

Abstract
Asymmetric scandium (III)-catalyzed rearrangement of 3-allyloxyflavones was utilized to prepare chiral, non-racemic 3,4-chromanediones in high yields and enantioselectivities. These synthetic intermediates have been further elaborated to novel heterocyclic frameworks including angular pyrazines and dihydropyrazines. The absolute configuration of rearrangement products was initially determined by a nonempirical analysis of circular dichroism (CD) using time-dependent density functional theory (TDDFT) calculations and verified by x-ray crystallography of a hydrazone derivative. Initial studies of the mechanism support an intramolecular rearrangement pathway that may proceed through a benzopyrylium intermediate.
Introduction
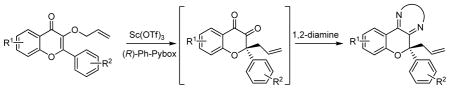
Enantioselective construction of quaternary stereogenic carbons is a significant challenge in organic chemistry.1 As part of our investigations concerning novel scaffolds, we encountered an interesting chemotype bearing two adjacent, fully substituted carbon centers present in cytotoxic, prenylated flavonoids including sanggenon A (1) and sanggenol F (2) (Figure 1).2 Further examination of their structures inspired the development of metal-catalyzed, asymmetric rearrangement 3, 4 of 3-allyloxyflavones 3 to 2-substituted 3,4-chromanediones 45 (Figure 2). Herein, we report the development of methodology to prepare chiral, non-racemic chromanediones using metal-catalyzed rearrangements6 of 3-allyloxyflavones and our initial studies to probe the reaction mechanism.
Figure 1.

Structures of sanggenon A and sanggenol F
Figure 2.

Asymmetric rearrangement of 3-allyloxyflavones
Results and Discussion
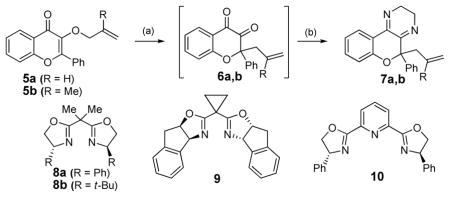
We began our investigation by evaluating reaction of 3-allyloxy flavone 5a with a series of Lewis acids (Table 1). Among a panel of metals evaluated, trifluoromethanesulfonate salts of Lewis acidic metals were found to catalyze the reaction of 5a to chromanedione 6a in low to moderate yields employing 10 mol% of catalyst (entries 1, 2, and 7). However, complete conversion of allyloxy flavone 5a was observed when 30 mol% of Sc(OTf)3 was employed (entry 8). For purification purposes, the reactive 1,2-dicarbonyls 6a were condensed with 1,2-ethylenediamine to afford dihydropyrazines 7.7
Table 1.
Lewis Acid-Catalyzed Rearrangementa
 | ||||||
|---|---|---|---|---|---|---|
| entry | catalyst | Equiv (mol%) | ligand | Allyloxy flavone | % conversion (isolated yield 7)b | erc |
| 1 | Lu(OTf)3 | 10 | none | 5a | 16 | - |
| 2 | Cu(OTf)2 | 10 | none | 5a | 50 | - |
| 3 | Cu(OTf)2 | 10 | 8a | 5a | 60 | 90:10 |
| 4 | Cu(OTf)2 | 10 | 8b | 5a | - d | - |
| 5 | Cu(OTf)2 | 10 | 9 | 5a | - d | - |
| 6 | Cu(OTf)2 | 10 | 10 | 5a | - d | - |
| 7 | Sc(OTf)3 | 10 | none | 5a | 38 | - |
| 8 | Sc(OTf)3 | 30 | none | 5a | 98 (95) | - |
| 9 | Sc(OTf)3 | 30 | 10 | 5a | 100 (98) | 97:3 |
| 10 | Sc(OTf)3 | 30 | 10 | 5b | 82 (57) | 98:2 |
Reaction conditions: (a) 0.16 mmol 3-allyloxyflavone, 0.02–0.05 mmol catalyst, 0.02–0.05 mmol chiral ligand, and 250 mg of activated 4Å MS in DCE (0.04M) for 12 h under Ar at 35 °C; (b) 0.40 mmol of 1,2-ethylenediamine at rt for 2 h.
Conversion determined by crude 1H NMR analysis and isolated yields after column chromatography on silica gel.
Determined by chiral HPLC analysis.7
No product isolated after column chromatography.
Encouraged by these preliminary results, we investigated use of chiral ligands for Sc(OTf)3 in the rearrangement. We found that the complex prepared using (R)-Ph-Pybox 10 as ligand8 led to good conversions and enantioselectivities to afford dihydropyrazines 7a,b when the internal position of the olefin was substituted with a hydrogen (Table 1, entry 9) or methyl group (entry 10). In preliminary studies, flavone ether substrates bearing disubstituted alkenes (e.g. Z or E crotyl ethers) were found to be sluggish in rearrangements in contrast to catalytic asymmetric Claisen rearrangements of 2-alkoxycarbonyl-substituted allyl vinyl ethers reported by Hiersemann and coworkers.3e,f Unfortunately, rearrangements did not proceed when different substituents including bromine, phenyl, or carboxylates occupied the 2-position of the olefin.7 Furthermore, complexes of ligands 8 and 9 with Sc(OTf)3 were found to be ineffective for the rearrangement.
Substrate Scope
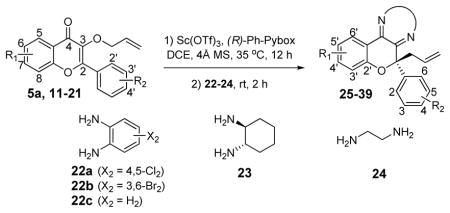
Asymmetric rearrangement of a number of 3-allyloxyflavone substrates with diverse aryl substituents is shown in Table 2. Neither the position nor the electronic nature of substituents affected yields and enantioselectivities of reactions. For example, an electron-rich substituted allyloxyflavone (p-MeO, entry 10) afforded the same selectivity and yields as an electron poor substrate (p-NO2, entry 14). Regarding the position of the substituents on the C-2 aryl ring, we found that the rearrangement tolerated the presence of electron-donating groups at C-2′ (entries 9 and 15). Use of 1,2-diamines that differed structurally and electronically in the condensation with the intermediate 3,4-chromanediones facilitated access to various heterocyclic structures including dihydropyrazines and pyrazines.
Table 2.
Rearrangement Substrate Scopea
 | |||||
|---|---|---|---|---|---|
| entry | substituents R1, R2 | diamine | % yield b | product | er |
| 1 | H, H (5a) | 22a | 78 | 25 | 93:7 |
| 2 | H, H (5a) | 22b | 98 | 26 | - d |
| 3 | H, H (5a) | 22c | 93 | 27 | 93:7 |
| 4 | H, H (5a) | 23 | 86 | 28 | >98:2 c |
| 5 | 5-OMe, H (11) | 22c | 91 | 29 | 97:3 |
| 6 | 6-OMe, H (12) | 22c | 90 | 30 | - e |
| 7 | 7-OMe, H (13) | 24 | 93 | 31 | 90:10 |
| 8 | 6-Me, H (14) | 24 | 88 | 32 | 96:4 |
| 9 | H, 2′-MeO (15) | 24 | 96 | 33 | 95:5 |
| 10 | H, 4′-MeO (16) | 24 | 94 | 34 | 98:2 |
| 11 | H, 4′-Me (17) | 24 | 94 | 35 | 96:4 |
| 12 | H, 4′-CF3 (18) | 24 | 86 | 36 | 95:5 |
| 13 | H, 4′-Br (19) | 24 | 93 | 37 | 91:9 |
| 14 | H, 4′-NO2 (20) | 24 | 92 | 38 | 96:4 |
| 15 | H, 2′-MeO-4′-Br (21) | 24 | 80 | 39 | 96:4 |
Reaction conditions: 0.16 mmol 3-allyloxyflavone, 0.05 mmol catalyst, 0.05 mmol (R)-Ph-Pybox, and 250 mg of activated 4Å MS in DCE (0.04 M) for 12 h under Ar at 35 °C, followed by reaction with 0.40 mmol of diamine at rt for 2 h.
Isolated yields after column chromatography on SiO2.
Dr value provided.
Separation of enantiomers via HPLC was not accomplished.
Not determined.
Absolute Configuration Assignment and Rationale
The absolute configuration of dichloropyrazine 25 was established by a nonempirical analysis of circular dichroism (CD) using time-dependent density functional theory (TDDFT) calculations.9 Prior to calculation of CD spectra, a conformational search using the MMFF94 was conducted on an arbitrarily chosen S absolute configuration of 25. The resulting 12 conformers within a 10 kcal/mol energy window were optimized at the DFT/B3LYP/6-31G(d) level of theory, leading to three stable conformers within 1.5 kcal/mol (Figure 3).7 CD theoretical calculations were carried out for these three conformers at the TDDFT/B3LYP/6-31G(d) level of theory and the final spectrum obtained as the weighted average based on Boltzmann populations (Figure 3). The theoretical and observed CD spectra showed good agreement including a negative band at around 390 nm and a positive band at around 330 nm, thus unambiguously establishing the absolute configuration of 25 as S. The absolute configuration of 7b produced from asymmetric rearrangement was studied in a similar manner (Table 1, entry 10).7 X-ray crystal structure analysis of a pyrazine-hydrazone 42 derived from (S)-(−)-27 (Scheme 1)7 independently confirmed the absolute configuration of rearrangement products as determined by CD calculations.
Figure 3.

Analysis of the absolute configuration of (−)-25. Top: the most stable conformer of (S)-25. Bottom: comparison of the calculated and the observed CD spectra of 25. (a) Calculated CD spectra at the TDDFT/B3LYP/6-31G(d) for (S)-25. The Boltzmann populations of each conformer are shown in parentheses. (b) The observed CD and (c) UV spectrum obtained as an acetonitrile solution (0.04 mM). The observed CD spectrum is normalized to 100%ee. UVλmax (ε): 225 (54000), 268 (25000), 382 (18900); CDλext (Δε): 236 (−12.0), 258 (−25.1), 278 (+6.7), 327 (+14.8), 382 (−14.5). [α]D25 = − 284.9 (c=1.2, CHCI3).7
Scheme 1.

Synthesis and X-ray crystal structure analysis of pyrazine-hydrazone 42
Mechanistic Studies
To rationalize the observed enantioselectivity, we modeled the transition state of the presumed octahedral intermediate obtained by bidentate coordination of the 3,4-chromanedione to the scandium (III)-(R)-Ph-Pybox (Figure 4).10 The transition model suggests that the rearrangement occurs on the Reface of the double bond due to steric hindrance of the phenyl groups of the Ph-Pybox ligand (Figure 4). Further experiments were conducted using the deuterium-labeled substrate 3-(1,1-dideuteuroallyl-oxy)-chromen-4-one 43. When 3-allyloxy flavone 43 was submitted to scandium (III)-catalyzed rearrangement, complete transfer of the deuterium to the terminal position of the olefin occurred. After condensation of the intermediate 3,4-chromanedione 44 with 1,2-dianiline 22a, the deuterated dihydropyrazine 45 was produced (eqn 1, Figure 5). This result indicates that the rearrangement process proceeds via an intramolecular pathway.
Figure 4.
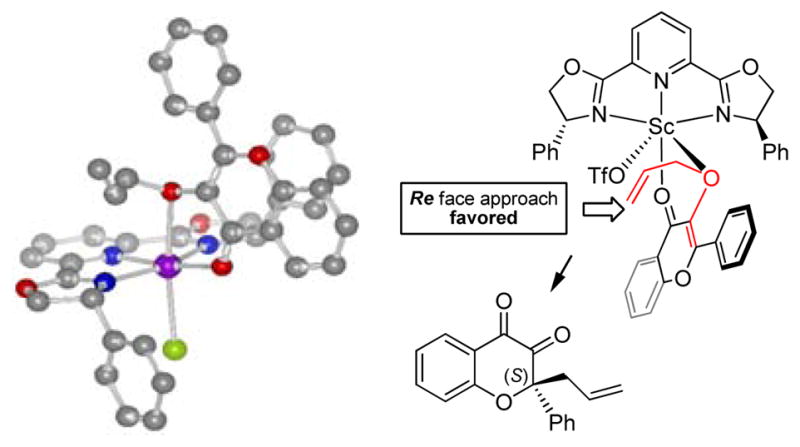
Proposed transition state model
Figure 5.

Deuterium-labeled substrates and crossover experiment
In order to rule out the existence of an intermolecular reaction pathway, a crossover experiment was also conducted (Figure 5, eqn 2).7 A 1:1 mixture of non-labeled allyloxyflavone 5a and deuterated 6-OMe derivative 46 subjected to the reaction conditions led to sole production of pyrazines 27 and 47. The absence of any observed allyl crossover is consistent with an intramolecular rearrangement process.
We also performed experiments to investigate possible mechanistic pathways. Crossover and deuterium-labeling experiments are consistent with asymmetric [3,3]-sigmatropic rearrangement3 of the scandium (III)-complexed flavone ether 48 to afford 3,4-chromanedione 44 (Figure 6). In an alternative pathway, the corresponding benzopyrylium11 49 derived from delocalization of the positive charge and aromatization may undergo a [2,3] sigmatropic rearrangement12 to 50 followed by a stereospecific [1,2]-allyl shift.13,14 In the latter case, the deuterium atoms would also be located on the terminal position of the double bond. In order to probe the latter mechanism, we prepared 4-siloxy-1-benzopyrylium salt 51 by treatment of 3-methoxyflavone with TBSOTf 15 and determined that it has a characteristic fluorescence emission (450 nm) upon excitation at 410 nm16 (Figure 7). Similar fluorescence emissions observed for the Sc(OTf)3-3-methoxyflavone complex 52 (excitation 400 nm) as well as the corresponding complex derived from 3-allyloxyflavone 5a7 support the involvement of benzopyrylium intermediates after Lewis acid activation and a plausible alternative to the [3,3] mechanism for rearrrangement of 3-allyloxyflavones. Comparison of 13C NMR spectra of 51 and complex 52 (Figure 8) also shows comparable downfield shifts for C-2 (3-methoxyflavone: 156 ppm; 51: 162 ppm; 52: 165 ppm), further supporting likely involvement of benzopyrylium intermediates in the asymmetric rearrangement of 3-allyloxyflavones.7
Figure 6.

Mechanistic Alternatives for the Asymmetric Rearrangement
Figure 7.

Overlay of fluorescence spectra of 51, 52, 53, and Sc(OTf)3 (3.0 × 10−4 M in CH2Cl2).
Figure 8.

Overlay of NMR spectra of 53, 51, and 52 in CD2Cl2, 52 with 4% by volume HPIF for solubility.
Conclusion
In summary, the asymmetric, scandium-catalyzed rearrangement of 3-allyloxyflavones has been utilized to prepare chiral, non-racemic 3,4-chromanediones in high yield and enantioselectivity. These reactive intermediates have been further elaborated to novel frameworks including angular pyrazines and dihydropyrazines. Initial mechanism studies support an intramolecular rearrangement pathway that may proceed through a benzopyrylium intermediate. Further applications of the methodology in both diversity- and target-oriented synthesis are currently under investigation and will be reported in due course.
Experimental Section
3-(Allyloxy)-2-phenyl-4H-chromen-4-one (5a)
To a suspension of commercially available 3-hydroxyflavone (1.00 g, 4.20 mmol, 1.0 equiv) in dry acetone (100 mL) was added at room temperature allyl bromide (0.54 mL, 6.30 mmol, 1.5 equiv, filtered through a plug of basic alumina), followed by K2CO3 (870.0 mg, 6.30 mmol, 1.5 equiv). The temperature was slowly increased to 65 °C and the reaction mixture was stirred overnight. The mixture was then cooled to room temperature and 30 mL of Et2O was added. After filtration of the salts through a pad of Celite®, the solvent was removed in vacuo and the crude product was purified by column chromatography on silica gel. The allyloxyflavone 5a was obtained as a white solid (1.14 g, 4.10 mmol, 97 %), after flash chromatography on silica gel (petroleum ether: ethyl acetate = 90: 10). M.p. (petroleum ether: Et2O) = 53–54 °C. 1H NMR (400 MHz, CDCl3) δ 8.27 (dd, J = 8.0, 1.7 Hz, 1H), 8.14-8.09 (m, 2H), 7.68 (ddd, J = 8.6, 7.1, 1.7 Hz, 1H), 7.56-7.49 (m, 4H), 7.41 (ddd, J = 8.1, 7.2, 1.0 Hz, 1H), 5.94 (tdd, J = 16.4, 10.3, 6.1 Hz, 1H), 5.28 (dd, J = 17.2, 1.5 Hz, 1H), 5.15 (dd, J = 10.3, 1.2 Hz, 1H), 4.65 (d, J = 6.1 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ175.1, 156.0, 155.2, 139.9, 133.5, 133.4, 131.0, 130.6, 128.7 (2C), 128.4 (2C), 125.8, 124.6, 124.1, 118.5, 118.0, 73.2. IR νmax (film): 3062, 2937, 1640, 1614, 1467, 1393, 1236, 1200, 1145, 993, 691 cm−1. HRMS (ESI+) m/z calculated for C18H15O3 279.1021 found 279.1016 (M+H).
General Procedure for Sc(OTf)3-(R)-Pybox-Ph-mediated Rearrangement
To a suspension of molecular sieves (4Å, 250.0 mg, flame-dried under high vacuum) was added, via cannula, a pre-stirred solution of Sc(OTf)3 (23 mg, 0.05 mmol, 0.30 equiv) and (R,R)-(+)-2,6-bis(4-phenyl-2-oxazolinyl)pyridine 10 (20 mg, 0.05 mmol, 0.33 equiv) in DCE (3 mL). After stirring the suspension at rt for 2 h, a solution of allyloxyflavone 5a (0.16 mmol, 1.0 equiv) in DCE (2 mL) was slowly added via cannula. The mixture was stirred at room temperature for 30 min and stirred overnight at 35 °C. 1,2-Ethylene diamine 24 (27 μL, 0.40 mmol, 2.50 equiv) was added in one portion and the mixture was allowed to stir for an additional 2 h at room temperature. After removal of the molecular sieves by filtration of the crude mixture through a pad of Celite, the solvent was evaporated in vacuo and the pyrazines 25 or dihydropyrazines 7 and 28 were isolated by flash column chromatography on silica gel.
(S)-5-Allyl-5-phenyl-3,5-dihydro-2H-chromeno[4,3-b] pyrazine (7a)
Purification on silica gel (petroleum ether: ethyl acetate = 80: 20) afforded dihydropyrazine 7a as a bright yellow oil (47 mg, 0.16 mmol, 98 %). [α]D25 (c 1.0, CHCl3) = + 52.3°. er = 93:7 (ChiralCel OD 1% IPA in hexane, retention time 4.58: 5.47 min, major: minor). 1H NMR (400 MHz, CDCl3) δ 7.82 (dd, J = 7.9, 1.5 Hz, 1H), 7.38 (ddd, J = 8.3, 7.2, 1.7 Hz, 1H), 7.30-7.15 (m, 5H), 7.13 (d, J = 8.3 Hz, 1H), 6.95 (app t, J = 7.6 Hz, 1H), 5.94-5.81 (m, 1H), 5.06 (app d, J = 15.9 Hz, 1H), 5.05 (app d, J = 10.6 Hz, 1H), 4.14-3.95 (m, 2H), 3.49-3.25 (m, 2H), 3.13 (dd, J = 14.7, 6.6 Hz, 1H), 2.92 (dd, J = 14.7, 7.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 159.0, 156.0, 149.6, 139.3, 133.1, 132.6, 128.4 (2C), 127.7, 126.0 (2C), 124.9, 122.0, 119.9, 118.4, 118.3, 85.6, 45.9, 44.4, 43.7. IR νmax (film): 3074, 2943, 2841, 1608, 1593, 1461, 1384, 1330, 1220, 1118, 993, 914, 703 cm−1. HRMS (ESI+) m/z calculated for C20H19N2O 303.1497 found 303.1492 (M+H).
(S)-5-(2-Methylallyl)-5-phenyl-3,5-dihydro-2H-chromeno[4,3-b]pyrazine (7b)
Dihydropyrazine 7b was obtained from the rearrangement of the methallyloxyflavone 5b (47 mg, 0.16 mmol, 1.0 equiv) after condensation of the intermediate 3,4-chromanedione 6b with 1,2-ethylene diamine 24 (27 μL, 0.40 mmol, 2.5 equiv). Purification on silica gel (petroleum ether: ethyl acetate = 80: 20) afforded the title compound 7b as a bright yellow oil (290 mg, .09 mmol, 57 %).[α]D25 (c 0.5, CHCl3) = + 16.8°. er = 98:2 (ChiralPak AD 1% IPA in hexane, retention time 6.05: 7.26 min, major: minor). 1H NMR (400 MHz, CDCl3) δ 7.83 (br d, J = 7.5 Hz, 1H), 7.39 (app t, J = 7.8 Hz, 1H), 7.30-7.16 (m, 5H), 7.13 (d, J = 8.3 Hz, 1H), 6.96 (app t, J = 7.6 Hz, 1H), 4.79 (br s, 1H), 4.57 (br s, 1H), 4.07 (ddd, J = 16.1, 5.2, 4.0 Hz, 1H), 3.96 (ddd, J = 15.3, 4.4, 1.8 Hz, 1H), 3.42 (ddd, J = 16.9, 15.2, 4.9 Hz, 1H), 3.30 (ddd, J = 16.8, 15.5, 4.9 Hz, 1H), 3.08 (d, J = 14.4 Hz, 1H), 2.95 (d, J = 14.4 Hz, 1H), 1.67 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 159.1, 155.9, 149.5, 140.5, 139.1, 133.0, 128.2 (2C), 127.5, 126.2 (2C), 124.9, 121.9, 120.0, 118.2, 115.7, 86.1, 46.2, 45.8, 44.6, 24.7. IR νmax (film): 3070, 2944, 2845, 1609, 1593, 1461, 1328, 1219, 1118, 994, 895, 699 cm−1. HRMS (ESI+) m/z calculated for C21H21N2O 317.1654 found 317.1679 (M+H).
(S)-6-Allyl-9,10-dichloro-6-phenyl-6H-chromeno[3,4-b]quinoxaline (25)
Pyrazine 25 was obtained using 4,5-dichloro-o-phenylenediamine 22a (71 mg, 0.40 mmol, 2.5 equiv) for the condensation step. Purification on silica gel (petroleum ether: dichloromethane = 80: 20) afforded the pyrazine 25 as a light yellow powder (52 mg, 0.12 mmol, 78 %). M.p. (petroleum ether: CH2Cl2) = 159–160 °C. [α]D25 (c 1.2, CHCl3) = − 245.0°. er = 93:7 (ChiralCel OD 0% IPA in hexane, retention time 9.78: 12.26 min, major: minor). 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1H), 8.24 (dd, J = 7.8, 1.7 Hz, 1H), 8.22 (s, 1H), 7.45 (ddd, J = 8.3, 7.3, 1.7 Hz, 1H), 7.37-7.31 (m, 2H), 7.25-7.12 (m, 4H), 7.09 (ddd, J = 7.8, 7.3, 1.1 Hz, 1H), 5.91 (app tdd, J = 17.1, 10.2, 6.9 Hz, 1H), 5.18 (dd, J = 17.2, 1.8 Hz, 1H), 5.06 (dd, J = 10.2, 2.0 Hz, 1H), 3.55 (dd, J = 14.6, 6.9 Hz, 1H), 3.23 (dd, J = 14.6, 7.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 156.1, 152.3, 144.5, 141.3, 141.0, 140.2, 134.6, 133.5 (2C), 132.8, 130.0, 129.7, 128.2 (2C), 127.6, 125.9 (2C), 125.7, 122.6, 120.7, 119.0, 118.3, 85.5, 45.5. IR νmax (film): 3076, 2918, 1609, 1586, 1560, 1486, 1467, 1453, 1339, 1223, 1182, 1152, 1108, 1028, 908, 884, 731, 700 cm−1. HRMS (ESI+) m/z calculated for C24H17Cl2N2O 419.0718 found 419.0676 (M+H).
(6S,7aS,11aS)-6-Allyl-6-phenyl-7a,8,9,10,11,11a-hexahydro-6H-chromeno[3,4-b]-quinoxaline (28)
Dihydropyrazine 28 was obtained using (1S, 2S)-cyclohexane-1,2-diamine 23 (46 mg, 0.40 mmol, 2.5 equiv) for the condensation step. Purification on silica gel (petroleum ether: ethyl acetate = 90: 10) afforded the title compound 28 as a bright yellow oil (49 mg, 0.14 mmol, 86 %). 1H NMR analysis of the crude showed only one diastereomer. [α]D25 (c 1.2, CHCl3) = − 103.1°. 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 7.8 Hz, 1H), 7.37 (app t, J = 7.8 Hz, 1H), 7.23 (s, 2H), 7.22 (d, J = 3.4 Hz, 2H), 7.21-7.15 (m, 1H), 7.13 (d, J = 8.3 Hz, 1H), 6.93 (app t, J = 7.6 Hz, 1H), 5.88 (dddd, J = 17.0, 10.5, 7.4, 6.6 Hz, 1H), 5.04 (d, J = 17.0 Hz, 1H), 5.03 (d, J = 10.4 Hz, 1H), 3.14 (dd, J = 14.6, 6.5 Hz, 1H), 2.95 (dd, J = 14.6, 7.4 Hz, 1H), 2.86 (dd, J = 11.6, 4.1 Hz, 1H), 2.76 (ddd, J = 15.2, 11.0, 4.1 Hz, 1H), 2.53 (br d, J = 13.2 Hz, 1H), 2.41 (br d, J = 11.3 Hz, 1H), 1.98-1.84 (m, 2H), 1.69-1.57 (m, 1H), 1.55-1.39 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 157.9, 156.1, 149.2, 139.2, 132.9 (2C), 128.3 (2C), 127.6, 126.0 (2C), 125.0, 121.8, 119.9, 118.3, 118.1, 85.5, 60.4, 59.1, 43.6, 33.9, 33.7, 25.6 (2C). IR νmax (film): 3073, 2933, 2857, 1606, 1589, 1574, 1462, 1448, 1321, 1257, 1221, 1055, 993, 916, 710 cm−1. HRMS (ESI+) m/z calculated for C24H25N2O 357.1967 found 357.1971 (M+H).
(S)-2-(6-phenyl-6H-chromeno[4,3-b]quinoxalin-6-yl)acetaldehyde (40)
Following a procedure published in the literature,17 aldehyde 40 was synthesized starting from pyrazine 27 (52 mg, 0.15 mmol, 1.0 equiv) and was obtained as a colorless oil (51 mg, 0.14 mmol, 97 %) after purification on silica gel (petroleum ether: Et2O = 70: 30). [α]D25 (c 1.0, CHCl3) = − 180.0°. 1H NMR (400 MHz, CDCl3) δ 9.86 (dd, J = 2.9, 1.9 Hz, 1H), 8.33 (d, J = 7.8 Hz, 1H), 8.17-8.12 (m, 2H), 7.83-7.73 (m, 2H), 7.43 (app t, J = 7.3 Hz, 1H), 7.31 (d, J = 7.7 Hz, 2H), 7.24-7.16 (m, 2H), 7.20 (d, J = 7.8 Hz, 2H), 7.12 (app t, J = 7.4 Hz, 1H), 3.88 (dd, J = 16.7, 1.9 Hz, 1H), 3.47 (dd, J = 16.6, 2.9 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 199.5, 155.2, 149.9, 143.4, 142.5, 141.2, 140.6, 133.0, 130.5, 129.6, 129.4, 129.3, 128.5 (2C), 128.1, 125.9 (2C), 125.7, 123.0, 121.5, 118.3, 83.5, 53.6. IR νmax (film): 3061, 2842, 2744, 1725, 1607, 1491, 1460, 1346, 1225, 1070, 704 cm−1. HRMS (ESI+) m/z calculated for C23H17N2O2 353.1290 found 353.1261 (M+H).
(S)-4-Benzyl-3-((E)-2-((S)-6-phenyl-6H-chromeno[4,3-b]quinoxalin-6-yl)ethylideneamino)oxazolidin-2-one (42)
Pyrazine-hydrazone 42 was prepared according to a procedure published in the literature18 starting from pyrazine-aldehyde 40 (51 mg, 0.14 mmol, 1.0 equiv) and hydrazine 41 (55 mg, 0.29 mmol, 2.0 equiv). After silica gel chromatography (petroleum ether: ethyl acetate = 60: 40), the title compound was obtained as colorless crystals (75 mg, 0.14 mmol, 99 %). M.p. (petroleum ether: Et2O) = 162–164°C. [α]D25 (c 1.2, CHCl3) = − 83.1°. 1H NMR (400 MHz, CDCl3) δ 8.31 (d, J = 7.9 Hz, 1H), 8.19 (d, J = 7.6 Hz, 1H), 8.13 (d, J = 7.8 Hz, 1H), 8.06 (app t, J = 5.4 Hz, 1H), 7.81-7.71 (m, 2H), 7.42 (app t, J = 7.3 Hz, 1H), 7.39 (d, J = 7.6 Hz, 2H), 7.25-7.13 (m, 5H), 7.18 (d, J = 7.5 Hz, 2H), 7.10 (app t, J = 7.5 Hz, 1H), 6.90 (d, J = 7.3 Hz, 2H), 4.22 (dd, J = 8.2, 3.9 Hz, 1H), 4.18 (dd, J = 16.2, 7.9 Hz, 1H), 4.04 (dd, J = 8.2, 4.2 Hz, 1H), 3.93 (dd, J = 15.1, 5.0 Hz, 1H), 3.74 (dd, J = 14.8, 6.5 Hz, 1H), 2.91 (dd, J = 13.9, 2.8 Hz, 1H), 2.60 (dd, J = 13.9, 8.1 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 155.4, 153.9, 150.8, 150.3, 143.3, 142.4, 141.4, 140.9, 134.9, 133.0, 130.4, 129.4 (2C), 129.2, 129.1 (2C), 128.7 (2C), 128.4 (2C), 127.9, 127.1, 126.0 (2C), 125.6, 122.8, 121.3, 118.3, 84.7, 65.5, 56.8, 44.8, 36.1. IR νmax (film): 3058, 3015, 2921, 1771, 1604, 1555, 1491, 1459, 1401, 1347, 1211, 1087, 1029, 704 cm−1. HRMS (ESI+) m/z calculated for C33H27N4O3 527.2083 found 527.2102 (M+H).
General Procedure for the Preparation of Deuterated Allyloxyflavones 43 and 46
To a solution of commercially available 3-hydroxyflavone (1.00 g, 4.20 mmol, 1.0 equiv) and (1,1-d2-allyl)-alcohol19 (378 mg, 6.30 mmol, 1.50 equiv.) in dry THF (15 mL) was added triphenylphosphine (1.32 g, 5.04 mmol, 1.20 equiv). After complete dissolution of the phosphine, the temperature was brought to 0 °C and diisopropyl azodicarboxylate (DIAD, 1.0 mL, 5.04 mmol, 1.20 equiv) was added dropwise to the mixture via syringe. The reaction was stirred overnight at room temperature and quenched with sat. NaHCO3 solution. After separation of the layers and extraction of the aqueous phase with ether, the combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated. The crude was purified by column chromatography on silica gel (petroleum ether: ether = 50: 50) to afford deuterated allyloxy flavone 43 as a white solid (565 mg, 2.01 mmol, 48 %). Following the same procedure, the methoxy derivative 46 was prepared starting from commercially available 6-methoxyflavonol (536 mg, 2.00 mmol, 1.0 equiv) and was isolated as a white powder (186 mg, 0.60 mmol, 30 %) after purification on silica gel (petroleum ether: ether = 50: 50).
3-Methoxy-2-phenyl-4H-chromen-4-one (53)
To a solution of 3-hydroxyflavone (150 mg, 0.06 mmol) in dry acetone (6 mL) was added dimethyl sulfate (0.10 mL, 0.09 mmol) and K2CO3 (131 mg, 0.09 mmol), and the reaction mixture was refluxed overnight. The mixture was cooled to room temperature and filtered through a pad of Celite®. The solvent was removed in vacuo and the crude product purified by column chromatography on silica gel. 1H NMR (400 MHz, CD2Cl2) δ 8.21 (d, J = 8.1 Hz, 1H), 8.13-8.07 (m, 2H), 7.70 (dd, J = 8.6, 1.5 Hz, 1H), 7.59-7.50 (m, 4H), 7.41 (dd, J = 7.8 Hz, 1H), 3.89 (s, 3H). 13C (400 MHz, CD2Cl2) δ 175.3, 156.0, 155.8, 142.0, 134.0, 131.6, 131.2, 129.0, 129.0, 126.0, 125.2, 124.8, 118.6, 60.4. IRmax (film): 1640, 1614, 1467, 1383, 1213, 1147, 897, 759 cm−1. HRMS (ESI+) m/z calculated for C16H13O3 253.0865 found 253.0857 (M+H).
4-(Tert-butyldimethylsilyloxy)-3-methoxy-2-phenylchromenylium triflate salt (51):20
To a solution of 3-methoxy-2-phenyl-4H-chromen-4-one 53 (10.0 mg, 0.04 mmol) in CD2Cl2 (1.0 mL) was added TBSOTf (9.6 μL, 0.04 mmol). The reaction mixture was stirred at 40 °C for 0.5 h. The crude mixture was directly used for NMR and UV/fluorescence studies without further purification. 1H NMR (500 MHz, CD2Cl2) δ 8.38 (dd, J = 8.2, 1.6 Hz, 1H), 8.25 (d, J = 7.1 Hz, 2H), 7.95 (dd, J = 7.5 Hz, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.69-7.59 (m, 4H), 3.86 (s, 3H), 1.00 (s); 0.88 (s) (9H total), 0.46, 0.03 (s, 6H). 13C (500 MHz, CD2Cl2) δ 174.8, 162.0, 156.3, 140.9, 136.7, 133.5, 130.0, 129.8, 127.4, 126.1, 121.5, 119.1, 61.7, 26.0, 25.0, 18.6, −2.7, −4.0. IRmax (film): 3452 (br), 1736, 1245, 1186, 1029, 640 cm−1.
Supplementary Material
Acknowledgments
Financial support from the National Institutes of Health (P50 GM067041) and AstraZeneca is gratefully acknowledged. We thank Prof. John Snyder and Drs. Baudouin Gerard and Dr. Joshua Giguere (Boston University) for helpful discussions, Drs. John Goodell, Aaron Beeler, and Ms. Susan Cunningham for assistance with UPLC analysis, and Dr. Emil Lobkovsky (Cornell University) for X-ray crystal structure analysis.
Footnotes
Supporting Information Available: Complete experimental procedures and compound characterization data including is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Steven A, Overman LE. Angew Chem, Int Ed. 2007;46:5488–5508. doi: 10.1002/anie.200700612. [DOI] [PubMed] [Google Scholar]; (b) Cozzi PG, Hilgraf R, Zimmermann N. Eur J Org Chem. 2007;36:5969–5994. [Google Scholar]
- 2.(a) Hano Y, Kanzaki R, Fukai T, Nomura T. Heterocycles. 1997;45:867–874. [Google Scholar]; (b) Shi YQ, Fukai T, Ochiai M, Nomura T. Heterocycles. 2001;55:13–20. [Google Scholar]
- 3.For catalytic, asymmetric Claisen (CAC) rearrangements, see: Trost BM, Schroeder GM. J Am Chem Soc. 2000;122:3785–3786.Abraham L, Czerwonka R, Hiersemann M. Angew Chem, Int Ed. 2001;40:4700–4703. doi: 10.1002/1521-3773(20011217)40:24<4700::aid-anie4700>3.0.co;2-6.Uyeda C, Jacobsen EN. J Am Chem Soc. 2008;130:9228–9229. doi: 10.1021/ja803370x.Linton EC, Kozlowski MC. J Am Chem Soc. 2008;130:16162–16163. doi: 10.1021/ja807026z.Rehbein J, Leick S, Hiersemann MJ. Org Chem. 2009;74:1531–1540. doi: 10.1021/jo802303m.Rehbein J, Hiersemann MJ. Org Chem. 2009;74:4336–4342. doi: 10.1021/jo900635k.
- 4.For attempted [3,3] rearrangements of flavone allyl ethers, see: Heimann W, Bär H. Chem Ber. 1965;98:114–119.
- 5.(a) Vinot N, Maitte P. J Heterocyclic Chem. 1989;26:1013–1021. [Google Scholar]; (b) Brown PE, Clegg W, Islam Q, Steele JE. J Chem Soc, Perkin Trans 1. 1990:139–144. [Google Scholar]; (c) Hegab MI, Abdel-Megeid FME, Gad FA, Shiba SA, Sotofte I, Moller J, Senning A. Acta Chem Scand. 1999;53:284–290. [Google Scholar]
- 6.For asymmetric Nazarov cyclizations using scandium-Pybox complexes, see: Liang G, Trauner D. J Am Chem Soc. 2004;126:9544–9545. doi: 10.1021/ja0476664.
- 7.See Supporting Information for complete experimental details.
- 8.(a) Fukuzawa SI, Matsuzawa H, Metoki K. Synlett. 2001;5:709–711. [Google Scholar]; (b) Sauerland SJK, Kiljunen E, Koskinen AMP. Tet Lett. 2006;47:1291–1293. [Google Scholar]; (c) Evans DA, Fandrick KR, Song HJ, Scheidt KA, Xu R. J Am Chem Soc. 2007;129:10029–10041. doi: 10.1021/ja072976i. [DOI] [PubMed] [Google Scholar]; (d) Desimoni G, Faita G, Toscanini M, Boiocchi M. Chem Eur J. 2007;13:9478–9485. doi: 10.1002/chem.200700995. [DOI] [PubMed] [Google Scholar]
- 9.(a) Diedrich C, Grimme S. J Phys Chem A. 2003;107:2524–2539. [Google Scholar]; (b) Stephens PJ, Devlin FJ, Gasparrini F, Ciogli A, Spinelli D, Cosimelli B. J Org Chem. 2007;72:4707–4715. doi: 10.1021/jo070302k. [DOI] [PubMed] [Google Scholar]; (c) Bringmann G, Bruhn T, Maksimenka K, Hemberger Y. Eur J Org Chem. 2009;17:2717–2727. [Google Scholar]
- 10.CAChe 6.1.12.33 was used to perform MM2/MM3 energy minimizations. For related models, see: Evans DA, Masse CE, Wu J. Org Lett. 2002;4:3375–3378. doi: 10.1021/ol026488l.Abraham L, Körner M, Schwab P, Hiersemann M. Adv Synth Catal. 2004;346:1281–1294.Desimoni G, Faita G, Mella M, Piccinini F, Toscanini M. Eur J Org Chem. 2007;9:1529–1534.
- 11.Moncada MC, Pina F, Roque A, Parola AJ, Maestri M, Balzani V. Eur J Org Chem. 2004;2:304–312. [Google Scholar]
- 12.For [2,3]-Wittig rearrangement of silyl enol ethers derived from 3-allyloxy-4-chromanones, see: Sato Y, Fujisawa H, Mukaiyama T. Bull Chem Soc Jpn. 2006;8:1275–1287.
- 13.Drutu I, Krygowski ES, Wood JL. J Org Chem. 2001;66:7025–7029. doi: 10.1021/jo015741c. [DOI] [PubMed] [Google Scholar]
- 14.An alternative mechanism involving Prins cyclization of 49 is also possible but is less likely based on benzopyrylium stability and reversibility of the reaction, see: Olier C, Kaafarani M, Gastaldi S, Bertrand MP. Tetrahedron. 2010;66:413–445.
- 15.Lee YG, Ishimaru K, Iwasaki H, Ohkata K, Akiba K. J Org Chem. 1991;56:2058–2066. [Google Scholar]
- 16.Fluorescence emission of benzopyryliums: Alluis B, Dangles O. Helv Chim Acta. 1999;82:2201–2212.Drabent R, Pliszka B, Huszcza-Ciokowska G, Smyk B. Spectrosc Lett. 2007;40:165–182.
- 17.Yu W, Mei Y, Kang Y, Hua Z, Jin Z. Org Lett. 2004;6:3217–3219. doi: 10.1021/ol0400342. [DOI] [PubMed] [Google Scholar]
- 18.Friestad GK, Marié JC, Suh Y, Qin J. J Org Chem. 2006;71:7016–7027. doi: 10.1021/jo061158q. [DOI] [PubMed] [Google Scholar]
- 19.Krompiec S, Kuźnik N, Urbala M, Rzepa J. J Mol Catal A: Chem. 2006:198–209. [Google Scholar]
- 20.Lee YG, Ishimaru K, Iwasaki H, Ohkata K, Akiba K. J Org Chem. 1991;56:2058–2066. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.