: 10.1002/hep.20384. [DOI] [PubMed] [Google Scholar]
- 32.Cook RT. Alcohol abuse, alcoholism and damage to the immune system-a review. Alcohol Clin. Exp. Res. 1998;22:1927–42. [PubMed] [Google Scholar]
- 33.Cerny A, Chisari FV. Pathogenesis of chronic hepatitis C: immunological features of hepatic injury and viral persistence. Hepatology. 1999;30:595–601. doi: 10.1002/hep.510300312. [DOI] [PubMed] [Google Scholar]
- 34.Piasecki BA, et al. Influence of alcohol use, race, and viral coinfections on spontaneous HCV clearance in a US veteran population. Hepatology. 2004;40:892–9. doi: 10.1002/hep.20384. [DOI] [PubMed] [Google Scholar]
- 35.Cook RT. Alcohol abuse, alcoholism and damage to the immune system: a review. Alcohol Clin. Exp. Res. 1998;22:1927–42. [PubMed] [Google Scholar]
- 36.Pianko S, Patella S, Ostapowicz G. Fas mediated hepatocyte apoptosis is increased by hepatitis C virus infection and alcohol consumption and may be associated with hepatic fibrosis: mechanisms of liver cell injury in chronic hepatitis C virus infection. J Viral Hepat. 2001;8:406–13. doi: 10.1046/j.1365-2893.2001.00316.x. [DOI] [PubMed] [Google Scholar]
- 37.Gressner OA, Gressner AM. Connective tissue growth factor: a fibrogenic master switch in fibrotic liver diseases. Liver Int. 2008;28:1065–79. doi: 10.1111/j.1478-3231.2008.01826.x. [DOI] [PubMed] [Google Scholar]
- 38.Hartland SN, et al. Active matrix metalloproteinase-2 promotes apoptosis of hepatic stellate cells via the cleavage of cellular N-cadherin. Liver Int. 2009;29:966–78. doi: 10.1111/j.1478-3231.2009.02070.x. [DOI] [PubMed] [Google Scholar]
- 39.Kendall TJ, et al. Neurotrophin receptor signaling regulates hepatic myofibroblast proliferation and apoptosis in recovery from rodent liver fibrosis. Hepatology. 2009;49:901–10. doi: 10.1002/hep.22701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.