


Abstract
We describe an algorithm for gene identification in DNA sequences derived from shotgun sequencing of microbial communities. Accurate ab initio gene prediction in a short nucleotide sequence of anonymous origin is hampered by uncertainty in model parameters. While several machine learning approaches could be proposed to bypass this difficulty, one effective method is to estimate parameters from dependencies, formed in evolution, between frequencies of oligonucleotides in protein-coding regions and genome nucleotide composition. Original version of the method was proposed in 1999 and has been used since for (i) reconstructing codon frequency vector needed for gene finding in viral genomes and (ii) initializing parameters of self-training gene finding algorithms. With advent of new prokaryotic genomes en masse it became possible to enhance the original approach by using direct polynomial and logistic approximations of oligonucleotide frequencies, as well as by separating models for bacteria and archaea. These advances have increased the accuracy of model reconstruction and, subsequently, gene prediction. We describe the refined method and assess its accuracy on known prokaryotic genomes split into short sequences. Also, we show that as a result of application of the new method, several thousands of new genes could be added to existing annotations of several human and mouse gut metagenomes.
INTRODUCTION
A metagenomic sample is a heterogeneous mixture of rather short sequences originated from a shotgun sequencing of a microbial community. A vast majority (99%) of microbial species in a given community are likely to be non-cultivable (1). Many protein-coding regions in a new metagenome are likely to code for barely detectable homologs of already known proteins. Therefore, along with comparative genomic methods that rely on sequence similarity search, ab initio methods able to identify genes having no similarity to ones existing in databases are vitally important tools of metagenomic sequence analysis. Sequence similarity-based methods possess high specificity and ability to characterize function of predicted genes (2–5). Ab initio gene finders exhibit high sensitivity along with sufficiently high specificity. The standard tools for ab initio prokaryotic gene prediction such as EasyGene (6), GeneMarkS (7) or Glimmer (8) were not designed to work with short sequence fragments from unknown genomes. However, a special method for assignment of parameters of a gene finder, the ‘heuristic model’ method, designed for accurate gene finding in short prokaryotic sequences with anonymous origin was proposed 4 years prior to the advent of metagenomics (9).
The idea was to bypass traditional ways of parameter estimation such as supervised training on a set of validated genes or unsupervised training on an anonymous sequence supposed to contain a large enough number of genes. It was proposed to use dependencies, apparently formed in evolution, between codon frequencies and genome nucleotide composition. Therefore, the vector of codon frequencies, critical for the model parameterization, could be derived from frequencies of nucleotides observed in a short sequence. This ‘heuristic model’ method has been used for (i) reconstructing codon frequency vector for gene finding in viral genomes (10) and (ii) initializing the algorithms for iterative parameters estimation for prokaryotic as well as eukaryotic gene finders (7,11–12). Recently, several new methods for ab initio gene finding in metagenomic sequences have been developed (13–15). Particularly, the authors of MetaGene (14) saw a significant potential in the ‘heuristic model’ method (9); they have extended the method to use of di-codon frequencies. The authors of new tools have shown that their performance is comparable to performance of the original ‘heuristic model’ method (Supplementary Table S3 in (14)) (16).
In this article, we describe further improvement of the ‘heuristic model’ method. A key observation made upon analysis of 17 genomes (9) was that frequencies of nucleotides in the three codon positions depend linearly, though with distinctly different slope coefficients, on global nucleotide frequencies. In turn, due to the second Chargaff rule (17), this observation means that nucleotide frequencies in the three codon positions depend linearly on genomic GC content. These linear functions were used to reconstruct codon frequencies in the whole genome using information derived from its short sequence fragment and to derive parameters of the ‘heuristic’ second-order Markov models [the Heuristic ALgorithm (HAL)-99 models] for a gene finding algorithm. Gene finding with ‘heuristic’ models was proved to be effective for viral genomes (10,18) as well as for metagenomic sequences (Nikos Kyrpides, personal communication).
With hundreds of new prokaryotic genomes available, it is now possible to enhance the original approach and to utilize direct polynomial and logistic approximations of oligonucleotide frequencies. Also, the analysis of a larger set of genomic sequences has shown that patterns of dependence of codon frequencies from nucleotide frequencies are distinctly different in the two domains of life, bacteria and archaea. Interestingly, distinctly different patterns of the dependence of codon frequencies from genome nucleotide composition have also been observed in mesophilic and thermophilic species. Thus, for gene finding in a short sequence, it is worthwhile to make a simultaneous use of two models, bacterial and archaeal, or mesophilic and thermophilic.
We have assessed the accuracy of a hidden Markov model (HMM) based gene finder, GeneMark.hmm, using the new models on the sets of short sequences obtained by splitting known genomes into equal length fragments (ranging from 72 to 1100 nt). The results demonstrate a higher accuracy in comparison with several other existing methods as well as with the use of original heuristic models.
Application of whole-genome shotgun sequencing to studies of mixed microbial communities, such as gut microbiota of human and mouse have a potential to reveal details of a large picture of the host metabolism combining microbial and mammalian elements. It is estimated that human intestinal microbiota consists of 1013–1014 microorganisms. This microbiome should contain at least 100 times as many genes as a human genome per se. Still, due to diversity of the microbiome, metagenomic data sets consist mainly of unassembled single-read sequences. We have applied the new method to the sequences of human and mouse gut microbial communities (19–20). We detected a large number of protein-coding regions not yet annotated; for a significant fraction of the protein products of newly predicted genes, we found homologs among known proteins. Notably, identification of incomplete genes carries valuable information for reconstruction of metabolic networks and signaling pathways. Since a number of protein-coding regions in a metagenome may be counted by millions (4), improving accuracy of gene finding by a percentage point would affect accurate prediction of tens of thousands of genes of the organisms constituting microbial communities. Therefore, development of accurate metagenome-specific methods is of critical importance for quality analysis of sequence data produced by the next generation sequencing technologies (21).
MATERIALS
Sequence data of 582 complete prokaryotic genomes (534 bacteria and 48 archaea; genetic code 11) were from the NCBI RefSeq database. Length of the shortest genome in the sample, Nanoarchaeum equitans (22), was 490 kb. Genome GC contents varied from 16.6% to 74.9%. The data on optimal growth temperature for 357 prokaryotic species (Supplementary Table S1) was from the NCBI Entrez genome database (23). Metagenomic sequence data and annotation for human and mouse gut microbiomes were from the JGI IMG/M database (24).
Test sets
For assessment of gene prediction accuracy, we used fragments from whole genomes of 29 bacterial and 15 archaeal species (containing 50 microbial chromosomes) listed in Supplementary Table S2. The genomic sequences were split into equal length non-overlapping fragments, with length ranging from 72 to 1100 nt; fragment annotations were derived from corresponding RefSeq records. To retain genes with most reliable annotation, fragments overlapping annotated hypothetical genes were discarded.
METHODS
Heuristic method of model parameters derivation
A conventional ab initio gene finding algorithm employs a probabilistic model of genomic sequence containing protein- and non-coding regions. Gene prediction accuracy critically depends on precision of the estimation of model parameters that are genome specific. The number of parameters of the probabilistic model of a protein-coding region, a three-periodic Markov chain model (25) increases exponentially (∼4N) with the Markov chain order N. The higher the model order, the larger the size of a set of training sequences required for parameter estimation without over-fitting, e.g. in practice, estimation of parameters of the fifth-order model is made on a set of verified protein-coding sequences with total length of 400 000 nt. Note that in our observations even if a larger training set is available, models with an order higher than five did not make a noticeable difference in power of discrimination between coding and non-coding regions (26).
Metagenomic sequence data, mixtures of shotgun sequences from numerous members of microbial communities, are populated with short sequences (with length ≤400 nt). The task is to identify a complete or incomplete protein-coding region residing in a short fragment. A gene finding algorithm, e.g. GeneMark.hmm, could be applied to solve this task should we know or are able to derive the genome-specific model parameters. However, the fact that the genomic context of the short fragment is missing precludes the use of standard approaches for parameter estimation. In a previous work (9), we proposed a method to infer parameters of the three-periodic second-order Markov model for gene finding in a short (e.g. 400 nt) sequence fragment of unknown origin. First, we have identified dependencies that link the nucleotide composition of a genome with the genome-specific codon frequencies. These dependencies are apparently the strongest determinants of a genome-wide synonymous codon usage pattern (27–28). Second, nucleotide frequencies observed in a short DNA fragment served as estimates of global nucleotide frequencies in the whole genome, the source of the short fragment. Then, starting from estimated values of global nucleotide frequencies we reconstructed the genome-specific codon frequencies.
In more details, in the first step, analysis of genomes with known annotation, by taking one genome at a time, we determined frequencies of occurrence of each of the 61 codons in a genome-wide set of annotated protein-coding regions. The codon frequency data determines 12 genome-specific positional frequencies f1X, f2X, and f3X, where X = A, C, G, T in the three codon positions. For a sample of known genomes, r = 1, 2, … R with observed fkX, k = 1, 2, 3 the fkX values were approximated by linear regression on the global nucleotide frequency fX, X = A, C, G, T. Initially, in 1999, the analysis was done for 17 completely sequenced genomes [Figure 1 in (9), see also (29)].
Figure 1.

Observed frequencies of 4 nt in the three codon positions (first: green; second: blue; third: red) as functions of genome GC content for 319 bacterial genomes. Nucleotides G and T have more contrast in frequencies in the first and second position in comparison with A and C. Frequencies in the third codon position are most sensitive to genome GC content.
Now, with many more sequenced genomes available, the linear regression analysis was done for 319 bacterial genomes (Figure 1) as well as for 38 archaeal genomes (Table 1). Graphs in Figure 1 look different from graphs in Figure 1 in (9) for the following reasons. The global nucleotide frequency variable strongly correlates with the genome GC content. The second Chargaff rule states that at a whole-genome level, nucleotide frequencies, fX, X = A, C, G, T in a single DNA strand are such that fA ∼ fT and fG ∼ fC. Therefore, four nucleotide frequencies observed in whole genomes can be derived from a single parameter, the GC content; if s is a genomic GC content, fG + fC, then frequencies of nucleotides fG = fC = s/2 and fA = fT = (1 − s)/2. Thus, new graphs of positional nucleotide frequencies (Figure1) were plotted as functions of genomic GC content.
Table 1.
Values of slopes of linear regression lines (such as in Figure1) showing slope values for frequencies of nucleotides in the three codon positions for bacterial (B) and archaeal species (A) and the same for mesophilic (M) and thermophilic (T) species
| Nucleotide type | Archaea/ bacteria | Codon position |
Mesophilic/ thermophilic | Codon position |
||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 1 | 2 | 3 | |||
| A | B | −0.43 | −0.34 | −0.91 | M | −0.44 | −0.34 | −0.92 |
| A | −0.50 | −0.29 | −0.97 | T | −0.55 | −0.32 | −0.92 | |
| C | B | 0.40 | 0.25 | 1.07 | M | 0.40 | 0.25 | 1.07 |
| A | 0.38 | 0.21 | 1.04 | T | 0.51 | 0.25 | 1.01 | |
| T | B | −0.25 | −0.11 | −0.93 | M | −0.25 | −0.11 | −0.93 |
| A | −0.24 | −0.10 | −0.86 | T | −0.25 | −0.15 | −0.81 | |
| G | B | 0.28 | 0.20 | 0.78 | M | 0.28 | 0.20 | 0.78 |
| A | 0.36 | 0.19 | 0.79 | T | 0.30 | 0.22 | 0.72 | |
The table shows almost identical sets of slope values for bacterial and mesophilic divisions. Slope values of archaeal and thermophilic divisions are distinctly different.
Further, the s value determined for a short genomic fragment is used as predictor of positional nucleotide frequencies fkX, where k = 1, 2, 3 and X = A, C, G, T. Assuming that a codon frequency, fXYZ, is proportional to product f1Xf2Yf3Z we could obtain an initial approximation of codon frequency f′XYZ. Additional correction comes from the value of predicted frequency of encoded amino acid α, fα (s) determined by linear regression of frequencies of amino acid α observed in corresponding proteomes with respect to the genomic GC contents. To give an example, for alanine with four synonymous codons, predicted frequency fGCT of codon GCT is:
| (1) |
Note that the left part of the formula does not change in further iterations (i.e. by substituting thus found fGCT into right part of the equation).
Finally, it was shown that all parameters of the three-periodic Markov chain model of a protein-coding region could be determined as functions of the set of predicted codon frequencies (9). A model of non-coding region was defined as the multinomial model, the zero-order Markov model. GC content of non-coding regions was observed to have strong correlation with the genome-wide GC content (Figure 2). Therefore, nucleotide frequencies observed in a relatively short DNA fragment are accepted as estimates of four parameters of the non-coding region model. Thus parameterized models of protein- and non-coding regions are ready for use in a gene finding program such as GeneMark.hmm (7,30).
Figure 2.

Dependence of GC content of genomic functional regions on genome-wide GC content. Protein-coding and non-coding regions were identified by GeneMarkS in randomly selected 155 bacterial and 16 archaeal genomes; tRNA genes by tRNAScan-SE, while rRNA genes were selected as annotated in RefSeq. Triangles and circles with species names indicate GC content of tRNA and rRNA genes of archaeal thermophiles.
Refined methods for estimation of parameters of the model of protein-coding regions
With hundreds of prokaryotic genomes sequenced and annotated, it is possible to use non-linear (polynomial or logistic) regression to more precisely determine the dependence of codon frequencies on genome GC content. To choose the order of regression polynomial, we recall the observed linearity in dependence of frequencies of nucleotides in the three codon positions on genome GC content; product of three linear functions is natural to approximate by the third-order polynomial A + Bs + Cs2 + Ds3; the least squares method is applied to estimate the four coefficients.
A logistic function 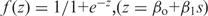 could approximate observed codon frequencies scaled with respect to the minimum and maximum values: f scaled = ( f − f
min)/( f
max − f
min); this approach was used earlier (14). A generalized linear regression function glmfit from the MatLab Statistics Toolbox was used to determine β0 and β1 parameters from the equation
could approximate observed codon frequencies scaled with respect to the minimum and maximum values: f scaled = ( f − f
min)/( f
max − f
min); this approach was used earlier (14). A generalized linear regression function glmfit from the MatLab Statistics Toolbox was used to determine β0 and β1 parameters from the equation 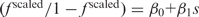 . For a given s, codon frequency was determined as follows. With
. For a given s, codon frequency was determined as follows. With 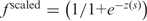 , predicted codon frequency was determined as f(s) = f
scaled
* ( f
max − f
min) + f
min. Frequencies of 64 nucleotide triplets residing in each of two other reading frames could be reconstructed by either one of the two regression approaches outlined above. The three vectors of triplet frequencies thus reconstructed for a short sequence S with respect to its GC content are sufficient for computing parameters of the second-order three-periodic Markov chain model, the model of protein-coding region in an unknown genome sequence S came from.
, predicted codon frequency was determined as f(s) = f
scaled
* ( f
max − f
min) + f
min. Frequencies of 64 nucleotide triplets residing in each of two other reading frames could be reconstructed by either one of the two regression approaches outlined above. The three vectors of triplet frequencies thus reconstructed for a short sequence S with respect to its GC content are sufficient for computing parameters of the second-order three-periodic Markov chain model, the model of protein-coding region in an unknown genome sequence S came from.
Summarizing the options described above, parameters of the three-periodic second-order Markov chain could be determined by several alternative techniques: (i) reconstructing codon frequencies from predicted nucleotide frequencies in the three codon positions, with subsequent derivation of triplet frequencies in the second and third frame (9), the technique named above HAL-99; (ii) reconstructing codon frequencies by the third-order polynomial functions, with derivation of triplet frequencies in two other frames as in HAL-99, C-3 technique; (iii) reconstructing frequencies of K-mers, K=3, 4, 5, 6 in the three frames with the K-order polynomial regression, K–K techniques; and (iv) reconstructing frequencies of K-mers, K=3, 4, 5, 6 in the three frames with the logistic regression, K–L techniques.
We show examples of typical regression graphs for codons AAT, GCC, TTG and CGT frequencies observed in bacterial genomes (Figure 3); the regression curves were produced by the HAL-99, C-3 and 3-L techniques. Codon AAT is A and T ‘rich’. As a rule, frequencies of eight out of 64 AT rich codons show monotonous decrease over the whole GC range with a rather small variation in any given GC content (Figure 3a). The codon GCC frequency, as well as frequency of other seven GC rich codons increases as genome GC content grows (Figure 3b). Frequencies of codons with mixed composition, such as TTG and CGT (Figure 3c and d) show more variation particularly in the mid GC range, and the task of approximation of these frequencies by a function of single variable is more challenging. It was reported that in genomes with the same GC content, the differences in codon frequencies correlate with differences in optimal growth temperature, t (31). These observations motivate introduction of yet another technique, designated as the C-M technique using approximation of codon frequencies by a function of two variables 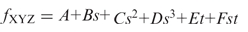 , the sequence GC content, s, and the temperature of microbiome habitat, t, with parameters determined by multiple regression (Figure 4).
, the sequence GC content, s, and the temperature of microbiome habitat, t, with parameters determined by multiple regression (Figure 4).
Figure 3.

Characteristic cases of codon frequency dependence on genome GC content. Each panel shows observed frequencies of a given codon in 319 bacterial genomes. Mesophilic, psychrophilic and thermophilic species are shown as light blue, dark blue and purple dots, respectively. Three techniques of approximating dependence of codon frequency from genome GC content are illustrated: 1999 heuristic model (HAL-99, black dotted line); logistic regression (3-L, green dotted line); and order three polynomial regression (C-3, red dotted line). Plots for 61 codons are available at http://exon.gatech.edu/GeneMark/metagenome/Training/PlotPDF/BAC2D.pdf.
Figure 4.

Result of multiple regression polynomial fitting of codon frequency as a function of both genomic GC content and optimal growth temperature. Note that the scales in Z-axes are not the same. Frequency of AAT mostly depends on genomic GC content, adding one more predictor variable explained just additional 1% variance (R2 value increased from 96% to 97%). Frequency of AGG largely depends on the optimal growth temperature; 30% of variance was explained by the temperature predictor. (R2 value increased to 31% from 1%.) The surface plot indicates a codon frequency change by color, from low (blue) to high (red). Plots for all 61 codons are available at http://exon.gatech.edu/GeneMark/metagenome/Training/PlotPDF/BAC3Dmulti.pdf.
Dual mode of using heuristic models
Linear trends in frequencies of nucleotides in the three codon positions with respect to genome GC content have been observed to be different in bacteria and archaea (Table 1). Therefore, two distinct heuristic models could be built, one for bacterial and another for archaeal sequences. Notably, no pre-processing is needed to identify a domain of life the short sequence fragment represents. The bacterial and archaeal heuristic models can be used in the GeneMark.hmm algorithm simultaneously (Figure 5), similarly to the simultaneous use of typical and atypical gene models (30). A protein-coding region, if present in the sequence, is supposed to be recognized by either bacterial or archaeal model.
Figure 5.

Hidden states diagram of the generalized hidden Markov model (HMM) used in the GeneMark.hmm algorithm; this is the case of using bacterial and archaeal model pair (a similar diagram would be valid for use of mesophilic and thermophilic model pair).
Alternatively, all prokaryotic species could be divided into mesophilic and thermophilic (310 mesophilic and 47 thermophilic in our reference set of sequenced genomes). Then, application of regression analysis of nucleotide frequencies in the three codon positions produced once again two distinct sets of 12 linear functions (Table 1). The two heuristic models (built for mesophiles and thermophiles) could also be used simultaneously in GeneMark.hmm. However, such a dual model seems to be less effective for practical use, as the temperature of a microbiome habitat is supposed to be known and one of the models could be chosen a priori.
In the Results section, we designate the model pairs by suffix BA or TM, e.g. 3-3BA stands for use a pair of bacterial and archaeal models derived by the third-order polynomial approximation of triplet frequencies.
Length distributions for partial and complete genes
An average gene length in a prokaryotic genome is about 900 nt. In a metagenomic sequence shorter than 900 nt, it is more likely to observe a part of a gene than a complete gene. To account for the frequent occurrence of partial coding sequences (CDS), we have to modify a formula for the gene length frequency distribution used in GeneMark.hmm for gene finding in complete genomes. This distribution is approximated by p(d) = Nc(d/dc)2exp(−d/dc), the γ distribution formula with two parameters (30). Also, the length distribution of non-coding regions is approximated by exponential distribution p(d)=Nnexp(−d/dn). Parameters, dc and dn are estimated by fitting to empirical distributions of gene length in known genomes. It was observed that values of dc and dn vary little among different prokaryotic species. Therefore, values of these parameters in the algorithm were given as default values: dc = 300 and dn = 150. The formula for length distribution of protein-coding regions in short metagenomic sequences is p(d) = Np(d2 + dcd +2d2c)exp(−d/dc), with parameters Np and dc. Corresponding graphs of theoretical and observed length distributions are shown in Figure 6. To avoid predicting too short partial genes, we have effectively defined 60 nt as minimum length of a predicted gene by setting p(l ≤ 60) = 0.
Figure 6.

Length distributions of coding and non-coding regions observed and expected in 700-nt long fragments of E. coli K12 genome. An average E. coli gene length is about 900 nt. Therefore, some of the 700-nt fragments are 100% coding, hence the peak of frequency of partial CDS length (light blue) at 700-nt point. Similarly, the frequency of length of non-coding region has two peaks at 15 and 700 nt. (a) Complete CDS length distribution is approximated by function g(d) = Nc(d/dc)2exp(−d/dc), dc = 300; (b) Non-coding region length distribution is approximated by function f(d)=Nnexp(−d/dn), dn = 150; (c) Partial CDS length distribution is approximated by function p(d) = Np(p(d) = Np(d2 + dcd +2d2c)exp(−d/dc), dc = 300.
RESULTS
Choice of parameters of length distributions
To analyze how accuracy of GeneMark.hmm depends on dc and dn values, we used sets of 700-nt long fragments of Escherichia coli and Bacillus subtilis genomes; the model used in the runs was the C-3BA one. Sensitivity (Sn) and specificity (Sp) were determined by comparison of gene predictions with fragments annotation. A prediction was accounted as a true positive if locations of the predicted and annotated 3′-ends matched or for partial genes without 3′-ends there was a match between predicted and annotated reading frames. The values of dc could vary from 100 to 800, while values of dn varied from 100 to 300. Particularly, dependence of Sn and Sp for dc = 800 while dn varied from 100 to 300 as indicated by blue line in Figure 7; similarly, dependence of Sn and Sp for dn = 100 while dc is varied from 100 to 800 as indicated by purple line. The dc, dn setting used for analysis of complete genomes (300, 150) is indicated by red dot. Combining larger dc (800) and smaller dn (100) leads to a substantial increase of Sp and a slight decrease of Sn. This result is due to the decrease in number of predicted short genes, many of them not matching annotation. To facilitate comparison of average values S = (Sn + Sp)/2, produced by the program runs with different dc and dn values, the constant S level lines (with slope −1) were plotted in Figure 7a and b. Performance (Sn, Sp) of MetaGene and MetaGeneAnnotator (with default parameters) was depicted for each of the two genomes as well; one can see that the performance is high, though it can be outperformed, especially in the E. coli, by GeneMark.hmm with a wide range of parameters dc and dn. As the result of modeling, we have used dc = 800 and dn = 100 in further analysis of artificial and real metagenomic sequences.
Figure 7.

Values of Sn and Sp obtained upon variations of parameters dn and dc. Light blue dots represent Sn and Sp values obtained for each of 1491 combinations of (dn, dc) parameters. Blue and purple lines correspond to variation of dn with dc = 800 and variation of dc with dn = 100, respectively. Red dots correspond to (dn, dc) setting (150, 300) that is used by default for complete genomes. Also shown are the highest Sn and the highest Sp (blue squares), the highest (Sn + Sp)/2 (yellow triangles). Use of pair of models, the native model (derived by the GeneMarkS from a complete genome) and the heuristic model HAL-99, produced the Sn and Sp values shown by orange diamonds. The Sn and Sp of the MetaGene and MetaGeneAnnotator predictions are shown by green and blue dots, respectively.
Tests on sequences with fixed length
We used the GeneMark.hmm program with the pairs of heuristic models, bacterial and archaeal (or mesophilic and thermophilic) derived by methods described above to analyze sequence fragments with fixed length, from 50 microbial chromosomes (Supplementary Table S2). All models were tested on sets of fragments with length of 400 and 700 nt; moreover, the models with highest performance were tested on sets of fragments with shorter (down to 72 nt) and longer (up to 1100 nt) lengths. Performance characteristics of different models are shown in Table 2 (with more details provided in Supplementary Tables S3–S6). Observed values of (Sn + Sp)/2 were clustered between 94.5% and 96.5% for 700-nt long fragments and between 93.5% and 96.0% for 400-nt long fragments. Interestingly, among the triplet-based models, C-3BA, C-3MT, 3-3BA and 3-LBA, the codon frequency derived models, C-3BA and C-3MT, demonstrated higher performance than 3-3BA and 3-LBA models, where frequencies of triplets as functions of GC content are independently approximated in each frame. Use of higher order Markov models: the third order, 4-4BA, the fourth order, 5-5BA, and the fifth order, 6-6BA and 6-LBA, resulted in similar performance, with differences in (Sn + Sp)/2 values <0.3%; this performance level is comparable to performance of the second-order models C-3BA and C-3MT. Still, a slightly higher (Sn + Sp)/2 for 700- and 400-nt long fragments was achieved with the use of 6-LBA heuristic model containing a pair of the fifth-order model, bacterial and archaeal, with parameters obtained by logistic regression approximation of hexamer frequencies. Note that the MetaGene authors found performance of MetaGene on 700-nt fragments comparable to performance of GeneMark.hmm with HAL-99 model (Supplementary Table S3 in 14). This result corresponds to our observations as well (Table 2).
Table 2.
Accuracy of gene prediction in 700- and 400-nt long fragments from 50 microbial chromosomes (listed in Supplementary Table S2)
| Program | Model | Sn | Sp | (Sn + Sp)/2 |
|---|---|---|---|---|
| 700 nt | ||||
| GeneMark.hmm | HAL-99 | 94.93 | 94.28 | 94.61 |
| C-3BA | 96.84 | 95.17 | 96.01 | |
| C-3MT | 96.86 | 95.04 | 95.95 | |
| C-MBA | 97.00 | 93.77 | 95.39 | |
| 3-3BA | 96.51 | 94.18 | 95.35 | |
| 3-LBA | 96.69 | 94.19 | 95.44 | |
| 4-4BA | 97.23 | 94.83 | 96.03 | |
| 5-5BA | 97.25 | 94.91 | 96.08 | |
| 6-6BA | 97.04 | 94.99 | 96.02 | |
| 6-LBA | 97.42 | 94.89 | 96.16 | |
| MetaGene | 97.57 | 92.36 | 94.97 | |
| MetaGeneAnnotator | 97.49 | 93.60 | 95.55 | |
| 400 nt | ||||
| GeneMark.hmm | HAL-99 | 93.81 | 93.38 | 93.59 |
| C-3BA | 96.24 | 94.80 | 95.52 | |
| C-3MT | 96.32 | 94.72 | 95.52 | |
| C-MBA | 96.34 | 93.31 | 94.83 | |
| 3-3BA | 95.64 | 93.85 | 94.74 | |
| 3-LBA | 95.97 | 93.77 | 94.87 | |
| 4-4BA | 96.70 | 94.57 | 95.63 | |
| 5-5BA | 96.75 | 94.66 | 95.70 | |
| 6-6BA | 96.49 | 94.77 | 95.63 | |
| 6-LBA | 96.99 | 94.63 | 95.81 | |
| MetaGene | 97.22 | 91.08 | 94.15 | |
| MetaGeneAnnotator | 97.15 | 92.35 | 94.75 | |
Values of length distribution parameters: dn = 100 and dc = 800.
The use of models utilizing higher order oligonuclotides brought in a marginal improvement of (Sn + Sp)/2 for gene prediction in 400- and 700-nt fragments in comparison with the codon-based models, e.g. C-3BA and C-3MT (Table 2, Supplementary Tables S3–S6). This observation is in agreement with findings of other authors that use of the fifth-order Markov chains and/or di-codon frequencies leads to a slight increase in gene prediction accuracy (13–15). In order to determine accuracy of gene prediction in fragments with length other than 400 and 700 nt, the particular values used in tests by several authors, we have derived from the 50 microbial chromosomes, 11 additional test sets with fragment lengths varying from 72 to 1100 nt (Table 3). Here, in comparison of MetaGene and MetaGeneAnnotator with GeneMark.hmm using HAL-1999, C-3BA and 6-LBA models, we see that GeneMark.hmm with 6-LBA model performs marginally better in terms of Sn and Sp average. Yet, MetaGene shows higher Sn for all the 13 test sets, while C-3BA model shows higher Sp for fragment length longer than 200 nt. For better visualization, we show the programs’ performance as functions of fragment length for the sequence sets with fragment length ≥100 nt (Figure 8). Notably, since the second-order C-3BA model is very close to the 6-LBA model in terms of performance, we use the C-3BA model in several applications discussed below along with the 6-LBA model (Tables 2–3, Supplementary Tables S3–S6).
Table 3.
Gene prediction accuracy of GeneMark.hmm with three different heuristic models, as well as MetaGene and MetaGeneAnnotator observed on the sets of sequence fragments with length from 72 to 1100 nt fragments from 50 microbial chromosomes
| Length | 1999 HAL | MetaGene | MetaGeneAnnotator | C-3BA | 6-LBA | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 72 | Sn | 64.5 | 72.8 | n/a | n/a | 84.2 | 83.1 | 77.8 | 81.7 | 81.2 | 84.0 |
| Sp | 81.1 | n/a | 82.1 | 85.5 | 86.8 | ||||||
| 96 | Sn | 77.0 | 80.8 | n/a | n/a | 90.6 | 87.3 | 85.9 | 87.3 | 88.6 | 89.1 |
| Sp | 84.6 | n/a | 84.0 | 88.7 | 89.6 | ||||||
| 100 | Sn | 78.4 | 81.8 | 91.2 | 87.8 | 90.9 | 87.8 | 87.0 | 88.1 | 89.4 | 89.7 |
| Sp | 85.1 | 84.5 | 84.6 | 89.2 | 90.0 | ||||||
| 200 | Sn | 90.7 | 90.8 | 95.7 | 92.0 | 95.6 | 92.5 | 94.3 | 93.9 | 95.6 | 94.6 |
| Sp | 90.9 | 88.3 | 89.5 | 93.4 | 93.6 | ||||||
| 300 | Sn | 92.7 | 92.5 | 96.8 | 93.3 | 96.7 | 93.9 | 95.5 | 94.8 | 96.4 | 95.2 |
| Sp | 92.3 | 89.9 | 91.1 | 94.1 | 94.0 | ||||||
| 400 | Sn | 93.9 | 93.6 | 97.3 | 94.1 | 97.2 | 94.7 | 96.3 | 95.5 | 97.0 | 95.8 |
| Sp | 93.3 | 90.9 | 92.2 | 94.7 | 94.5 | ||||||
| 500 | Sn | 94.4 | 94.2 | 97.5 | 94.5 | 97.4 | 95.2 | 96.6 | 95.8 | 97.2 | 96.0 |
| Sp | 93.9 | 91.5 | 92.9 | 95.0 | 94.8 | ||||||
| 600 | Sn | 94.8 | 94.4 | 97.6 | 94.7 | 97.5 | 95.4 | 96.9 | 95.9 | 97.5 | 96.1 |
| Sp | 94.0 | 91.9 | 93.3 | 95.0 | 94.7 | ||||||
| 700 | Sn | 95.0 | 94.6 | 97.6 | 94.9 | 97.5 | 95.5 | 96.9 | 96.0 | 97.4 | 96.1 |
| Sp | 94.2 | 92.2 | 93.4 | 95.0 | 94.8 | ||||||
| 800 | Sn | 95.2 | 94.8 | 97.7 | 95.0 | 97.6 | 95.6 | 97.0 | 96.1 | 97.6 | 96.2 |
| Sp | 94.3 | 92.4 | 93.6 | 95.1 | 94.8 | ||||||
| 900 | Sn | 95.4 | 94.9 | 97.7 | 95.1 | 97.7 | 95.8 | 97.1 | 96.1 | 97.6 | 96.2 |
| Sp | 94.4 | 92.5 | 93.8 | 95.1 | 94.7 | ||||||
| 1000 | Sn | 95.5 | 95.0 | 97.9 | 95.3 | 97.8 | 95.8 | 97.2 | 96.2 | 97.7 | 96.3 |
| Sp | 94.5 | 92.8 | 93.9 | 95.2 | 94.8 | ||||||
| 1100 | Sn | 95.7 | 95.1 | 97.8 | 95.3 | 97.7 | 95.9 | 97.3 | 96.2 | 97.7 | 96.2 |
| Sp | 94.5 | 92.9 | 94.0 | 95.2 | 94.7 | ||||||
The best numbers are in bold.
Figure 8.

Gene prediction accuracy of GeneMark.hmm with three different heuristic models, as well as MetaGene and MetaGeneAnnotator observed on the sets of sequence fragments with length from 100 to 1100 nt from 50 microbial chromosomes.
Inferring origin of genes and sequence fragments
Upon analysis of short sequence fragments from 50 microbial chromosomes, a run of GeneMark.hmm with bacterial and archaeal model pair not only produced a list of predicted genes but also an indication of a likely origin of each gene (Supplementary Tables S7 and S8). We have seen that a vast majority of genes in bacterial (archaeal) sequence fragments was predicted by the bacterial (archaeal) model. Similarly, a vast majority of genes in thermophilic (mesophilic) sequence fragments were predicted by thermophilic (mesophilic) model. Interestingly, for the thermophilic bacteria Thermotoga maritima (with optimal growth temperature of 80°C) the archaeal model predicted 3137 out of a total of 3225 fragmented genes, corroborating the findings made in the original T. maritima genome paper (32) of massive horizontal influx of genes transferred from archaeal species (33). On the other hand, a vast majority of genes in Methanosarcina acetivorans, identified in many sources as mesophilic archaea, were predicted by the thermophilic model. This result corresponds to observations that M. acetivorans is able to live in deep sea hydrothermal vents. Similar observations were made for bacteria Aquifex aeolicus (34) living in high temperature, as well as for low temperature archaeal species such as Haloarcula, Halobacterium and Methanosphaera (Supplementary Tables S7 and S8).
In short fragments, one rarely sees more than one gene per fragment; therefore, a gene characterization could normally be extended to the whole sequence fragment. Rare cases, when there are several genes in a metagenomic fragment each predicted by different models are worthwhile to set aside as candidates for case study of horizontal gene transfer. Throughout, in the test set of 700-nt long fragments, with a total of 31 584 archaeal (136 210 bacterial) fragments, GeneMark.hmm with C-3BA model misclassified 2757 fragments as bacterial type (16 284 fragments as archaeal type); thus archaeal fragments were identified correctly in 91.27% of cases and bacterial fragments were identified correctly in 88.04% of cases (Supplementary Table S7, column C-3BA). Similar analysis for a set of 400-nt long fragments resulted in 89.92% correct predictions for archaea and 87.26% for bacteria (Supplementary Table S8, column C-3BA). Note that a life domain classification within a metagenomic gene finder was first proposed by Noguchi et al. (14). The difference with the method they used is rather technical; domain recognition in GeneMark.hmm is embedded in the Viterbi algorithm that assigns the most likely type of a hidden state, bacterial or archaeal (thermophilic or mesophilic), to predicted coding region.
Analysis of sequences from human and mouse gut microbiomes
We used GeneMark.hmm with C-3BA model to predict genes in metagenomic sequences from two human and five mouse gut microbiomes (Table 4). In these sequence sets, we have identified 11 865 genes that were not annotated earlier. Protein products of 1984 genes (in human samples) and 3435 genes (in mouse samples) had similarity to known proteins detectable by BLASTP with E-value threshold 10−5. Protein functions that could be assigned to the 50 longest genes predicted in the gut microbiomes derived sequences are listed in Supplementary Table S9. A relative proportion of new genes in the mouse gut metagenomic sequences is about three times higher than that in human; the mere numbers are about or larger 50% of the number of initially annotated genes. Interestingly, 17% (15%) of the metagenomic sequences in human Subject 7 (8) could be mapped to known genomes of bacteria and archaea (Supplementary Tables S10–S12) by the BLASTN search with E-value threshold 10−13. However, in the metagenomic sequences from mice guts, we were not able to identify DNA sequence fragments highly similar to a sequence in already sequenced genomes (with threshold 10−13). Still, for less stringent threshold 10−5, we observed dozens of fragments with similarity to genomes of known species in each mouse gut metagenomic sample. Typical situations that are prone to errors in annotation are illustrated in Figure 9: short genes could be missed (Figure 9a). Some genes could be omitted due to artifacts, such as erroneous extension of the 5′-end of a gene to the longest possible start (Figure 9b); such an extension may overlap a real gene in the opposite strand and this real gene will be missed in annotation.
Table 4.
Results of analysis of metagenomic sequences from human and mouse gut microbiomes. Annotation coordinates were retrieved from JGI IMG/M database (24)
| Methods | Microbiome size (bp) | Number of annotated genes | Number of predicted genes | Number of missed genes | Missed genes (%)a | Number of novel genes | Novel genes (%)a | (Missed + Novel)/2 (%) | Novel genes that have hit to nr (%) |
|---|---|---|---|---|---|---|---|---|---|
| human_sub7 | |||||||||
| MetaGene | 15,817,685 | 20523 | 22 271 | 893 | 4.4 | 2641 | 11.9 | 8.1 | 34.6 |
| MetaGeneAnnotator | 22 164 | 755 | 3.7 | 2396 | 10.8 | 7.2 | 40.5 | ||
| GeneMark.hmm with C-3BA model | 21 941 | 730 | 3.6 | 2148 | 9.8 | 6.7 | 40.7 | ||
| human_sub8 | |||||||||
| MetaGene | 20 486 813 | 25 980 | 27 750 | 1223 | 4.7 | 2993 | 10.8 | 7.7 | 38.2 |
| MetaGeneAnnotator | 27 707 | 971 | 3.7 | 2698 | 9.7 | 6.7 | 41.7 | ||
| GeneMark.hmm with C-3BA model | 27 589 | 840 | 3.2 | 2449 | 8.9 | 6.1 | 45.3 | ||
| mouse_lean1 | |||||||||
| MetaGene | 2 234 664 | 2935 | 4579 | 244 | 8.3 | 1888 | 41.2 | 24.8 | 40.6 |
| MetaGeneAnnotator | 4417 | 216 | 7.4 | 1698 | 38.4 | 22.9 | 44.0 | ||
| GeneMark.hmm with C-3BA model | 4279 | 236 | 8.0 | 1580 | 36.9 | 22.5 | 47.6 | ||
| mouse_lean2 | |||||||||
| MetaGene | 2 133 081 | 2782 | 4279 | 296 | 10.6 | 1793 | 41.9 | 26.3 | 32.1 |
| MetaGeneAnnotator | 4152 | 265 | 9.5 | 1635 | 39.4 | 24.5 | 35.7 | ||
| GeneMark.hmm with C-3BA model | 3950 | 264 | 9.5 | 1432 | 36.3 | 22.9 | 43.9 | ||
| mouse_lean3 | |||||||||
| MetaGene | 2 143 888 | 2793 | 4262 | 202 | 7.2 | 1671 | 39.2 | 23.2 | 38.7 |
| MetaGeneAnnotator | 4198 | 188 | 6.7 | 1593 | 37.9 | 22.3 | 42.8 | ||
| GeneMark.hmm with C-3BA model | 3971 | 195 | 7.0 | 1373 | 34.6 | 20.8 | 47.0 | ||
| mouse_ob1 | |||||||||
| MetaGene | 2 359 017 | 3051 | 4698 | 218 | 7.1 | 1865 | 39.7 | 23.4 | 38.8 |
| MetaGeneAnnotator | 4626 | 196 | 6.4 | 1771 | 38.3 | 22.4 | 43.2 | ||
| GeneMark.hmm with C-3BA model | 4432 | 213 | 7.0 | 1594 | 36.0 | 21.5 | 47.7 | ||
| mouse_ob2 | |||||||||
| MetaGene | 1 841 347 | 2331 | 3675 | 192 | 8.2 | 1536 | 41.8 | 25.0 | 37.2 |
| MetaGeneAnnotator | 3599 | 172 | 7.4 | 1440 | 40.0 | 23.7 | 42.8 | ||
| GeneMark.hmm with C-3BA model | 3444 | 176 | 7.6 | 1289 | 37.4 | 22.5 | 50.4 | ||
Note that the total numbers of genes annotated in JGI IMG/M are different from the number of genes given in original publications (19). This is because JGI IMG/M used YACOP, a combination of several gene finding methods, namely Critica, Glimmer and ZCURVE (38), while BLASTX and BLASTP were used in original publications to identify genes in metagenomic sequences of human and mouse microbiomes. Annotation was not readily available in the original publications. aPercentage values are computed with respect to the number of annotated genes.
Figure 9.

Genome Browser view for two sequences from Subject 7 human microbiome. The C-3BA model was used to predict coding regions. (a) The first and third genes shown in panel ‘Predicted coding regions’ were not previously annotated. Protein products of both predicted genes have sequence similarity to proteins in the nr database with E-value of 8e-44 and 2e-35, respectively. (b) In a 2649-nt microbiome sequence, a single partial gene was annotated in positive strand in frame +3, starting from nucleotide position 39. New to annotation, three genes were predicted in frames −3, +1 and +3, respectively. Sequences analyzed can be found in Microbiome DB: http://exon.gatech.edu/cgi-bin/gbrowse/microbiome_human_sub7/?name=hgutS7_s7_164312; http://exon.gatech.edu/cgi-bin/gbrowse/microbiome_human_sub7/?name=hgutS7_s7_179818_3.
The whole set of gene predictions is available at (http://exon.gatech.edu/GeneMark/metagenome/database); it was also visualized in a genome browser utilizing the GBrowse program (35).
Web interface and downloads
We have designed a web site providing access to the new program for gene prediction in metagenomic sequences: http://exon.gatech.edu/GeneMark/metagenome. Running time of GeneMark.hmm with the 6-LBA models on the Sargasso Sea environmental sample with size 1.045 GB was 88 s. The program is available for download for academic use. For reference purposes, we have also provided an interface to the database of genome-wide codon frequencies observed in genomes used in the training set.
DISCUSSION
Back in 1999, upon analysis of 17 prokaryotic genomes, we have determined that genome-wide 61 codon frequencies could be approximated by functions of genome-wide nucleotide frequencies (9), the functions of a single parameter, genomic GC content. This critical observation strongly suggested that genomic GC content is the major factor influencing genome-wide codon usage pattern. This was a conclusion formulated upon introduction of the heuristic models (9). Moreover, it soon received further support by results independently obtained by other authors (27–28). The major focus of the current study is on further developing the heuristic models and on their applications to gene finding in metagenomic sequences. Therefore, we had to leave aside intriguing questions on (i) possible evolutionary mechanisms that formed the dependence of codon usage pattern on genome GC content and (ii) how could this dependence evolve differently in the domains of bacteria and archaea, or in the classes of mesophilic and thermophilic species.
Notably, both divides, either by phylogeny (bacteria versus archaea) or by the optimal growth temperature (mesophiles versus thermophiles), have produced similar results in terms of accuracy of gene finding in short sequences. Use of the bacteria and archaeal model pairs is a natural choice, since the origin of a short sequence is not known a priori. The second pair of models, mesophilic and thermophilic, may have less frequent use since the temperature of microbiome habitat is known and the model can be chosen a priori.
The ability to identify a sequence origin in terms of bacterial or archaeal domain appears to be an added value benefit since the algorithm automatically identifies the model, bacterial or archaeal that best fits the gene sequence and ‘is attached to’ the most likely type of a hidden state. Domain classification was shown to be correct for 88.04% of 700-nt long bacterial fragments and for 91.27% of 700-nt long archaeal fragments (Supplementary Tables S7 and S8). Notably, genes horizontally transferred between the two domains should be responsible for a fraction of misclassification errors.
The results indicate that gene prediction in fragmented sequences of prokaryotic genomes has the same rate of success as in complete prokaryotic genomes. This result is rather surprising as the complete genomes provide a context for each individual sequence fragment and offer much larger sets of sequence data for model training. However, most of prokaryotic genomes are heterogeneous in terms of GC content. Parameters of a conventional model used in a genomic gene finder are defined for the genome as a whole and the accuracy may slightly suffer in regions whose local GC content deviates from the average one. Derivation of model parameters for each short sequence individually, as it is done for metagenomic sequences, tunes up parameters for each sequence with regard to its GC content. Therefore, short sequences as targets for gene prediction have advantages as well.
Existence of a difference between GC content of protein- and non-coding regions is a well-known fact. However, the nearly constant value of this difference among genomes ranging wide in GC content is an interesting observation (Figure 2). Notably, RNA genes have been observed to be uniformly GC rich regardless of genome GC content (Figure 2); hence, tRNA genes could be easily detected in AT-rich genomes as local regions with a sharp GC content elevation. GC content of protein-coding genes does not correlate with temperature of the species habitat. Still, it is the RNA genes that show temperature-dependent composition. RNA genes in genomes of thermophilic species (genomes that could be either AT or GC rich) have a significantly higher GC content than RNA genes in genomes of mesophilic species (Figure 2). Frequencies of nucleotides in the three codon positions in protein coding regions of mesophilic and thermophilic species show difference in patterns of dependence on genome GC content. Similarly to inferring a domain of origin, bacterial or archaeal, for a gene within the gene finding algorithm with bacterial and archaeal model pair, a pair of heuristic models derived for mesophilic and thermophilic species could be used to for inferring mesophilic or thermophilic origin for an individual gene.
We should mention that the sets of bacterial and mesophilic species used in this study well overlap each other; 301 out of 319 species in the bacterial set are mesophilic. Hence, bacterial and mesophilic protein-coding regions exhibit a similar dependence of frequencies of nucleotides in the three codon positions on genome GC content (Table 1). On the other hand, although the set of 38 archaeal species contains 23 thermophiles and overlaps significantly with the set of 47 thermophilic species in this study, most archaeal and thermophilic regression slope coefficients (Table 1) are distinctly different.
We should note that frameshifts in protein-coding regions, caused by sequencing errors, are more frequent in metagenomes than in complete genomes. It was shown (36) that performance of all current methods for metagenome gene finding including GeneMark.hmm with the original HAL-99 models is sensitive to presence of frameshifts. The new heuristic models make no exception, and sensitivity to sequence errors has roughly the same pattern as one already reported for HAL-99 (36). Additionally, as a separate project we have developed a new algorithm and software tool for frameshift identification (37) that could be combined with the heuristic models and used for frameshift detection in metagenomic sequences.
In conclusion, we should say that we have presented here methods of reconstruction of codon and oligomer frequencies that have led to new heuristic models for gene finding in short sequences. We have shown that use of the new models in GeneMark.hmm resulted in more accurate gene predictions than use of heuristic model HAL-99, developed earlier. The gene prediction accuracy was shown to be higher than that of MetaGene and MetaGeneAnnotator (Table 2).
The HAL-99 models have been used in gene prediction and annotation since 1999. They were used in ab initio prokaryotic and eukaryotic gene finders GeneMarkS and GeneMark-ES to initiate unsupervised training for complete and nearly complete genomes (7,11–12). Particularly, HAL-99 models were used in ab initio gene prediction and annotation in viral genomes (10) and in metagenomic sequences within the pipeline of DOE Joint Genome Institute (Nikos Kyrpides, personal communication).
FUNDING
Georgia Tech School of Biology; US National Institute of Health (grant HG00783 to M.B.). Funding for open access charge: National Institute of Health (grant HG00783 to M.B.).
Conflict of interest statement. None declared.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENT
We wish to thank Konstantinos Mavromatis, Natalia Ivanova and Nikos Kyrpides for interest to the project and useful discussions.
REFERENCES
- 1.Chen K, Pachter L. Bioinformatics for whole-genome shotgun sequencing of microbial communities. PLoS Comput. Biol. 2005;1:106–112. doi: 10.1371/journal.pcbi.0010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu D, Paulsen I, Nelson KE, Nelson W, et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- 3.Krause L, Diaz NN, Bartels D, Edwards RA, Puhler A, Rohwer F, Meyer F, Stoye J. Finding novel genes in bacterial communities isolated from the environment. Bioinformatics. 2006;22:e281–289. doi: 10.1093/bioinformatics/btl247. [DOI] [PubMed] [Google Scholar]
- 4.Yooseph S, Sutton G, Rusch DB, Halpern AL, Williamson SJ, Remington K, Eisen JA, Heidelberg KB, Manning G, Li W, et al. The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families. PLoS Biol. 2007;5:e16. doi: 10.1371/journal.pbio.0050016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yooseph S, Li W, Sutton G. Gene identification and protein classification in microbial metagenomic sequence data via incremental clustering. BMC Bioinformatics. 2008;9:182. doi: 10.1186/1471-2105-9-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larsen TS, Krogh A. EasyGene – a prokaryotic gene finder that ranks ORFs by statistical significance. BMC Bioinformatics. 2003;4:15. doi: 10.1186/1471-2105-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29:2607–2618. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Besemer J, Borodovsky M. Heuristic approach to deriving models for gene finding. Nucleic Acids Res. 1999;27:3911–3920. doi: 10.1093/nar/27.19.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mills R, Rozanov M, Lomsadze A, Tatusova T, Borodovsky M. Improving gene annotation of complete viral genomes. Nucleic Acids Res. 2003;31:7041–7055. doi: 10.1093/nar/gkg878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005;33:6494–6506. doi: 10.1093/nar/gki937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008;18:1979–1990. doi: 10.1101/gr.081612.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noguchi H, Taniguchi T, Itoh T. MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res. 2008;15:387–396. doi: 10.1093/dnares/dsn027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noguchi H, Park J, Takagi T. MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006;34:5623–5630. doi: 10.1093/nar/gkl723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoff KJ, Tech M, Lingner T, Daniel R, Morgenstern B, Meinicke P. Gene prediction in metagenomic fragments: a large scale machine learning approach. BMC Bioinformatics. 2008;9:217. doi: 10.1186/1471-2105-9-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoff KJ, Lingner T, Meinicke P, Tech M. Orphelia: predicting genes in metagenomic sequencing reads. Nucleic Acids Res. 2009;37:W101–105. doi: 10.1093/nar/gkp327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rudner R, Karkas JD, Chargaff E. Separation of B. subtilis DNA into complementary strands. 3. Direct analysis. Proc. Natl Acad. Sci. USA. 1968;60:921–922. doi: 10.1073/pnas.60.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kattenhorn LM, Mills R, Wagner M, Lomsadze A, Makeev V, Borodovsky M, Ploegh HL, Kessler BM. Identification of proteins associated with murine cytomegalovirus virions. J. Virol. 2004;78:11187–11197. doi: 10.1128/JVI.78.20.11187-11197.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gill SR, Pop M, DeBoy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turnbaugh PJ. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 21.Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–141. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 22.Randau L, Munch R, Hohn MJ, Jahn D, Soll D. Nanoarchaeum equitans creates functional tRNAs from separate genes for their 5′- and 3′-halves. Nature. 2005;433:537–541. doi: 10.1038/nature03233. [DOI] [PubMed] [Google Scholar]
- 23.Sayers EW, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2009;37:D5–15. doi: 10.1093/nar/gkn741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markowitz VM, Ivanova NN, Szeto E, Palaniappan K, Chu K, Dalevi D, Chen IM, Grechkin Y, Dubchak I, Anderson I, et al. IMG/M: a data management and analysis system for metagenomes. Nucleic Acids Res. 2008;36:D534–538. doi: 10.1093/nar/gkm869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Borodovsky M, Mcininch J. Genmark – parallel gene recognition for both DNA strands. Comput. Chem. 1993;17:123–133. [Google Scholar]
- 26.Azad RK, Borodovsky M. Effects of choice of DNA sequence model structure on gene identification accuracy. Bioinformatics. 2004;20:993–1005. doi: 10.1093/bioinformatics/bth028. [DOI] [PubMed] [Google Scholar]
- 27.Knight RD, Freeland SJ, Landweber LF. A simple model based on mutation and selection explains trends in codon and amino-acid usage and GC composition within and across genomes. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-4-research0010. research0010.0011–0010.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen SL, Lee W, Hottes AK, Shapiro L, McAdams HH. Codon usage between genomes is constrained by genome-wide mutational processes. Proc. Natl Acad. Sci. USA. 2004;101:3480–3485. doi: 10.1073/pnas.0307827100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gorban AN, Zinovyev AY. The mystery of two straight lines in bacterial genome statistics. Bull. Math. Biol. 2007;69:2429–2442. doi: 10.1007/s11538-007-9229-6. [DOI] [PubMed] [Google Scholar]
- 30.Lukashin AV, Borodovsky M. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 1998;26:1107–1115. doi: 10.1093/nar/26.4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lobry JR, Necsulea A. Synonymous codon usage and its potential link with optimal growth temperature in prokaryotes. Gene. 2006;385:128–136. doi: 10.1016/j.gene.2006.05.033. [DOI] [PubMed] [Google Scholar]
- 32.Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Nelson WC, Ketchum KA, et al. Evidence for lateral gene transfer between archaea and bacteria from genome sequence of Thermotoga maritima. Nature. 1999;399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- 33.Zavala A, Naya H, Romero H, Musto H. Trends in codon and amino acid usage in Thermotoga maritima. J. Mol. Evol. 2002;54:563–568. doi: 10.1007/s00239-001-0040-y. [DOI] [PubMed] [Google Scholar]
- 34.Basak S, Banerjee T, Gupta SK, Ghosh TC. Investigation on the causes of codon and amino acid usages variation between thermophilic Aquifex aeolicus and mesophilic Bacillus subtilis. J. Biomol. Struct. Dyn. 2004;22:205–214. doi: 10.1080/07391102.2004.10506996. [DOI] [PubMed] [Google Scholar]
- 35.Stein LD, Mungall C, Shu S, Caudy M, Mangone M, Day A, Nickerson E, Stajich JE, Harris TW, Arva A, et al. The generic genome browser: a building block for a model organism system database. Genome Res. 2002;12:1599–1610. doi: 10.1101/gr.403602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoff KJ. The effect of sequencing errors on metagenomic gene prediction. BMC Genomics. 2009;10:520. doi: 10.1186/1471-2164-10-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Antonov I, Borodovsky M. GeneTack: Frameshift identification in protein coding sequences by the Viterbi algorithm. J. Bioinform. Comput. Biol. 2010;8:1–17. doi: 10.1142/s0219720010004847. [DOI] [PubMed] [Google Scholar]
- 38.Tech M, Merkl R. YACOP: enhanced gene prediction obtained by a combination of existing methods. In Silico Biol. 2003;3:441–451. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.