

Abstract
The small G-protein NRAS is mutated in 22% of human melanomas, whereas the related proteins, KRAS and HRAS are mutated in only 2% and 1% of melanomas respectively. We have developed a mouse models of melanoma in which Cre recombinase/loxP technology is used to drive inducible expression of G12VKRAS in the melanocytic lineage. The mice develop skin hyper-pigmentation, nevi and tumors that bear many of the cardinal histopathology features and molecular characteristics of human melanoma. These tumors invade and destroy the underlying muscles and cells derived from them can grow as subcutaneous tumors and colonise the lungs of nude mice. These data establish that oncogenic KRAS can be a founder event in melanomagenesis.
Keywords: Oncogenic RAS, nevi, melanoma, transgenic mice
Introduction
The RAS proteins are components of a conserved signal transduction cascade that regulates cell responses to their environment (1, 2). These small guanine nucleotide binding proteins are activated downstream of growth factor, cytokine and hormone receptors. Specifically, they are activated by guanine-nucleotide exchange factors (GNEFs), which replace GDP for GTP, and inactivated by GTPase-activating proteins (GAPs), which stimulate RAS GTPase activity, promoting GTP hydrolysis and returning RAS to the GDP-bound state. RAS activates several effector proteins, including phosphoinositide 3-kinase (PI3K), the RAF protein kinases, and the Ral guanine-nucleotide exchange factor (2) to regulate the activity of signalling pathways that control proliferation, senescence, survival and differentiation. There are three RAS genes in mammals, HRAS, KRAS and NRAS and the proteins they encode serve overlapping but non-conserved functions. Thus, whilst Hras or Nras null mice are viable, Kras null mice suffer embryonic lethality (3, 4).
In addition to its critical functions in normal cells, RAS proteins are important in cancer. Approximately 30% of human tumors harbor somatic gain-of-function mutations in one of the RAS genes, most commonly at the codons for glycine 12 (G12), glycine 13 (G13) or glutamine 61 (Q61) (5). Through different mechanisms the mutations impair the GTPase activity of RAS, trapping it in the active GTP-bound state and promoting tumorigenesis (2). RAS mutations are found in about 25% of melanomas, a form of skin cancer that arises from melanocytes, specialised pigment cells that provide skin and hair tone and protection from ultraviolet (UV) radiation. In most western societies, melanoma incidence is doubling every decade and due to a lack of effective therapies, metastatic malignant melanoma has a particularly poor outcome (6).
NRAS is mutated in 22% of human melanomas whereas KRAS and HRAS are mutated in 2% and 1% of cases respectively. Over 80% of the NRAS and HRAS mutations involve Q61, whereas ~77% of the KRAS mutants involve G12 (www.sanger.ac.uk/genetics/CGP/cosmic/). It is unclear why NRAS mutations predominate in melanoma, whereas in cancers such as pancreatic carcinoma and colorectal cancer KRAS mutations predominate. It is possibly that codon Q61 of NRAS is a mutation hotspot in melanoma, but this is difficult to consolidate based on DNA sequence conservation. Alternatively, NRAS may be expressed at levels that are optimal for oncogenic transformation if a mutation occurs, or it may possess distinct biological properties. Accordingly, G12VNRAS and G12VKRAS both activate RAF and induce anchorage independent growth in melanocytes in vitro, but NRAS-transformed melanocytes are more tumorigenic than KRAS-transformed melanocytes in mice (7). Thus, KRAS may be a weaker oncogene than NRAS in melanocytes.
Several transgenic mouse melanoma models driven by RAS have been developed, but in the absence of additional events, the induction of melanoma in these models is generally inefficient. Thus, constitutive or inducible expression of G12VHRAS only induces tumor formation efficiently when Cdkn2a (p16INK4a) or Tp53 are deleted (8-10) or the mice are exposed to UV light (11). The tumors are dermal, display variable pigmentation and do not metastasize. Similarly, conditional expression of G12VHRAS in melanocytes using Cre-recombinase/LoxP technology only induces melanoma efficiently when p16INK4a is deleted (12). The tumors were heavily pigmented and also non-metastatic. Further, constitutively expression of Q61KNRAS in melanocytes using a melanocyte-specific tyrosinase promoter only induces tumors efficiently when p16INK4a was deleted and these tumors were pigmented, frequently ulcerated and tumor cells invaded and colonized the epidermis and epidermal/dermal junctions (13). They invaded the reticular dermis, the subcutis, the underlying muscles and in some mice spread to the lymph nodes, liver and lungs.
These data show that HRAS and NRAS can induce melanomagenesis in mice, albeit inefficiently. Since no such studies are reported for KRAS, in this study we used Cre-recombinase/LoxP technology induce expression of G12VKRAS in melanocytes to determine it can also induce melanomagenesis in mice.
Materials and Methods
Transgenic mouse procedures
Transgenic mouse procedures were performed under Home Office license authority with local Ethics Committee approval. Transgenic β-actin::LSL::G12VKRAS and Tyr::CreERT2 mice, and the primers required for their genotyping have been described (14, 15). The mice were treated with tamoxifen and samples were prepared for DNA genotyping and RT-PCR analysis as described (16). Cell lines for use in intravital imaging were prepared and analysed as described (16). For allograft tumor growth, 1×106 KRASG12V-melanoma cells were injected subcutaneously into the flanks of nude mice in 100μl PBS. Tumor size was measured using calipers and volume was calculated using the formula length × width × depth × 0.5. For western blotting protein extracts were prepared using NP40 buffer (17) and the following antibodies: mouse anti-c-myc and rabbit anti-ERK2 (Santa Cruz Biotechnology, Heidelberg, Germany), and rabbit anti-Tp53 (NCL-p53-CM5p; Novocastra, Newcastle, United Kingdom).
To sequence Tp53 mRNA, representative 8μm frozen sections from 4 tumours were prepared and microdissected as described (18) to provide samples with >90% tumor cells from which cDNA was prepared as described (19). The entire Tp53 cDNA was amplified using eight paired-primers (supplementary Table 1) for sequencing using Big Dye(Applied Biosystems, Foster City, CA) as described (20). Mutation analysis was performed using Mutation surveyor software™ (SoftGenetics LLC, State College, PA). All samples were sequenced in duplicate from separately microdissected samples.
Immunohistochemistry and immunofluorescence
Immunohistochemistry analysis was as described (16) using rabbit polyclonal antibody Z0331 (1/800) for S100 (Dako; Glostrup, Denmark), rat monoclonal antibody M7249 (1/25) for Ki67 (Dako; Glostrup, Denmark) and rabbit polyclonal antibody p16INK4a (1/25)(Santa Cruz Biotechnology, Heidelberg, Germany). S100 antigen retrieval was performed using Labvision pre-treatment module citrate pH6 for 30 min (Labvision, Runcorn, United Kingdom) or by microwave (p16INK4a, Ki67) in Dako pH6 buffer (Dako) for 18 min. Antibody binding was detected using the Rabbit enVision Peroxidase kit, and the AEC substrate chromogen (S100) and the Vectastain Elite ABC kit (p16INK4a, Ki-67), or with AK-5001 rabbit IgG Vectastain ABC-AP kit (S100) and rat IgG Vectastain ABC-AP (Ki67), with SK-5300 Vector Blue substrate or DAB substrate (all from Vector, Petersborough, United Kingdom). Negative controls for each run included appropriate non-immune sera (X0902, Dako; 012-000-120, Jackson ImmunoResearch).
Immunofluorescence was performed with mouse antibody MP-606-PG1 for S100 (Menarini, Florence, Italy) and a rabbit antibody M-157, SC-1207 for p16INK4a (Santa Cruz Biotechnology). Antigen were retrieved using Menarini Antigen access unit, with Super buffer pH9.5. Primary antibody incubation was overnight and antibody binding was detected with AlexaFluor488 goat anti-rabbit IgG (A11008, Invitrogen) and AlexaFluor555 goat anti-mouse IgG (A21424, Invitrogen) for p16INK4a and S100 respectively. Slides were counterstained with DAPI. Negative controls were included for each immunohistochemical run using an appropriate non-immune serum IgG (N1698, X0902, Dako).
Results
To develop a KRAS-driven mouse model of melanoma, we crossed β-actin::LSL::G12VKRAS and Tyr::CreERT2 mice. In β-actin::LSL::G12VKRAS mice, G12VKRAS is downstream of a β-actin promoter, but its expression is blocked by an intervening LoxP-STOP-LoxP (LSL) cassette that can be removed by Cre recombinase (Supplemental Fig 1)(14). In Tyr::CreERT2 mice, a tamoxifen (TM)-activated version of Cre recombinase (CreERT2) is expressed using a melanocyte-specific tyrosinase promoter (15). The double transgenic mice thus allow exquisite temporal and spatial control of G12VKRAS expression, because CreERT2 is restricted to the melanocytes and only activated upon TM treatment, allowing the human acquisition of somatic mutations in KRAS to be mimicked in the mice.
Mice at 1-2 months of age were treated with topical application of TM to their shaved backs. Two to three months later the β-actin::LSL::G12VKRAS;Tyr::CreERT2 mice (henceforth G12VKRAS;CreERT2 mice) developed diffuse skin hyper-pigmentation on their backs, tails and ears (Fig 1A). Importantly, pigmentation was never seen in ethanol-treated G12VKRAS;CreERT2 mice, or in TM-treated β-actin::LSL::G12VKRAS or Tyr::CreERT2 mice (Fig 1A and data not shown). The TM-treated G12VKRAS;CreERT2 mice also developed several types of melanocytic lesion. Most of the animals developed multiple small lesions bearing a striking similarity to human blue nevi, predominantly within the TM-treated areas but also in the non-treated regions of the skin. In approximately 30% of the animals, these lesions were visible as dome-shaped lesions on the surface of the skin (Fig 1A, arrow) and were located in the superficial dermal layers (Fig 1B). More commonly (~80% of animals) the lesions were centred on the deeper regions of the dermo-hypodermal interface (Fig 1C) and were not obvious until post-mortem examination of the skin. These lesions were composed of epithelioid and dendritic melanocytes immersed in a collagenous stroma, occasionally forming vague nests and displaying low levels of pigmentation (Fig 1B, 1C). They did not invade the epidermis and no junctional components were observed (Fig 1B, 1C). Nuclear pleomorphism and atypia were not found and mitotic figures were absent. Ki67 staining was negligible (<1%)(Fig 1D). These lesions therefore demonstrated low levels of proliferation and overall did not exhibit any features of malignancy. Importantly however, no such lesions were observed in control animals either macroscopically or upon histological examination (Fig 1A; data not shown).
Figure 1. G12VKRAS induces benign melanocytic lesions in mice.

A. Left panel: Photograph showing hyper-pigmentation and small dome-shaped lesions (arrow) on the skin of TM-treated G12VKRAS;Tyr::CreERT2 (left) and control (right) mice. The fur was removed to reveal the skin. Upper right panel: photograph showing tail pigmentation on a TM-treated G12VKRAS;Tyr::CreERT2 (left) and control (right) mouse. Lower right panel: photograph showing ear pigmentation (*) and peri-orbital areas (arrowhead) of a TM-treated G12VKRAS;Tyr::CreERT2 (left) and control (right) mouse.
B. Photomicrograph of a haematoxylin and eosin (H&E)-stained paraffin section of a nevus near the epidermal-dermal junction. Note the pushing borders of the lesion.
C. Photomicrograph of an H&E-stained paraffin section of a nevus found within the dermo-hypodermal interface of a TM-treated G12VKRAS;Tyr::CreERT2 mouse.
D. Photomicrograph showing lack of Ki67 expression in a nevus from a TM-treated G12VKRAS;Tyr::CreERT2 mouse. The section is counterstained with haematoxylin (purple) and the nevus is devoid of mitotic figures.
Slightly different, but still relatively benign melanocytic lesion were observed in the periorbital areas of the TM-treated G12VKRAS;CreERT2 mice (Fig 1A, arrowheads; Fig 2A), but again not in the controls (Fig 1A). These darkly pigmented lesions were also first evident 2-3 months after TM-treatment and were characterized by an accumulation of heavily pigmented cells within the dermis, but again without epidermal or epidermal-dermal junction involvement (Fig 2B). These lesions were significantly larger than the blue nevi and possessed occasional extensions into the subcutaneous tissue, but without displaying overt infiltrating or destructive growth patterns. They were also composed of admixed heavily melanin-laden epithelioid (Fig 2C, arrows) and less pigmented spindle/dendritic cells (Fig 2C, arrowheads). The nuclei of these cells were slightly enlarged, but were still uniform and possessed small discrete nucleoli. Overt nuclear atypia and nuclear pleomorphism were not observed. Mitotic figures were scant at <1 per 10 high power fields (HPFs) and restricted to the superficial areas of the lesions. These histological features are consistent with a diagnosis of pigmented epithelioid melanocytoma, a melanocytic lesion characterized by local aggressiveness and borderline metastatic potential in humans (21).
Figure 2. G12VKRAS induces pigmented epithelioid melanocytoma in mice.
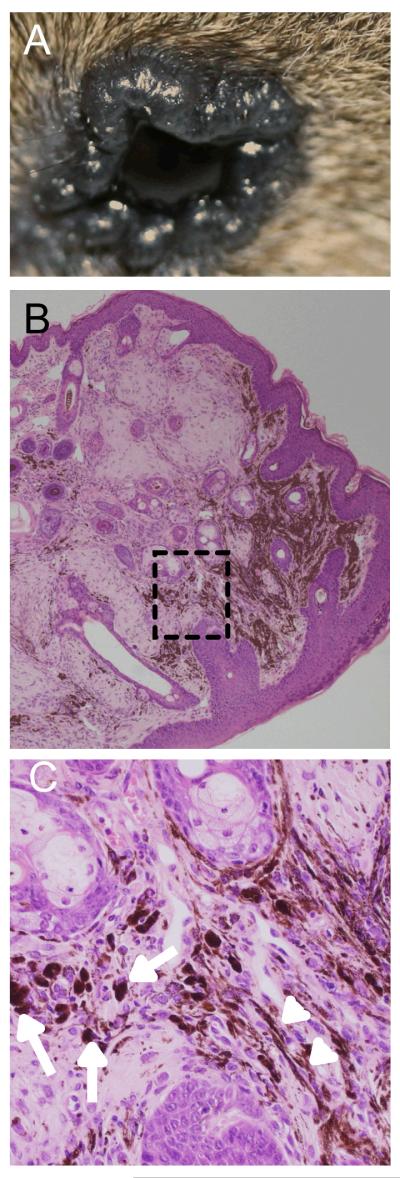
A. Photograph showing an elevated dark lesion in the peri-orbital area of a TM-treated G12VKRAS;Tyr::CreERT2 mouse
B. Low power magnification of an H&E-stained paraffin section of a pigmented epithelioid melanocytoma from the peri-orbital area of a TM-treated G12VKRAS;Tyr::CreERT2 mouse. Note the absence of a junctional component.
C. High power magnification photomicrograph of the boxed area in B. Note the presence of heavily melanin-laden epithelioid cells (arrows) and less pigmented spindle cells (arrow heads).
Importantly, all of the mice eventually developed large, rapidly growing tumors, either within the TM-treated areas of skin (seen in 85% of the mice; Fig 3A), and/or in the perianal region (seen in 79% of the mice; Fig 3B). These tumors arose between 1 and 10 months of TM-treatment with a median time of 4 months (Fig 3C). Ultimately, one or other of these lesions necessitated sacrifice of the animals. The TM-treated skin lesions were hypo-pigmented, asymmetrical and dome shaped (Fig 3A). The perianal lesions were ostensibly black, but also dome-shaped, asymmetrical (Fig 3B) and often ulcerated. Histologically, the superficial aspects of the perianal lesions were similar in appearance to the periorbital lesions. They lacked epithelial or junctional components and were composed of heavily pigmented epithelioid cells mixed with less pigmented spindle/dendritic cells (Fig 3D). However, in their deeper aspects the perianal lesions were composed largely of hypo/amelanotic spindle cells that exhibited conspicuous nuclear pleomorphism with large nucleoli (Fig 4A) and nuclear pseudo inclusions (Fig 4B). The lesions displayed diffuse positivity for Ki67 (Fig 4C; supplemental Fig 2), which correlated with high mitotic activity and an average of 6 mitoses/10 HPFs both in the superficial and deep aspects of the lesions (Fig 4D, arrows).
Figure 3. G12VKRAS induces tumors in mice.

A. Photograph showing a dorsally located tumor found on the TM-treated skin of a G12VKRAS;Tyr::CreERT2 mouse.
B. Photograph showing large pigmented perianal lesion in a TM-treated G12VKRAS;Tyr::CreERT2 mouse (left panel) compared to a WTKras control mouse (right panel).
C. Kaplan-Meier plot showing tumor-free survival (%) in TM-treated G12VKRAS;Tyr::CreERT2 mice (14 animals) compared to TM-treated β-actin::LSL::G12VKRAS mice (5 animals) TM-treated Tyr::CreERT2 mice (5 animals) and EtOH-treated G12VKRAS::Tyr::CreERT2 mice (5 animals).
D. Low power magnification of an H&E-stained paraffin section of a perianal lesion in a TM-treated G12VKRAS;Tyr::CreERT2 mouse. Note the absence of a junctional component.
Figure 4. Characterisation of tumors induced by G12VKRAS.

A. A deep area from a perianal melanocytic lesion predominantly composed of hypo/amelanotic spindle cells exhibiting conspicuous nuclear pleomorphism and large nucleoli (arrowheads).
B. A deep area from a perianal melanocytic lesions composed of a mixture of spindle and epithelioid cells displaying the presence of pigmented cells. The arrow highlights an example of a nuclear pseudo-inclusion.
C. Ki67 positive staining within the deeper aspects of the neoplastic lesion.
D. Photomicrographs of mitotic figures (arrows) found within the deeper aspects of a perianal lesion.
The TM-treated skin lesions lacked the superficial pigmented cells seen in the perianal lesions, but were otherwise very similar to their deeper aspects (summarised in supplemental Table 2). They were asymmetrical, grossly infiltrative and destructive, and often ulcerated. Generally they were composed largely of invasive hypo/amelanotic spindle cells with atypical and markedly pleomorphic nuclei, large nucleoli, pseudo-nuclear inclusions and diffuse Ki67 positivity (Supplemental Table 2). Compared to the perianal lesions, they appeared more aggressive, with higher mitotic activity (9 mitoses/10 HPFs; Supplemental Table 2) and mitotic figures promptly found in their deep aspects. Consistent with their higher proliferative fraction, they reached tumor burden limits within 3 weeks of first presentation, whereas the perianal lesions took up to 10 weeks to reach tumor burden limits.
Using PCR, we provide clear evidence that the β-actin::LSL::G12VKRAS locus was rearrangement in both the TM-treated skin and perianal tumors (Fig 5A, tumors A and B respectively). Unlike previously described RAS-driven models of melanomagenesis, in our studies tumor induction was efficient without the need for further manipulation of the mice, so we examined p16INK4a and Tp53 status in these tumors. Using double-staining immunofluorescence with the melanocytic marker S100, we show that p16INK4a is expressed in the cancer cells in the tumors in our mice (Fig 5B). We also performed complete sequencing of the Tp53 cDNA from these tumors and did not detect any mutations (data not shown). Further, by western blot we show that Tp53 protein was not detectable in cell lines derived from the tumors, but that it responded normally and was stabilised when the cells were treated with the DNA damaging agent doxorubicin (Fig 5C). Together these data show that p16INK4a and Tp53 are intact in the G12VKRAS-driven tumors.
Figure 5. Molecular characterisation of melanocytic lesions.

A. PCR-mediated genotyping for recombined G12VKRAS (lox-G12VKRAS; 240bp) from a dorsally located (Tumor A) and perianal (Tumor B) tumor. Kidney DNA was used as a negative control and Tyr::CreERT2 is present in all three samples.
B. Double immunofluorescence staining for p16INK4a (p16; green) and S100 (red) in a G12VKRAS-driven tumor. DAPI nuclear staining (blue) reveals the nuclei and a merged image is shown.
C. Western blot for Tp53 in three G12VKRAS-driven tumor cell lines (#8352, #5954, #4755) treated with DMSO (control) or doxorubicin (1μM; 8hr). Erk2 is used as a loading control.
D. RT-PCR analysis for expression of Tyrosinase (Tyr.), Dct, Pax3, Silver (Si/gp100) and Gapdh (loading control). Tumors were taken from a section of TM-treated skin (Tumor A) or from the perianal region (Tumor B). The indicated tissues were also analysed from at TM-treated G12VKRAS;CreERT2 (G12VKRAS) or control (WT) mouse.
The histopathological features of the deeper aspects of the TM-treated skin and perianal lesions were remarkably similar and both stained positive for S100 (supplemental Table 2). However, our attempts to stain these tumors with other melanocytic markers, including HMB45/gp100, Melan A and Mitf were unsuccessful because the antibodies were raised against human antigens and do not appear to have sufficient sensitivity and specificity for mouse tissues. Furthermore, using IHC it was recently shown that human spindle cell melanomas frequently lack expression of melanocytic markers other than S100 (22). We therefore used RT-PCR to investigate expression of melanocytic genes in the tumors because it is more sensitive. Tyrosinase, Dct, Pax3 and silver (which encodes HMB45/gp100) were all strongly expressed in the tumors from both the TM-treated skin and perianal regions (Fig 5D, tumors A and B respectively). As a control, we show that these genes were not expressed in brain or kidney from TM-treated animals. We also examined these markers in the benign lesions in the mice. We show expression of Dct and, albeit more weakly, of tyrosinase, Pax3 and silver in the periorbital lesions from TM-treated G12VKRAS;CreERT2 mice, but not in the eyelids of wild-type animals (Fig 5D). We also observed expression of tyrosinase, Dct, Pax3 and silver in the nevus-rich skin from TM-treated G12VKRAS;CreERT2 mice but not, or at considerably lower levels, in the skin from wild-type mice (Fig 5D).
Together, these data confirm that the benign lesions derive from melanocytic cells and provide evidence to support a diagnosis of melanoma in the TM-treated skin and perianal tumors. Although these tumors display many features that are used to differentiate malignant from benign melanocytic lesions in human pathology (Supplemental Table 2), we did not observe metastatic deposits in our mice upon post-mortem examination (data not shown). However, the cells from these lesions did invade the subcutaneous tissue and the skeletal muscle fibres in a destructive growth fashion (Figure 6A). Therefore to confirm the tumorgenicitiy of the cancer cells, we show that cell lines developed from the lesions grew as subcutaneous tumors in nude mice (Fig 6B). We also engineered these cells to express firefly luciferase and injected them into the tail veins of nude mice. One month after injection, intra-vital imaging indicated that melanoma cells had colonized the lungs of all (5/5) of the recipient animals (Fig 6C), and multiple tumor foci were observed in the lungs of these mice upon post-mortem examination (Fig 6D, arrows). Importantly, IHC revealed that the sub-cutaneous lesions and the lung lesions both contained metastatic deposits harbouring cells with morphological features similar to those in the original tumors (supplementary Fig 3). Thus, despite the lack of metastatic deposits in situ, these data confirm the malignant potential of the cancer cells in these tumors.
Figure 6. G12VKRAS-driven tumors have metastatic potential.

A. Photomicrograph showing deeper aspects of a perianal melanocytic lesion from a TM-treated G12VKRAS;Tyr::CreERT2 mouse with melanocytes infiltrating skeletal muscle.
B. Growth of G12VKRAS-driven tumor cells as sub-cutaneous tumors in the flanks of nude mice. Error bars represent SEM.
C. Intra vital imaging of mice 1 month after tail vein injection of G12VKRAS/luciferase-expressing melanoma cells (left panel) or PBS (right panel).
D. Photograph of lungs from mice 1 month after tail vein injection of G12VKRAS/luciferase-expressing melanoma cells (left panel) or PBS (right panel). Several tumor nodules are highlighted (arrows).
Discussion
Here we describe a mouse model of melanoma driven by oncogenic KRAS. We show that G12VKRAS expression in the melanocytes induces rapid onset skin hyper-pigmentation, followed by induction of two types of benign melanocytic lesion that are characterized by an accumulation of pigmented epithelioid and spindle cells. Despite the obvious expansion of pigmented cells, these lesions were benign and possessed low levels of proliferation. They were largely (but not exclusively) restricted to the TM-treated areas of skin where they displayed the characteristic histological features of human blue nevi, or they occurred on the eyelids, where they resembled pigmented epithelioid melanocytomas. They were restricted to the dermal layers of the skin and did not possess epidermal or junctional components, presumably reflecting the fact that mouse melanocytes, unlike human melanocytes, are largely restricted to the dermis. We note that although TM was topically applied to the skin on backs of the mice, they mounted a systemic response, developing pigmented lesions at distant sites. This is similar to the responses we observed when oncogenic Braf was expressed in mouse melanocytes (16) and presumably occurs because TM is absorbed through the dermis or ingested during post-treatment grooming.
All of the mice developed aggressive tumors on the TM-treated skin, or in their perianal regions. Blue nevi were common at the periphery of these tumors, but as with melanoma induced by oncogenic Braf (16), it is not possible to prove that the tumors emerged directly from these nevi or if they developed de novo and their juxtaposition was circumstantial. We confirmed the tumorigenicity of the lesions by showing that cells derived from them grew as subcutaneous tumors in nude mice and their metastatic potential was confirmed by their locally invasive and destructive growth patterns, their high proliferation indices, and their ability to colonise the lungs of recipient mice following tail vein injections. The median latency for tumor induction was 4 months after G12VKRAS expression, but tumors occurred as early as 1 month and within 10 months all mice had tumors on at least one site; 64% of the mice developed tumors on both sites. Generally the tumors on the two sites were similar and displayed the hallmark features of malignant melanocytic tumors in humans, including asymmetry, infiltrative and destructive growth patterns and areas of ulceration. In agreement with studies showing that oncogenic RAS suppresses tyrosinase expression (23), the tumors were generally hypo/amelanotic. The cells were generally spindle shaped with atypical, pleomorphic nuclei, large nucleoli and pseudo-nuclear inclusions. They expressed S100, tyrosinase Dct, Pax3 and silver, markers of the melanocytic lineage and importantly, the β-actin::LSL::G12VKRAS locus had been recombined in these tumors. This provides strong circumstantial evidence to support a diagnosis of metastatic melanoma driven by oncogenic KRAS for both types of tumor.
Despite the general overall similarity of the tumors on the two sites, some subtle differences were seen. There were pigmented cells in the superficial aspects of the perianal tumors that were not seen in the TM-treated skin tumors. The perianal tumors were also generally less aggressive. They had fewer proliferating cells and it took them three times as long as the TM-treated skin tumors to reach tumor burden limits. It is unclear what governs these differences or why distinct types of lesions develop in the various sites. It may stem from differences in melanocyte distribution or type in the perianal and periorbital regions due to the transition from skin to non-squamous epithelium, or it may be due to the presence of distinct microenvironments caused by differences in growth factor production by the epithelial cells lining the mucosa and skin, or differences in the types of fibroblasts or inflammatory cells found locally. Thus cell-intrinsic or cell-extrinsic factors could mediate these differences, but the mechanism is currently unclear.
We note several differences between our model and the RAS-driven models previously reported. In particular, our mice appear to succumb to a progressive phenotype and a broader spectrum of melanocytic lesions. Skin hyper-pigmentation was seen when G12VHRAS or Q61LNRAS were constitutively expressed using a tyrosinase enhancer/promoter (13, 24) but not when G12VHRAS was expressed using a doxycyclin- or TM-inducible system (10, 12). We also report induction of nevi – only previously reported in a tyrosinase-driven G12VHRAS model (24) – and, unique for Ras-driven models, pigmented epithelioid melanocytomas, although oncogenic Braf induces both types lesion (16). The most significant difference in our model is its penetrance. All of our mice developed melanoma within 10 months without the loss of p16INK4a or Tp53, or the exposure to UV light required by previous models (8, 9, 12, 13). Intriguingly, p16INK4a remains intact in the tumors induced by G12VHRAS combined with UV light (11) and we show that p16INK4a and Tp53 also remain intact in the tumors in our mice and therefore conclude that loss of these tumor suppressors is not required for G12VKRAS-induced melanomagenesis in this model.
The differences between the various mouse models presumably reflect transgene design. Many of the previous models used the tyrosinase promoter (8, 10, 12, 13, 24), which is activated early and so would be drive constitutive oncogene expression throughout melanocyte development. This approach does not therefore mimic the acquisition of somatic mutations that occurs in humans and may allow melanocytes to adapt to the oncogene during their development. Furthermore, different tyrosinase enhancer elements could regulate subtle differences in expression or, as has even been suggested (12), expression in distinct sub-populations of melanocytes. This could explain why Powell et al reported a constitutive Tyr::HRAS model that gives hyper-pigmentation and nevi (24), whereas Chin et al reported another Tyr::HRAS model that does not develop these lesions (9). To overcome some of these concerns, we used a ubiquitous promoter with exquisite control over timing of expression and observed the broader spectrum of skin lesions than previously reported. Surprisingly however, we recently reported that expression of G12DKras using the endogenous mouse gene to ensure physiological expression levels induced some skin darkening but no nevi, and tumors only when a second genetic manipulation (expression of kinase-dead BRAF) was also engineered (25).
The data above suggest that the timing and levels of expression, and possibly also the precise mutation and RAS isoform involved are important factors in determining whether tumors are induced in RAS-driven models. Importantly, we present a model of melnomagenesis driven by oncogenic KRAS that is 100% penetrant without the need for additional genetic manipulation or exposure to UV light. Despite this high level of penetrance, we argue that G12VKRAS alone is not sufficient to induce melanoma even in this model. The progressive nature of the mouse response and the long latency required for tumor induction suggests that additional genetic events are likely to be required and we are currently attempting to indentify those events. KRAS is reported to be less oncogenic than NRAS in melanocytes in vitro (7) and KRAS mutations are less common than NRAS or HRAS mutations in human melanoma. It has been suggested that this is because KRAS is less efficient at blocking Myc phosphorylation by GSK3 (26) and although we show that Myc is expressed in the tumors from our mice (supplemental Fig 4), we have yet to investigate its phosphorylation status. Alternatively, it has been suggested that unlike HRAS and NRAS which are in lipid rafts, KRAS is disordered in the plasma membrane and so less able to activate PI3K/AKT (26). However, our data now establish that G12VKRAS can transform melanocytes in a non-sensitised background in vivo, suggesting that its oncogenicity may be context specific.
Thus, we have developed a mouse model of melanoma driven by oncogenic KRAS that is initiated by a single, inducible genetic event. We establish that oncogenic KRAS can induce nevi and be a founder event in melanomagenesis. The lesions we observe resemble those of amelanotic/oligomelanotic malignant metastatic melanoma in humans and the model is relevant because although KRAS mutations are rare in human melanoma, when present they usually (>75%) involve a G12 substitution (www.sanger.ac.uk/genetics/CGP/cosmic/). This tractable model is therefore a powerful tool in the study of KRAS-driven melanoma.
Supplementary Material
Acknowledgements
The Institute of Cancer Research and Cancer Research UK (C107/A10433) funded this work. Felipe Geyer, Maryou Lambros and Jorge Reis-Filho are funded in part by Breakthrough Breast Cancer. We acknowledge NHS funding to the NIHR Biomedical Research Centre. We thank Prof Anton Berns (NKI, Amsterdam) for β-actin::LSL::G12VKRAS mice, Dr Kay Savage (ICR) and Ms Annette Lane (ICR) for histology and immunohistochemistry, and Ms Alice Smith (ICR) for DNA sequencing.
References
- 1.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 2.Young A, Lyons J, Miller AL, Phan VT, Alarcon IR, McCormick F. Ras signaling and therapies. Adv Cancer Res. 2009;102:1–17. doi: 10.1016/S0065-230X(09)02001-6. [DOI] [PubMed] [Google Scholar]
- 3.Johnson L, Greenbaum D, Cichowski K, et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997;11:2468–81. doi: 10.1101/gad.11.19.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koera K, Nakamura K, Nakao K, et al. K-ras is essential for the development of the mouse embryo. Oncogene. 1997;15:1151–9. doi: 10.1038/sj.onc.1201284. [DOI] [PubMed] [Google Scholar]
- 5.Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–9. [PubMed] [Google Scholar]
- 6.Cho YR, Chiang MP. Epidemiology, staging (new system), and prognosis of cutaneous melanoma. Clin Plast Surg. 37:47–53. doi: 10.1016/j.cps.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Whitwam T, Vanbrocklin MW, Russo ME, et al. Differential oncogenic potential of activated RAS isoforms in melanocytes. Oncogene. 2007;26:4563–70. doi: 10.1038/sj.onc.1210239. [DOI] [PubMed] [Google Scholar]
- 8.Bardeesy N, Bastian BC, Hezel A, Pinkel D, DePinho RA, Chin L. Dual inactivation of RB and p53 pathways in RAS-induced melanomas. Mol Cell Biol. 2001;21:2144–53. doi: 10.1128/MCB.21.6.2144-2153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chin L, Pomerantz J, Polsky D, et al. Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes Dev. 1997;11:2822–34. doi: 10.1101/gad.11.21.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chin L, Tam A, Pomerantz J, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–72. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- 11.Hacker E, Irwin N, Muller HK, et al. Neonatal ultraviolet radiation exposure is critical for malignant melanoma induction in pigmented Tpras transgenic mice. J Invest Dermatol. 2005;125:1074–7. doi: 10.1111/j.0022-202X.2005.23917.x. [DOI] [PubMed] [Google Scholar]
- 12.Huijbers IJ, Krimpenfort P, Chomez P, et al. An inducible mouse model of melanoma expressing a defined tumor antigen. Cancer Res. 2006;66:3278–86. doi: 10.1158/0008-5472.CAN-05-3216. [DOI] [PubMed] [Google Scholar]
- 13.Ackermann J, Frutschi M, Kaloulis K, McKee T, Trumpp A, Beermann F. Metastasizing melanoma formation caused by expression of activated N-RasQ61K on an INK4a-deficient background. Cancer Res. 2005;65:4005–11. doi: 10.1158/0008-5472.CAN-04-2970. [DOI] [PubMed] [Google Scholar]
- 14.Meuwissen R, Linn SC, van der Valk M, Mooi WJ, Berns A. Mouse model for lung tumorigenesis through Cre/lox controlled sporadic activation of the K-Ras oncogene. Oncogene. 2001;20:6551–8. doi: 10.1038/sj.onc.1204837. [DOI] [PubMed] [Google Scholar]
- 15.Yajima I, Belloir E, Bourgeois Y, Kumasaka M, Delmas V, Larue L. Spatiotemporal gene control by the Cre-ERT2 system in melanocytes. Genesis. 2006;44:34–43. doi: 10.1002/gene.20182. [DOI] [PubMed] [Google Scholar]
- 16.Dhomen N, Reis-Filho JS, da Rocha Dias S, et al. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;15:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 17.Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–67. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 18.Natrajan R, Weigelt B, Mackay A, et al. An integrative genomic and transcriptomic analysis reveals molecular pathways and networks regulated by copy number aberrations in basal-like, HER2 and luminal cancers. Breast Cancer Res Treat. 2009 doi: 10.1007/s10549-009-0501-3. [DOI] [PubMed] [Google Scholar]
- 19.Natrajan R, Lambros MB, Rodriguez-Pinilla SM, et al. Tiling path genomic profiling of grade 3 invasive ductal breast cancers. Clin Cancer Res. 2009;15:2711–22. doi: 10.1158/1078-0432.CCR-08-1878. [DOI] [PubMed] [Google Scholar]
- 20.Reis-Filho JS, Pinheiro C, Lambros MB, et al. EGFR amplification and lack of activating mutations in metaplastic breast carcinomas. J Pathol. 2006;209:445–53. doi: 10.1002/path.2004. [DOI] [PubMed] [Google Scholar]
- 21.Zembowicz A, Carney JA, Mihm MC. Pigmented epithelioid melanocytoma: a low-grade melanocytic tumor with metastatic potential indistinguishable from animal-type melanoma and epithelioid blue nevus. Am J Surg Pathol. 2004;28:31–40. doi: 10.1097/00000478-200401000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Ohsie SJ, Sarantopoulos GP, Cochran AJ, Binder SW. Immunohistochemical characteristics of melanoma. J Cutan Pathol. 2008;35:433–44. doi: 10.1111/j.1600-0560.2007.00891.x. [DOI] [PubMed] [Google Scholar]
- 23.Englaro W, Bertolotto C, Buscá R, et al. Inhibition of the Mitogen-activated Protein Kinase Pathway Triggers B16 Melanoma Cell Differentiation. J Biol Chem. 1998;273:9966–70. doi: 10.1074/jbc.273.16.9966. [DOI] [PubMed] [Google Scholar]
- 24.Powell MB, Hyman P, Bell OD, et al. Hyperpigmentation and melanocytic hyperplasia in transgenic mice expressing the human T24 Ha-ras gene regulated by a mouse tyrosinase promoter. Mol Carcinog. 1995;12:82–90. doi: 10.1002/mc.2940120205. [DOI] [PubMed] [Google Scholar]
- 25.Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–21. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prior IA, Hancock JF. Compartmentalization of Ras proteins. J Cell Sci. 2001;114:1603–8. doi: 10.1242/jcs.114.9.1603. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.