

Abstract
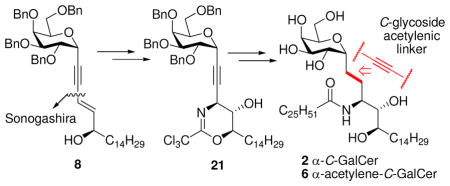
Stereocontrolled syntheses of α-C-GalCer (2) and its α-C-acetylenic analogue 6 were accomplished in high efficiency by a convergent construction strategy from 1-hexadecene and D-galactose. The key transformations include Sonogashira coupling, Sharpless asymmetric epoxidation, and Et2AlCl-catalyzed cyclization of an epoxytrichloroacetimidate to generate protected dihydrooxazine 21.
KRN7000 [(2S,3S,4R)-1-O-(α-D-galactopyranosyl)-2-(N-hexacosanoylamino)-1,3,4-octadecanetriol, α-GalCer, 1, Figure 1], is a synthetic analogue of a glycolipid extracted from the marine sponge1 Agelas mauritianus during a screen of natural products possessing antitumor properties in mice by Kirin Pharmaceuticals.2 α-GalCer forms a complex with a glycoprotein in antigen presenting cells known as CD1d.3 Glycolipid presentation in CD1d-ligand complexes to the T cell receptor of invariant natural killer T (iNKT) cells gives a high affinity ternary complex4 that activates iNKT cells in mice and humans to secrete a complex mixture of cytokines. The production of pro-inflammatory T helper (Th1) type cytokines such as interferon γ (IFN-γ) is correlated with antitumor, antiviral/antibacterial, and adjuvant activities, whereas anti-inflammatory Th2 type cytokines (such as interleukins 4, 5, 10, and 13) are regulators of some autoimmune and inflammatory diseases.5 However, the simultaneous production of both types of conflicting cytokine activities comprises the therapeutic potential of activated iNKT cells and interferes with a concerted biological outcome.6 The potential clinical utility of 1 is also limited by the long-term unresponsiveness (anergy) of iNKT cells when multiple doses of 1 are administered.
Figure 1.

Structures of glycolipids 1–6.
α-C-Glycoside analogues of of α-GalCer are expected to be long lived because they are resistant to α-glycosidase activity. Moreover, replacing the glycosidic oxygen atom with a methylene group removes a hydrogen-bonding acceptor site.7 Thus α-C-GalCer analogues may bind less tightly to CD1d, which may be a factor in determining the type of cytokine release. An isosteric C-glycoside analogue (2, Figure 1) was found to be active in mice in vivo, with a biased induction of Th1 responses compared to 1.8 In addition, 2 produced a long-term production of IFN-γ in mice, suggesting that the α-C-GalCer/CD1d complex is more stable in antigen presenting cells in vivo than the KRN7000/CD1d complex.8d,e We found that nonisosteric α-C-GalCer analogue 3, in which the glycosidic oxygen was deleted, induced an even higher Th1-type cytokine response than 1 and 2 in human iNKT cells in vitro.9 Interestingly, other α-C-glycoside homologues that contain a 3-carbon linker (4) were inactive.8b GCK127 (5), an analogue with an E-alkene linker, not only exhibited activity in mice, but also induced a potent stimulatory activity against human iNKT cells, which was ascribed to the preservation of an ~170° dihedral angle in the linker region between the galactose and the ceramide (Gal-C1-O1-phytosphingosine C1′-phytosphingosine C2′).10 These studies indicate that α-C-GalCer analogues are useful for presentation by CD1d to iNKT cells and have potential immunotherapeutical applications compared with 1, the most commonly used ligand.
In order to make larger quantities of 2 available to the immunology community,11 diverse synthetic approaches toward this important synthetic target have been developed.8,12 However, there remains a need for efficient stereoselective methods for the preparation of 2. We report a concise convergent synthesis of 2 from readily available, inexpensive starting materials. In addition, the synthetic route to 2 reported here permits modification of the linker region, which appears to be critical for Th1 vs. Th2 selectivity. We also report the synthesis of 6, which contains an acetylenic moiety and also preserves an ~170° dihedral angle in the linker.
As shown in the retrosynthetic analysis (Scheme 1), we envisioned that the three contiguous stereogenic centers in the phytosphingosine moiety can be accessed from epoxy alcohol 7 after reaction with trichloroacetonitrile to give 8, followed by a Lewis acid catalyzed epoxide opening at the propargylic carbon. The requisite epoxide 7 could be furnished by Sharpless asymmetric epoxidation (SAE)13 of 9, which in turn could be obtained from 10 via Sonogashira cross-coupling14 between two building blocks, 11 and 12 (or 13). 11 can be assembled via α-C-ethynylation of 14a (accessible from D-galactose; see Supporting Iinformation), and 12 can be prepared via Takai olefination15 of aldehyde 15, which can be made from 1-hexadecene (16).
Scheme 1.

Retrosynthetic Plan
As shown in Scheme 2, Sharpless asymmetric dihydroxylation of 16 with AD-mix-β provided the desired diol 17 (85% ee) in almost quantitative yield.16 Alternatively, the use of the ligand (DHQD)2AQN, which was reported to have a higher enantioselectivity than the PHAL-based ligand in an aliphatic system,17 delivered 17 in a lower yield (71%) and slightly higher ee value (89% ee). Diol 17 was converted to its p-methoxybenzylidene (PMB) acetal, which was reduced with DIBAL-H to give alcohol 18 (85%, two steps).18 The use of a protocol with sodium hypochlorite catalyzed by TEMPO19 gave the desired aldehyde 15 in 62% yield without any erosion of the ee value.20 Since an (E)-l-iodoalkene was required, we used the Takai reaction,15 which is known to be highly E stereoselective. Condensation of aldehyde 15 with iodoform in the presence of chromium(II) chloride yielded the expected (E)-vinyl iodide 13 in 70% yield when the Evans modification21 was used. Deprotection of the PMB group using I2 in MeOH22 afforded vinyl iodide 12 (75%).
Scheme 2.

Synthesis of Vinyl Iodide 12
Initially, α-C-ethynylgalactoside 11 was prepared by reaction of 1-acetoxy-2,3,4,6-tetra-O-benzyl-D-galactopyranoside (14b,c) with ethynyl precursor 19 in the presence of TMSOTf followed by desilylation of 20.23 However, we subsequently found that methyl β-D-galactosylpyranoside (14a), which is crystalline, can also react with 19 under the same conditions (Scheme 3). This reaction proceeded with very high α-stereoselectivity; no corresponding β-anomer was found by 1H NMR. Its efficiency in the preparation of 11 is comparable to that of acetate 14c. The two-step yield of 11 from 14c was 54%.23a,c Furthermore, 14c must be prepared from 14a in two additional steps (~67% overall yield).24 Thus our two-step yield of 11 from 14a (37%) is not only comparable to that from 14c but also offers the advantage that 14a can be prepared from the very inexpensive D-galactose as reported in the Supporting Information.
Scheme 3.

Synthesis of 2 and 6
With an efficient synthesis of the two building blocks 11 and 12 established, we directed our efforts toward Sonogashira coupling (Scheme 3).14 An initial trial of cross-coupling [Pd(PPh3)4, CuI (0.5 equiv), iPr2NEt (6 equiv), THF] between PMB-protected alcohol 13 and alkyne 11 in THF provided enyne 10 in 56% yield. During deprotection of the PMB group of 10 with DDQ, the hydroxy group was oxidized to the corresponding ketone. However, when free alcohol 12 was coupled with 10 in the presence of Pd(Ph3P)4 and CuI in CH3CN/Et3N (5:1) the yield of enynol 9 improved to 88%. Use of Pd(PPh3)2Cl2 as a precatalyst [Pd(PPh3)2Cl2, CuI, CH3CN/Et3N (5:1)] afforded 9 in about the same yield as obtained with Pd(Ph3P)4.
Catalytic or substoichometric SAE of 9 was ineffective, producing little conversion after 20 h at −20 °C. The reaction using 4 equiv of cumene hydroperoxide as the epoxidizing agent catalyzed by 2.5 equiv of Ti(OiPr)4 and 2.6 equiv of D-(−)-DIPT provided propargylic epoxy alcohol 7 in high yield (84%) and excellent diastereoselectivity (>95%).25 Chelation-controlled opening of 2,3-epoxy alcohol 7 with NaN3 and in the presence of NH4Cl in aqueous MeOH under reflux failed to provide the desired azido diol, delivering instead a complex mixture.26 Et2AlCl-catalyzed cyclization27 of trichloroacetimidate 8, prepared by reaction of 7 with 6.0 equiv of trichloroacetonitrile in the presence of 3.5 equiv of DBU,27d gave dihydrooxazine 21 in a two-step yield of 67%. It is noteworthy that BF3.Et2O also catalyzed cyclization of 8 to 21; however, we obtained a mixture of 21 and its hydrolysis product 22 in a ratio of 1:1 (30% vs 27%, respectively).
Acid hydrolysis of 21 provided trichloroacetimide 22, which was treated with ethanolic NaOH to deliver amine 23 in 68% overall yield. Reaction of amine 23 with n-hexacosanoyl chloride12 gave amide 24 in 73% yield. Catalytic hydrogenation (Pd/C, H2, EtOH/TFA)10a of the linking triple bonds, together with global removal of the benzyl protecting groups, afforded the target α-C-glycoside 2.28 However, attempted reduction using Pearlman’s catalyst [H2, Pd(OH)2, CH2Cl2/MeOH] resulted in incomplete saturation of the acetylenic group. The preparation of 6 from 24 was achieved by BF3.OEt2/EtSH deprotection of the benzyl groups,29 leaving the acetylenic moiety intact, in almost quantitative yield.
In conclusion, a convergent and stereoselective synthetic route to α-C-GalCer (2) and its analogue 6 containing an acetylenic linker was accomplished. Notable features include the concise formation of three contiguous stereogenic centers in the phytosphingosine moiety by Sonogashira cross-coupling followed by Sharpless asymmetric epoxidation and Et2AlCl-catalyzed cyclization of an epoxytrichloroacetimidate intermediate. This convergent construction from simple starting materials (10 steps from 14a with 6.5% overall yield) permits the preparation of analogues with variations in the linker area.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health Grant HL-083187. Z. L. thanks a Doctoral Student Research Grant from the CUNY Graduate Center.
Footnotes
Supporting Information Available: Experimental procedures as well as 1H and 13C NMR spectra for all new compounds and synthetic 2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Since mammalian and plant glycosphingolipids generally are β-anomers, it is surprising that sponges would produce α-glycosphingolipids. It is likely that 1was actually derived from Novosphingobium bacteria that colonized A. mauritianus.
- 2.(a) Natori A, Koezuka Y, Higa T. Tetrahedron Lett. 1993;34:5591. [Google Scholar]; (b) Kobayshi E, Motoki K, Uchida T, Fukushima H, Koezuka Y. Oncol Res. 1995;7:529. [PubMed] [Google Scholar]
- 3.CD1d molecules, which are expressed by professional antigen-presenting cells such as dendritic cells, macrophages, and B cells, recognize various lipid and glycolipid antigens. Bendelac A, Savage PB, Teyton L. Annu Rev Immunol. 2007;35:297. doi: 10.1146/annurev.immunol.25.022106.141711.
- 4.Invariant NKT cells are a subset of T lymphocytes that recognize glycolipid antigens and are dependent on CD1d as the antigen presenting molecule to their T cell receptor (TCR). In the glycolipid-CD1d complex, the two long hydrocarbon chains of α-GalCer are buried in the two long apolar channels of CD1d, and the carbohydrate head group protrudes above the surface toward the TCR of iNKT cells.7
- 5.Savage PB, Teyton L, Bendelac A. Chem Soc Rev. 2006;35:771. doi: 10.1039/b510638a. [DOI] [PubMed] [Google Scholar]
- 6.(a) Miyamoto K, Miyake S, Yamamura T. Nature. 2001;413:531. doi: 10.1038/35097097. [DOI] [PubMed] [Google Scholar]; (b) Van Kaer L. Nat Rev Immunol. 2005;5:31. doi: 10.1038/nri1531. [DOI] [PubMed] [Google Scholar]
- 7.(a) Borg NA, Wun KS, Kjer-Nielsen L, Wilce MCJ, Pellicci DG, Koh R, Besra GS, Bharadwaj M, Godfrey DI, McCluskey J, Rossjohn J. Nature. 2007;448:44. doi: 10.1038/nature05907. [DOI] [PubMed] [Google Scholar]; (b) Koch M, Stronge VS, Shepherd D, Gadola SD, Mathew B, Ritter G, Fersht AR, Besra GS, Schmidt RR, Jones EY, Cerundolo V. Nat Immunol. 2005;6:819. doi: 10.1038/ni1225. [DOI] [PubMed] [Google Scholar]; (c) Schiefner A, Fujio M, Wu D, Wong CH, Wilson IA. J Mol Biol. 2009;394:71. doi: 10.1016/j.jmb.2009.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Yang G, Schmieg J, Tsuji M, Franck RW. Angew Chem, Int Ed Engl. 2004;43:3818. doi: 10.1002/anie.200454215. [DOI] [PubMed] [Google Scholar]; (b) Chen G, Schmieg J, Tsuji M, Franck RW. Org Lett. 2004;6:4077. doi: 10.1021/ol0482137. [DOI] [PubMed] [Google Scholar]; (c) Pu J, Franck RW. Tetrahedron. 2008;64:8618. doi: 10.1016/j.tet.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Franck RW, Tsuji M. Acc Chem Res. 2006;39:692. doi: 10.1021/ar050006z. [DOI] [PubMed] [Google Scholar]; (d) Schmieg J, Yang G, Franck RW, Tsuji M. J Biomed Biotechnol. 2010;2010:283612. doi: 10.1155/2010/283612. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sullivan BA, Nagarajan NA, Wingender G, Wang J, Scott I, Tsuji M, Franck RW, Porcelli SA, Zajonc DM, Kronenberg M. J Immunol. 2010;184:141. doi: 10.4049/jimmunol.0902880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu X, Song L, Metelitsa LS, Bittman R. ChemBioChem. 2006;7:1750. doi: 10.1002/cbic.200600197. [DOI] [PubMed] [Google Scholar]
- 10.(a) Chen G, Chien M, Tsuji M, Franck RW. ChemBioChem. 2006;7:1017. doi: 10.1002/cbic.200500386. [DOI] [PubMed] [Google Scholar]; (b) Li X, Chen G, Garcia-Navarro R, Franck RW, Tsuji M. Immunology. 2009;127:216. doi: 10.1111/j.1365-2567.2008.02943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li X, Shiratsuchi T, Chen G, Dellabona P, Casorati G, Franck RW, Tsuji M. J Immunol. 2009;183:4415. doi: 10.4049/jimmunol.0901021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The Tetramer Core Facility funded by NIH provides reagents for NKT cell activation, including CD1d ligands, to approved investigators.
- 12.Wipf P, Pierce JG. Org Lett. 2006;8:3375. doi: 10.1021/ol0613057. [DOI] [PubMed] [Google Scholar]
- 13.For a review of SAE, see: Katsuki T, Martin VS. Org React. 1996;48:1.
- 14.For a review, see: Sonogashira K. J Organomet Chem. 2002;653:46.
- 15.Takai K, Nitta K, Utimoto K. J Am Chem Soc. 1986;108:7408. doi: 10.1021/ja00279a068. [DOI] [PubMed] [Google Scholar]
- 16.The chiral purity of 17was enriched to >95% by recrystallization from EtOAc, and this high chiral purity diol was used in the following reaction. See: He L, Byun HS, Bittman R. J Org Chem. 2000;65:7618. doi: 10.1021/jo001225v.
- 17.Eecker H, Sharpless KB. Angew Chem Int Ed Engl. 1996;35:448. [Google Scholar]
- 18.Synthesis of primary alcohol 18was readily achieved by following a reported synthesis of a shorter chain analogue from 1-heptene. Smith AB, Chen SSY, Nelson FC, Reichert JM, Salvatore BA. J Am Chem Soc. 1997;119:10935.
- 19.Jurczak J, Gryko D, Kobrzycka E, Gruza H, Prokopowiczit P. Tetrahedron. 1998;54:6051. [Google Scholar]
- 20.15was reduced back to 18by NaBH4 and subsequently converted to the (S)-MTPA ester using (R)-MTPA chloride. The 1H NMR spectrum of the MTPA ester exhibited no signals of the other isomer; see the Supporting Information.
- 21.Evans DA, Ng HP. Tetrahedron Lett. 1993;34:2229. [Google Scholar]
- 22.Vaino AR, Szarek WA. Synlett. 1995:1157. [Google Scholar]
- 23.Nishikawa T, Koide Y, Kajii S, Wada K, Ishikawa M, Isobe M. Org Biomol Chem. 2005;3:687. doi: 10.1039/b414905j.Dondoni A, Moriotti G, Marra A. J Org Chem. 2002;67:4475. doi: 10.1021/jo020054m.(c) The stereoselectivity of the C-glycosidation was found to be independent of the anomeric configuration of the acetate.23a,b
- 24.Kulkarni SS, Gervay-Hague J. Org Lett. 2006;8:5765. doi: 10.1021/ol062354m. [DOI] [PubMed] [Google Scholar]
- 25.7was converted to the (S)-MTPA ester using using (R)-MTPA chloride. The de value was determined by analyzing the 1H NMR spectrum of the (S)-MTPA ester of 7.
- 26.Two mechanisms may be responsible for the failure: (1) Intramolecular 1,3-dipolar cycloaddition of the propargyl azide followed by MeOH attack on the resulting strained bicyclic triazole or (2) a triaza-Cope rearrangement followed by cyclization of the resulting allenyl azide to triazafulvene and reaction with MeOH. See: Banert K. Chem Ber. 1989;122:911.Banert K. Chem Ber. 1989;122:1963.
- 27.For Lewis acid catalyzed cyclization of 2,3-epoxytrichloroacetimidates, see: Schmidt U, Respondek M, Lieberknecht A, Werner J, Fisher P. Synthesis. 1989:256.Hatakeyama S, Matsumoto H, Fukuyama H, Mukugi Y, Irie H. J Org Chem. 1997;62:2275. doi: 10.1021/jo9618278.Lu X, Sun C, Valentine WJ, ES, Liu J, Tigyi G, Bittman R. J Org Chem. 2009;74:3192. doi: 10.1021/jo900023u.(d) The preparation of 8in the presence of a catalytic amount of DBU27b was ineffective.
- 28.The spectral data of 2matched well with the previously reported material in 500-MHz 1H and 125-MHz 13C NMR spectra and optical rotation (see Table S1, Supporting Information).8,12
- 29.Xu J, Egger A, Bernet B, Vasella A. Helv Chim Acta. 1996;79:2004. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.