

Abstract
Interferon-gamma (IFN-γ) regulates various immune responses that are often critical for vaccine-induced protection. In order to annotate the IFN-γ-related gene interaction network from a large amount of IFN-γ research reported in the literature, a literature-based discovery approach was applied with a combination of natural language processing (NLP) and network centrality analysis. The interaction network of human IFN-γ (Gene symbol: IFNG) and its vaccine-specific subnetwork were automatically extracted using abstracts from all articles in PubMed. Four network centrality metrics were further calculated to rank the genes in the constructed networks. The resulting generic IFNG network contains 1060 genes and 26313 interactions among these genes. The vaccine-specific subnetwork contains 102 genes and 154 interactions. Fifty six genes such as TNF, NFKB1, IL2, IL6, and MAPK8 were ranked among the top 25 by at least one of the centrality methods in one or both networks. Gene enrichment analysis indicated that these genes were classified in various immune mechanisms such as response to extracellular stimulus, lymphocyte activation, and regulation of apoptosis. Literature evidence was manually curated for the IFN-γ relatedness of 56 genes and vaccine development relatedness for 52 genes. This study also generated many new hypotheses worth further experimental studies.
1. Introduction
In 1965 Wheelock et al. first reported Interferon-gamma-(IFN-γ-) like virus inhibitor induced in supernatant fluid of cultures of fresh human leukocytes following incubation with phytohemagglutinin [1]. In early 1970s, IFN-γ was further studied, and its name was eventually designated. IFN-γ is the only type II IFN family member. It is secreted by activated immune cells—primarily T and NK cells, but also B-cells, NKT cells, and professional antigen presenting cells. IFN-γ has been widely studied and found critical in anti-infectious host defense, inflammatory conditions, cancer, and autoimmune diseases [1, 2]. The most striking phenotype from mice lacking either IFN-γ or its receptor has increased susceptibility to the infections of bacterial and viral pathogens [3]. IFN-γ is also critical for tumor immunosurveillance as assessed using spontaneous, transplantable, and chemical carcinogen-induced experimental tumors. Additionally, IFN-γ is found important in leukocyte homing, cellular adhesion, immunoglobulin class switching, T helper cell polarity, antigen presentation, cell cycle arrest and apoptosis, neutrophil trafficking, and NK cell activation [1, 4, 5].
The induction of IFN-γ response is critical for successful development of vaccines against various viruses and intracellular bacteria, for example, human immunodeficiency virus (HIV) [6, 7], Mycobacterium tuberculosis [8–10], Leishmania spp. [11, 12], and Brucella spp. [13, 14]. The IFN-γ analysis is widely used for the quantification and characterization of the HIV-specific CD8+ T cell responses [6]. It is a marker used as a representative function of cytotoxic T cells to quantify the HIV-specific cellular immune response. IFN-γ is required for protection against mycobacterial infection [15]. M. tuberculosis-stimulated whole-blood production of IFN-γ, although imperfect, is the best available correlate of protective immunity to M. tuberculosis in humans [8]. In humans, complete IFN-γR deficiency is associated with frequent infection and ultimately death from the attenuated M. tuberculosis BCG vaccine [16]. The inability to secrete IFN-γ or the development of auto-antibodies neutralizing endogenous IFN-γ resulted in the death of a patient by overwhelming mycobacterium infection [17].
Today IFN-γ is ranked as one of the most important endogenous regulators of immune responses. Thousands of relevant papers have been published. However, a comprehensive understanding of how it works and what other factors it interacts with is still largely unclear. Although IFN-γ is essential for protective immunity, animal and human studies have found that IFN-γ alone is not sufficient for the prevention of TB disease [8]. Therefore, it would be very interesting to investigate what other genes or gene interaction networks are needed to stimulate protective immunity. However, due to so-complicated roles of IFN-γ in different conditions, it is challenging to annotate the interaction network of IFN-γ such that it becomes increasingly suitable to interpret its role in various diseases [1].
One of the greatest challenges that the researchers in the biomedical domain face is that most of the knowledge remains hidden in the unstructured text of the published articles. Currently, there are over 19 million publications indexed in PubMed (http://www.ncbi.nlm.nih.gov/pubmed/) and both the total number of publications and the growth rate of the number of publications are increasing exponentially [18, 19]. Given the current amount and the growth rate of the biomedical literature, it is difficult or impossible for biomedical scientists to keep up with the relevant publications. For example, a search in PubMed for “ifn-gamma OR interferon-gamma” returned 75464 articles as of October, 2009. Even if a researcher is only interested in the relatedness of IFN-γ to vaccine development and restricts his search to “vaccine AND (ifn-gamma OR interferon-gamma)”, the number of articles retrieved was 7536, which is still too high for reading manually. There are a number of manually curated databases that store protein interactions, such as the Molecular INTeraction database (MINT) [20], the Biomolecular Interaction Network Database (BIND) [21], and the Human Protein Reference Database (HPRD) [22]. Many databases also summarize results from publications about gene-disease relationships, such as the Online Mendelian Inheritance in Man (OMIM) [23], the Brucella Bioinformatics Portal (BBP) [24], and the Pathogen-Host Interaction Data Integration and Analysis System (PHIDIAS) [25]. However, it usually takes a lot of time and effort before new discoveries are included in these databases.
To systemically analyze the network of IFN-γ with other genes, an internally developed literature-based discovery approach based on literature mining and network centrality analysis was applied [26]. This literature-based discovery methodology in [26] was shown to be effective in identifying prostate cancer-related genes. To discover genes relevant to IFN-γ and vaccine development, IFN-γ was used as the single seed gene, and all the article abstracts available in PubMed were used as the text knowledge source. The interactions of IFN-γ and its neighbors from abstracts in PubMed were first extracted using a natural language processing (NLP) and machine learning (ML) based method. Two gene interaction networks were eventually built using the automatically extracted interactions. The first network is the generic IFN-γ (IFNG) network, which is the network of interactions of IFN-γ and its neighbors. The second network is the vaccine-specific subgraph of the first network, which is built using only the interactions that are extracted from vaccine relevant sentences. Next, the topologies of the networks were analyzed using the degree, eigenvector, betweenness, and closeness network centrality measures.
To the best of our knowledge, this is the first study that integrates text mining with network analysis in the vaccine informatics domain. The literature-based discovery approach that we have introduced in [26] has been successfully adapted and expanded to discover genes related to IFN-γ and vaccine development. The literature-mined IFN-γ and IFNG-vaccine-mediated networks were systematically analyzed using network centrality metrics. The results support our hypothesis that the central genes in the two IFN-γ networks are related to the functions of IFN-γ and part of the gene list is important for vaccine development. Many predicted genes and gene networks are good candidates for further IFN-γ and vaccine development studies. In this paper, we describe the overall method design and the results.
2. Methods
The high-level system description for predicting IFN-γ and vaccine-associated genes is shown in Figure 1. The approach is described in more detail in the following subsections.
Figure 1.
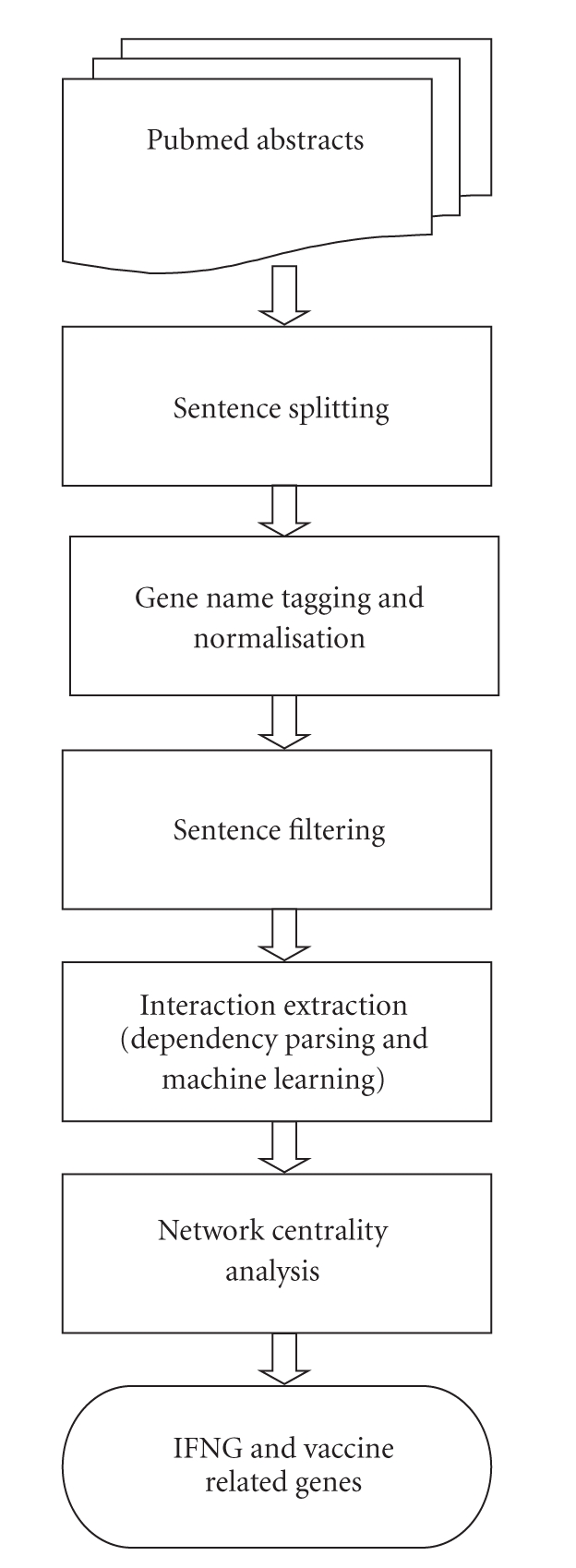
Description of the literature-based discovery system for identifying IFN-γ and vaccine-related genes.
2.1. Literature Corpus
To construct the literature-mined IFN-γ gene interaction network, all article abstracts available in PubMed are used. The sentences of the abstracts were obtained from the BioNLP database in the National Center for Integrative Biomedical Informatics (NCIBI; http://ncibi.org/), which were generated using the MxTerminator [27] sentence boundary detection tool.
2.2. Gene Name Identification and Normalization
Genia Tagger (http://www-tsujii.is.s.u-tokyo.ac.jp/GENIA/tagger/), whose developers report an F-score performance of 71.37% for biological named entity recognition [28], was used to identify the gene names in the sentences. Consider the example sentence “[IL-2] and [IL-15] induced the production of [IL-17] and [IFN-γ] in a dose dependent manner by PBMCs” taken from the abstract of [29]. The gene names, which were correctly identified by Genia Tagger, are enclosed in square brackets.
One of the greatest challenges in biomedical text processing is that a gene might have several different synonyms. For example, the IFN-γ gene can occur in text as IFN-gamma, IFNG, IFNGamma, interferon-gamma, or interferon gamma. Similarly, the IL2 gene can occur in text as IL2, IL-2, or interleukin 2. If the gene names that correspond to the same gene are not normalized, each different synonym will be represented as a separate node in the gene-interaction network as shown in Figure 2. With five different synonyms for IFN-γ and three different synonyms for IL2, 15 different edges can be obtained although they actually represent the same edge (interaction). Therefore, a dictionary-based approach was used to normalize the gene names tagged by Genia Tagger so that each gene is represented by a single node in the interaction network. HUGO Gene Nomenclature Committee (HGNC) database (http://www.genenames.org/) [30] was used as the dictionary for gene names and their synonyms. As of October, 2009 the database contains 28240 approved gene records. Each tagged gene name was unified with its corresponding approved gene symbol. In the HGNC database, the official gene symbol for the IFN-γ gene is listed as IFNG, and the description is listed as “interferon, gamma”. The database does not include any synonyms for the gene. However, IFN-γ is frequently mentioned in text with the names that are shown in Figure 2. Therefore, we included these names to the HGNC dictionary as synonyms for IFN-γ.
Figure 2.

Gene name normalization example.
2.3. Sentence Filtering
The potential interaction sentences were selected from the abstracts in PubMed that have “human” in the MeSH heading, before applying the text mining method to extract the IFN-γ (IFNG) gene-interaction network from the literature. A list of 826 interaction keywords such as binds, bound, interacts, activates, inhibits, and phosphorylates was compiled from the literature (the list of interaction keywords is available at: http://clair.si.umich.edu/clair/ifngnet/interaction_keywords.txt). Our assumption is that a sentence that describes an interaction between a pair of genes should contain an interaction keyword and at least two distinct normalized gene names. The sentences that do not meet this requirement were filtered out.
The IFNG gene-interaction network was built in two steps. In the first step, the genes that interact with IFNG (or called the neighbors of IFNG) were extracted. The number of sentences that contain IFNG or one of its synonyms (case-insensitive match) and are from abstracts that have “human” in the MeSH headings is 73024. A filter program was further performed to filter out those sentences that do not have at least one interaction keyword and at least two distinct normalized gene names, one of which is IFNG. As a result, 26876 sentences were obtained with our interaction extraction module for identification of the genes that interact with IFNG. The interaction extraction module extracted 1059 neighbors of IFNG.
In the second step, the interactions among the neighbors of IFNG were extracted. There are over 9 million sentences that are from abstracts which have “human” in the MeSH headings and contain at least one of the IFNG neighbors or their synonyms. Out of these, the sentences for further processing by the interaction extraction module are those that have at least one interaction keyword, and at least two distinct normalized gene names, which were identified as neighbors of IFNG in the first step. In total, 422566 sentences met these criteria and were further processed by the interaction extraction module, which is described in the next subsection.
2.4. Gene Interaction Extraction from the Literature
The interaction extraction task was formulated as a classification task, where each sentence is classified as a possible interaction between a given gene pair. The support vector machines (SVMs) [31] were used as our classification algorithm with features extracted from the dependency parse trees of the sentences, which capture the semantic predicate-argument dependencies among the words. The Stanford Parser (http://nlp.stanford.edu/software/lex-parser.shtml) was used to obtain the dependency parse trees of the sentences [32].
Figure 3 shows the dependency parse tree for the example sentence “IL-2 and IL-15 induced the production of IL-17 and IFN-γ in a dose dependent manner by PBMCs”. The nodes of the tree represent the words of the sentence and the edges represent the types of the dependencies among the words. For example, “IL-2” is the noun subject “nsubj” of “induced”. There are four gene names in the sentence. The sentence describes an interaction between the gene pairs “IL-2 and IL-17”, “IL-2 and IFN-γ”, “IL-15 and IL-17”, and “IL-15 and IFN-γ”. It does not describe an interaction between the gene pairs “IL-2 and IL-15” and “IL-17 and IFN-γ”.
Figure 3.

The dependency parse tree of the sentence “IL-2 and IL-15 induced the production of IL-17 and IFN-γ in a dose dependent manner by PBMCs.”
The shortest path between each gene pair from the dependency tree of the sentence was used with SVM. The motivating assumption is that the path between two gene names in a dependency tree is a good description of the semantic relation between them in the corresponding sentence. For example, the path between the interacting gene pair “IL-2 and IL-17” is “nsubj induced dobj production prep_of” and the path between the noninteracting pair “IL-2 and IL-15” is “conj_and”.
The similarity between two dependency paths was indicated based on the word-based edit distance, which is defined as the minimum number of word insertion, deletion, or substitution operations needed to transform the first path to the second. For example, the edit distance between the paths “nsubj induced dobj production prep_of” and “conj_and” is five, since the first path can be transformed to the second one by deleting four words (nsubj, induced, dobj, and production) and substituting one word, that is, substituting prep_of with conj_and. The more similar two paths are (smaller edit distance), the more likely they belong to the same class; that is, either both describe or both do not describe an interaction for the corresponding gene pairs. The path edit distance measure between two paths pi and pj was converted into a path similarity function as follows:
| (1) |
This path similarity measure was integrated as a kernel function to SVM by plugging it in the SVMlight package (http://www.svmlight.joachims.org/) [31].
This interaction extraction approach was introduced in [33] and was shown that it achieves the state-of-the-art results (55.61% F-score performance for the AIMED data set (ftp://ftp.cs.utexas.edu/pub/mooney/bio-data/) and 84.96% F-score performance for the CB data set). We have successfully applied this approach to extract the interactions of the prostate cancer relevant genes in [26] and to provide annotations for the BioCreative Meta-Server by classifying abstracts as describing a protein interaction or not in [34]. To extract the interactions of IFNG and its neighbors, the system was trained by combining the AIMED and the CB data sets. The preprocessed data sets are available at http://clair.si.umich.edu/clair/biocreative/datasets/.
2.5. Network Centrality Analysis
Gene interactions can be represented as a network, where the genes are represented as nodes, and an interaction between a pair of genes is represented with an edge connecting the corresponding nodes. This representation allows the analysis of interactions from a graph theory and complex networks perspective, which can give biologists a variety of new insights. For example, Schwikowski et al. used a majority-rule method that assigns to a protein the function that occurs most commonly among its neighbors and reported an accuracy of 70% for the yeast protein interaction network [35]. Similarly, Spirin and Mirny used the protein interaction networks to discover molecular modules that function as a unit in certain biological processes by identifying subgraphs that are densely connected within themselves but sparsely connected with the rest of the network [36].
Another network feature that can reveal important principles underlying the biological systems is the centrality of a node, which defines the relative importance of the node in the graph. The importance of a node can be defined in different ways. Degree centrality is defined as the number of edges incident to the node (i.e., the number of neighbors that a node has) [37]. It measures the extent of influence that a node has on the network. The more neighbors a node has, the more important it is.
In degree centrality each neighbor contributes equally to the centrality of a node. However, all the connections of a node are not always equally important. This notion is defined as “prestige” in social networks. The prestige of a person does not only depend on the number of acquaintances he has but also on who his acquaintances are (i.e., how prestigious they are). Eigenvector centrality assigns each node a centrality that not only depends on the quantity of the connections but also on their importance. The eigenvector centrality of a node is proportional to the sum of the centralities of its neighbors [38].
Closeness centrality of a node is defined as the inverse sum of the distances from the node to the other nodes in the network [37]. The closer a node to the other nodes in the network, the more important it is.
Betweenness centrality of a node is defined as the proportion of the shortest paths between all the pairs of nodes in the network that pass through the node in interest [37]. A node is considered important if it occurs on many shortest paths between other nodes. This characterizes the control of a node over the information flow of the network.
Centrality measures have originally been developed and used in nonbiological domains. For example, the web pages in the popular search engine Google are ranked by using the Pagerank algorithm, which is based on eigenvector centrality [39]. A number of recent studies have successfully applied centrality measures in biological domains. For example, Jeong et al. used degree centrality to predict lethal mutations in the yeast protein interaction network [40]. They showed that the network is tolerant to random errors, whereas errors related to the most central proteins cause lethality. Similarly, Joy et al. [41] and Hahn and Kern [42] have found that there is an association between the betweenness centrality and the essentiality of a gene, where an essential gene is a gene that causes the organism to die when it malfunctions. Recently, we have applied centrality measures to predict genes relevant to prostate cancer [26]. We were able to identify genes, which are not marked as being related to prostate cancer by the curated databases such as the Online Mendelian Inheritance in Man (OMIM) and the Human Prostate Gene Database (PGDB) [43] even though there are recent articles that confirm the association of these genes with the disease.
In this study the IFNG interaction network was analyzed from graph centrality perspective. IFNG and its neighbors are represented as nodes and there is an edge between two genes if an interaction between them from the literature has been extracted. The gene names in the network are normalized and represented with their official HGNC symbols. The vaccine-specific subgraph of this network contains only the interactions that have been extracted from sentences that contain the term “vaccin”, which is the root form of the vaccine-related terms such as vaccine, vaccines, vaccination, and vaccinated. Therefore, the edges in this subgraph are all vaccine specific. Analysis of this IFNG-vaccine network helps us to understand the genes and interactions that play important roles in both the vaccine and IFNG network. Since IFNG is one of the most important immune factors and critical for vaccine development, we hypothesized that genes central in the generic IFNG and IFNG-vaccine networks might be important for vaccine development. The results presented in the next section support the hypothesis.
2.6. Gene Annotation Enrichment Analysis
The web-based DAVID bioinformatics program was used to perform the gene annotation enrichment analysis [44].
3. Results
3.1. Topological Properties of the Networks
Our program detected 1060 nodes (genes including IFNG and its neighbors) linked by 26313 edges (interactions) (Figure 4). Since all the genes in the IFNG network are connected to IFNG, the diameter of the network (the longest of the shortest paths between the pairs of genes in the interaction network) is 2 and the average shortest path length (the average of the shortest paths between all genes in the network) is 1.95. The clustering coefficient of the network is 0.4933, which is an order of magnitude higher than the clustering coefficient of a random network with the same number of nodes (0.0473). The clustering coefficient [45] of a node describes how well connected a node's neighbors are and is defined as the number of connections between this node's neighbors divided by the number of possible connections between them. The clustering coefficient of a network is the average of the clustering coefficients of the nodes in the network. The IFNG network is a small-world network [45], characterized by having a small average shortest path length and a clustering coefficient that is significantly higher than that of a random network with the same number of nodes. The IFNG network is a scale-free network, which is characterized by having a power-law degree distribution, P(k) ~ k−γ, where P(k) is the probability that a randomly selected node will have a degree (i.e., number of connections) of k [46].
Figure 4.

Summary of the IFNG network and its vaccine-specific subnetwork.
In scale-free networks most nodes make only a few connections, while a small set of nodes (known as hubs) have very large number of links. This is different from random networks, which follow Poisson distribution, where majority of the nodes have degrees close to the average degree of the network. The exponent (γ) of the power-law degree distribution of the IFNG network is 2.15. The graph of the IFNG network is shown in Figure 1 of the supplementary material available online at doi:10.1155/2010/426479.
The IFNG and vaccine-associated network (IFNG-vaccine network) is a much smaller subset of the generic IFNG network. This small subnetwork contains 102 genes and 154 interactions (Figure 4). Since the IFNG-vaccine network is built by removing the edges that are not associated with “vaccine” from the IFNG network, some of the genes that were connected in the IFNG network are not connected in the IFNG-vaccine network. In total, the IFNG-vaccine network contains 84 genes that are interconnected and 18 genes that are separated from this largest connected component of 84 genes (Figure 5). Also, the diameter of the IFNG-vaccine network and the average shortest path length are larger than those of the IFNG network. The diameter of the IFNG-vaccine network is 9 and the average shortest path length is 3.55. The IFNG-vaccine network still possesses the small-world property with a relatively small average shortest path length and a clustering coefficient (0.2218) that is significantly higher than the clustering coefficient of a random network with the same number of nodes (0.0388). The network is scale-free with a power-law degree distribution with exponent 2.37. The small-world and scale-free characteristics of the generic IFNG and the IFNG-vaccine networks are consistent with the topological properties of previously studied biological networks [26, 40, 47, 48] and nonbiological networks such as the Internet [49] and social networks [45].
Figure 5.

The graph of the IFNG-vaccine network extracted from the literature. The network consists of 102 nodes (genes) and 154 edges (interactions). All the edges in the network are associated with the term “vaccine” and its variants. The purple nodes are the genes that are central in both the generic and the IFNG-vaccine networks. The red nodes are the genes that are central only in the IFNG-vaccine network. The green nodes are the genes that are central only in the generic IFNG network. The rest of the nodes are shown in yellow.
3.2. Lists of Genes Are Predicted and Sorted by Centrality Analyses
All the genes in the two networks (generic IFNG network and IFNG-vaccine network) are sorted based on centrality analyses. Supplementary File 1 lists the rankings of all the genes in the generic IFNG network and Supplementary File 2 lists the rankings of all the genes in the IFNG-vaccine network. IFNG is not included in these rankings, since it is trivially ranked highest by all the centrality measures in both networks due to the fact that the networks are specific to IFNG. The most central genes (the genes ranked among the top 25 by at least one of the centrality measures) are analyzed in more detail in Table 1. These genes (a total of 56 genes) are predicted to be associated with IFNG and relevant for vaccine development. Literature evidence was manually curated for the IFNG association (IFNG-Ref column in Table 1) and the vaccine development relatedness (Vaccine-Ref column in Table 1) of these genes.
Table 1.
Predicted 56 genes related to IFN-γ and vaccine networks.
| Gene | Generic IFNG Network | IFNG-vaccine Network | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| D | E | B | C | IFNG-Ref | D | E | B | C | Vaccine-Ref | |
| TNF | 1 | 1 | 1 | 1 | 3132506 | 2 | 3 | 2 | 7 | 16446013 |
| NFKB1 | 2 | 2 | 2 | 2 | 9888423 | — | 23 | — | — | 16971487 |
| IL6 | 3 | 3 | 3 | 3 | 1719090 | 3 | 4 | 7 | 3 | 10225849 |
| IL8 | 4 | 5 | 6 | 4 | 8473010 | 10 | 13 | 10 | 9 | 11378044 |
| IL10 | 5 | 4 | 4 | 5 | 8102388 | 6 | 8 | 11 | 2 | 10930151 |
| IL4 | 6 | 6 | 5 | 6 | 2136895 | 4 | 2 | 4 | 4 | 8519092 |
| MAPK1 | 7 | 9 | 9 | 7 | 15307176 | — | — | — | — | 19428911 |
| IL2 | 8 | 7 | 8 | 8 | 6429853 | 1 | 1 | 1 | 1 | 8459207 |
| VEGFA* | 9 | 10 | 10 | 9 | 12816689 | — | — | — | — | 17502972 |
| TP53* | 10 | 8 | 7 | 10 | 16391798 | — | — | — | — | 18846387 |
| BCL2* | 11 | 13 | 13 | 11 | 11064392 | — | — | — | — | 19389797 |
| AKT1* | 12 | 11 | 12 | 12 | 11135576 | — | — | — | — | 19107122 |
| MAPK8 | 13 | 14 | 14 | 13 | 18950753 | — | — | 15 | — | 19428911 |
| INS | 14 | 12 | 11 | 14 | 8383325 | — | — | — | 16 | 19203100 |
| MAPK14 | 15 | 15 | 18 | 15 | 10700460 | — | — | — | — | 19428911 |
| CSF2 | 16 | 18 | 17 | 16 | 11665752 | 7 | 6 | 6 | 6 | 19459853 |
| FAS | 17 | 17 | 16 | 17 | 10895367 | — | — | — | — | 15979942 |
| CCL2 | 18 | 19 | 19 | 18 | 9407497 | — | — | — | — | 19833737 |
| IFNA1 | 19 | 16 | 15 | 19 | 11449378 | — | — | — | 13 | 19667099 |
| EGFR* | 20 | 20 | 23 | 20 | 17362940 | — | — | — | — | 19178753 |
| JUND* | 21 | 21 | 22 | 21 | 10070035 | — | — | — | — | 19124729 |
| KITLG* | 22 | 24 | — | 22 | 7540064 | — | — | — | — | - |
| CCL5 | 23 | 23 | 21 | 23 | 8921438 | — | 24 | — | — | 15827150 |
| CD4 | 24 | 22 | 20 | 24 | 15173593 | 9 | 5 | 3 | 12 | 17298856 |
| EGF* | 25 | 25 | — | 25 | 18160214 | — | — | — | — | 16357522 |
| CRP | — | — | 24 | — | 10675363 | — | — | — | — | 16395099 |
| STAT3* | — | — | 25 | — | 7488223 | — | — | — | — | - |
| IL5 | — | — | — | — | 9432015 | 5 | 7 | 20 | 8 | 11138639 |
| IL13 | — | — | — | — | 12670721 | 8 | 9 | 5 | 5 | 12232042 |
| IL7 | — | — | — | — | 7594482 | 11 | 14 | 12 | 17 | 17496983 |
| EIF2AK2 | — | — | — | — | 11342638 | 12 | 10 | 8 | — | 19596385 |
| CD28 | — | — | — | — | 7634349 | 13 | 12 | — | — | 12594842 |
| HSPD1 | — | — | — | — | 12407015 | 14 | 19 | 16 | 14 | 12218165 |
| SILV | — | — | — | — | 11839572 | 15 | 20 | 17 | 23 | 11459172 |
| IL21 | — | — | — | — | 14657853 | 16 | 17 | — | — | 16785513 |
| IL18 | — | — | — | — | 8666798 | 17 | — | — | 10 | 19467215 |
| HBEGF | — | — | — | — | 9062364 | 18 | 25 | — | 21 | 10729731 |
| CD46 | — | — | — | — | 15307176 | 19 | 11 | 9 | — | 11757799 |
| CD40 | — | — | — | — | 7554483 | 20 | 16 | — | — | 11403919 |
| PSG2 | — | — | — | — | 2516715 | 21 | — | 22 | — | 11155821 |
| GAD1 | — | — | — | — | 9703171 | 22 | — | — | 18 | 12421990 |
| IL15 | — | — | — | — | 9834271 | 23 | — | — | 22 | 16785513 |
| C3 | — | — | — | — | 1337336 | 24 | 15 | — | — | 19477524 |
| PRF1 | — | — | — | — | 19651871 | 25 | 22 | 19 | — | 15214037 |
| ZAP70 | — | — | — | — | 11034358 | — | 18 | 23 | — | — |
| CD40LG | — | — | — | — | 10769003 | — | 21 | 18 | — | 11403919 |
| GNLY | — | — | — | — | 17382591 | — | — | 13 | 19 | 10644038 |
| PTPN11 | — | — | — | — | 12270932 | — | — | 14 | — | — |
| CD86 | — | — | — | — | 9836505 | — | — | 21 | — | 12594842 |
| CCR5 | — | — | — | — | 9616137 | — | — | 24 | — | 16672545 |
| HSPA4 | — | — | — | — | 18442794 | — | — | 25 | — | 11779704 |
| TPBG | — | — | — | — | 16630022 | — | — | — | 11 | 16630022 |
| KLK3 | — | — | — | — | 16000955 | — | — | — | 15 | 19171173 |
| CD8A | — | — | — | — | 1904117 | — | — | — | 20 | 18425263 |
| CD80 | — | — | — | — | 7537534 | — | — | — | 24 | 10498243 |
| LTA | — | — | — | — | 3102976 | — | — | — | 25 | 15908422 |
Note: The genes ranked among the top 25 by the centrality measures (D: Degree; E: Eigenvector; B: Betweenness; C: Closeness) in the generic IFNG network or the IFNG-vaccine network. The genes are represented with their official HGNC symbols. Literature evidences for the relatedness of the genes to IFNG (IFNG-Ref) and to vaccine development (Vaccine-Ref) are manually curated. “—” indicates that the gene is not ranked among the top 25 by the corresponding centrality measure in the corresponding network or no literature evidence was found.
It is interesting that in the generic IFNG network, all centrality measures find the same 23 genes among the top 25, although the ranking might change slightly (Table 1). For example, IL10 is ranked 5th by degree and closeness centralities, but 4th by eigenvector and betweenness centralities. Since all the genes in the generic IFNG network are connected to IFNG, the distance (shortest path length) between a pair of genes is at most two. In other words, the distance between a pair of genes is one if they are directly connected to each other and it is two if they are not directly connected to each other (i.e., they are connected through IFNG). Therefore, in this network, the more genes a gene is connected to (higher degree centrality), the less distant it is to the other genes (higher closeness centrality). So, the degree and closeness centralities produce the same rankings for the generic IFNG network. For the IFNG-vaccine network, the top 25 genes sorted based on centrality analyses overlapped with the sorted results from the generic IFNG network.
Three different levels of prediction are available based on the comparison between the generic IFNG network and the more specific IFNG-vaccine network.
(1) Genes Ranked High in Both Networks —
Thirteen genes were ranked among the top 25 in both networks by at least one of the centrality measures. Among these 13 genes, 8 genes are central by all centrality measures in both networks: TNF, IL6, IL8, IL10, IL4, IL2, CSF2, and CD4. These genes are well studied in both generic IFNG research and vaccine specific research. The ranking may change in both networks. For example, IL2 was ranked top 1 in the IFNG-vaccine network, while it was ranked top 7-8 in the generic IFNG network based on different centrality scores. This is probably due to the fact that the role of IL2 in vaccine research has widely been recognized and studied in more depth in the vaccine context.
Among the 13 genes in this group, five genes (NFKB1, MAPK8, INS, IFNA1, and CCL5) were ranked high in the IFNG network by all measures but only high in the IFNG-vaccine network by certain centrality measures. For example, MAPK8 (mitogen-activated protein kinase 8; Aliases: JNK, JNK1, SAPK1) was ranked high by all centrality metrics in the IFNG network, whereas it was ranked high by only the betweenness centrality metric in the IFNG-vaccine network (Table 1). The high betweenness score was reflected by the fact that MAPK8 connects the two genes (ZAP70 and MAPK1) to the rest of the network (Figure 5). In the generic IFNG network, 322 other genes are directly connected to MAPK8 (Figure 6). Many of these genes (e.g., NFKB1, IL4, and CD40) also exist in the IFNG-vaccine network (Figure 5) although they do not directly interact with MAPK8. However, the majority of these 322 genes (e.g., TLR4 and IL1B) are not in the IFNG-vaccine network. It is reasonable to suggest that many of these genes that were found in the IFNG-MAPK8 network (Figure 6) but not in the IFNG-vaccine network (Figure 5) may also be important for vaccine specific network through an interaction with MARK8. Therefore, the comparison between these two networks may lead to hypothesis of new genes involved in vaccine specific immune network, some of which deserve further experimental verifications.
Figure 6.

Interactions of MAPK8 with other genes in the generic IFNG network. MAPK8 is shown in purple. The two genes (ZAP70 and EIF2AK2) that MAPK8 also interacts in the IFNG-vaccine network are shown in red.
(2) Genes Ranked High in the Generic IFNG Network but Not in the IFNG-Vaccine Network —
In total 14 genes are included in this group. Nine out of these 14 genes were not found in the IFNG-vaccine network (Supplementary File 2). These genes are labeled with “∗” in Table 1. These genes have not been well studied in the vaccine context. However, since these genes are strongly associated with IFNG, it is likely that each of these genes may also play an important role in vaccine-induced protective immune network. For example, as one of the 14 genes, the serine/threonine kinase AKT1 is a key regulator of cell proliferation and death. AKT1 regulates lymphocyte apoptosis and Th1 cytokine propensity [50]. IFNG is a representative cytokine in Th1 response that is crucial for induction of vaccine-induced protection. Therefore, it is reasonable to hypothesize that AKT1 plays an important role in regulated vaccine-induced protective immune responses.
Among the 14 genes in this group, five genes (MAPK1, MAPK14, FAS, CCL2, and CRP) were found in the IFNG-vaccine network but not ranked high based on any centrality analysis. For example, FAS is a critical gene in regulation of programmed cell death through the FAS pathway. FAS (TNF receptor superfamily, member 6; Aliases: CD95, APO-1) has been found to play an important role in promoting an appropriate effector response following vaccinations against Helicobacter pylori [51], hepatitis C virus [52], and cancer [53]. Since FAS is well studied and ranked top in the generic IFNG network, more knowledge about its interactions with other genes shown from the generic IFNG network provides valuable basis for further analysis of FAS-related, vaccine-specific interaction network.
(3) Genes Ranked High in the IFNG-Vaccine Network but Not in the Generic IFNG Network —
In total, 29 genes that were ranked among the top 25 in the IFNG-vaccine network based on at least one of the centrality scores are not ranked among the top 25 in the generic IFNG network (Table 1). These genes may be more vaccine-specific and play relatively less important roles in many other IFNG-regulated immune systems (e.g., cell cycle). It is also possible that some of these genes are very important for other IFNG-related immune functions. In that case, the data for these genes obtained from vaccine research may provide supportive results for expanded studies. One important set of these 29 genes cover many interleukins including IL5, IL7, IL13, IL15, IL18, and IL21. For example, interleukin-18 (IL18) is a newly discovered cytokine with profound effects on T-cell activation. IL18 can possibly be used as a strong vaccine adjuvant [54]. The new knowledge obtained from IL18 in vaccine research may be applied to other IFNG-related immune systems.
3.3. Gene Annotation Enrichment Shows Various Immune Responses Regulated by IFN-γ
The 56 genes ranked among the top 25 by at least one of the centrality methods in one or both networks were used for gene enrichment analysis using DAVID [44]. These genes were classified in various immune mechanisms such as response to extracellular stimulus, lymphocyte activation, and regulation of apoptosis (Table 2). These gene annotation enrichment results are correlated with current knowledge about IFN-γ [1, 4, 5]. It further demonstrates the capability of our literature-based discovery approach in correctly extracting genes related to IFN-γ.
Table 2.
Gene annotation enrichment among top predicted genes in the generic IFNG and the IFNG-vaccine networks.
| Category | Term | Count | P-Value | FDR |
|---|---|---|---|---|
| GOTERM_BP_ALL | GO:0050896~response to stimulus | 43 | 2.99E − 22 | 5.71E − 19 |
| GOTERM_BP_ALL | GO:0007154~cell communication | 39 | 5.74E − 13 | 1.10E − 09 |
| GOTERM_BP_ALL | GO:0007165~signal transduction | 35 | 9.70E − 11 | 1.86E − 07 |
| GOTERM_BP_ALL | GO:0006950~response to stress | 29 | 7.14E − 20 | 1.37E − 16 |
| GOTERM_BP_ALL | GO:0030154~cell differentiation | 28 | 6.94E − 13 | 1.33E − 09 |
| GOTERM_BP_ALL | GO:0006952~defense response | 26 | 7.12E − 23 | 1.36E − 19 |
| GOTERM_BP_ALL | GO:0006955~immune response | 26 | 8.88E − 18 | 1.70E − 14 |
| GOTERM_BP_ALL | GO:0008283~cell proliferation | 23 | 9.37E − 16 | 1.70E − 12 |
| GOTERM_BP_ALL | GO:0008219~cell death | 23 | 2.28E − 15 | 4.46E − 12 |
| GOTERM_BP_ALL | GO:0006915~apoptosis | 22 | 9.38E − 15 | 1.78E − 11 |
| GOTERM_BP_ALL | GO:0007242~intracellular signaling cascade | 19 | 4.85E − 07 | 9.27E − 04 |
| GOTERM_BP_ALL | GO:0001775~cell activation | 18 | 5.86E − 19 | 1.12E − 15 |
| GOTERM_BP_ALL | GO:0006954~inflammatory response | 17 | 8.26E − 16 | 1.49E − 12 |
| GOTERM_BP_ALL | GO:0046649~lymphocyte activation | 14 | 1.77E − 14 | 3.38E − 11 |
| GOTERM_BP_ALL | GO:0006468~protein amino acid phosphorylation | 14 | 2.60E − 07 | 4.98E − 04 |
| GOTERM_BP_ALL | GO:0006807~nitrogen compound metabolic process | 13 | 4.47E − 08 | 8.56E − 05 |
| GOTERM_BP_ALL | GO:0042110~T cell activation | 12 | 9.02E − 14 | 1.73E − 10 |
| GOTERM_BP_ALL | GO:0048534~hemopoietic or lymphoid organ development | 12 | 4.57E − 11 | 8.74E − 08 |
| GOTERM_CC_ALL | GO:0005576~extracellular region | 29 | 5.33E − 18 | 8.27E − 15 |
| GOTERM_MF_ALL | GO:0005125~cytokine activity | 19 | 5.30E − 21 | 9.51E − 18 |
| GOTERM_MF_ALL | GO:0008083~growth factor activity | 12 | 6.77E − 12 | 1.21E − 08 |
| KEGG_PATHWAY | hsa04060:Cytokine-cytokine receptor interaction | 23 | 7.90E − 16 | 9.77E − 13 |
| KEGG_PATHWAY | hsa04620:Toll-like receptor signaling pathway | 13 | 3.12E − 10 | 3.91E − 07 |
| KEGG_PATHWAY | hsa04660:T cell receptor signaling pathway | 12 | 2.04E − 09 | 2.57E − 06 |
| KEGG_PATHWAY | hsa04630:Jak-STAT signaling pathway | 11 | 2.99E − 06 | 0.003745 |
4. Discussion
Our method is different from many other literature mining approaches. To extract the gene interactions from the text, an SVM classifier was used in our approach with features extracted from the dependency parse trees of the sentences [33]. A dependency parse tree captures the semantic predicate-argument relationships among the words of a sentence. Compared to the traditional cooccurrence and pattern-matching-based information extraction methods, our method allows us to make more syntax-aware inferences about the roles of the genes in a sentence. Our method of integrating literature mining with network analysis was first introduced in 2008 to study prostate cancer [26]. In that study, 15 genes related to prostate cancer were chosen as seed genes, and 48245 articles from PubMed Central (PMC) Open Access (http://ncbi.nlm.nih.gov/pmc/about/openftlist.html) were processed to build the network of their interactions. Genes that are not marked as being related to prostate cancer by the curated OMIM or PGDB [43] databases were identified even though there are recent articles that confirm their association to the disease. In this current study, only one gene (IFNG) was used as the seed gene, and 19 million papers in PubMed were analyzed. Therefore, our method of literature-based discovery can be generalized and used in different applications. Since the vaccine is emphasized, the vaccine term and its variation terms in our NLP analysis were used (act like gene in our approach). This approach is new in this type of analysis.
Our analysis discovered a large number of genes that interact with IFNG and genes important for both IFNG and vaccine. Many of these genes have been studied but never been collected for systematic network analysis. Current databases contain limited information about IFNG gene interaction network. The Michigan Molecular Interactions (MiMI) is a repository that includes interaction data from over 10 databases such as the Database of Interacting Proteins (DIP), the Human Protein Reference Database (HPRD), and the Biomolecular Interaction Network Database (BIND) [55]. As of October 2009, MiMI contains only 12 genes that interact with IFNG and 27 interactions among these genes. Our IFNG gene interaction network contains more than 80-fold of genes that interact with IFNG. While the correctness of all these interactions require further confirmation, our manual confirmation of selected 56 interactions (Table 1) has already demonstrated the power of our literature-based discovery method. Since IFNG is an important immune regulator for vaccine-induced protective immunity, the systematical analysis of vaccine-induced IFNG-regulated gene network is critical to understand vaccine-induced immune mechanism and support rational vaccine design. Our selective analyses of the IFNG-vaccine subnetwork showed that genes potentially important for vaccine research can be predicted. Many predicted genes and gene networks deserve further experimental verifications.
Our study demonstrated that MAPK8 is an important component of the generic IFNG network (Table 1, Figures 5 and 6). MAPK8 is a member of the mitogen-activated protein (MAP) kinase family. MAPK8 is important for many cellular processes such as cell proliferation, apoptosis, and differentiation. IFNG and MAPK8 regulate each other depending on different experimental conditions [56–60]. For example, the IFNG inhibits the activation of MAPK8 in macrophages and many other cells through the production of nitric oxide [56]. However, IFNG activates JNK activation and both contribute to apoptosis in lymphocyte cells through the regulation of the reactive oxygen species (ROS) production [57]. Meanwhile, the JNK stress-activated MAPK signal transduction pathway is required for IFNG production for T helper 1 (Th1) effector cells [58]. The inhibition of MAPK8 results in marked reduction of IFNG transcription in activated Jurkat T cells [59]. The activation of JNK pathway also mediates the production of IFNG in human breast tumor cells [60]. Our study shows that MAPK8 interacts with 322 genes which also interact individually with IFNG in the generic IFNG network (Figure 6). The finding of such a large number of interactive genes suggests that the interactions among MAPK8, IFNG, and the other genes may regulate many different biological processes. Based on our GO enrichment analysis (data not shown), the 322 genes that interact with both MAPK8 and IFNG (Figure 6) cover a variety of different biological processes, such as response to external stimulus, inflammatory response, cell proliferation, programmed cell death, and cytokine activity. It is interesting that only two genes (ZAP70 and EIF2AK2) among the 323 genes in the IFNG-MAPK8 network were found to connect to MAPK8 in the IFNG-vaccine network (Figure 5). Since many other genes (e.g., NFKB1, IL4, and CD40) in the IFNG-MAPK8 network also exist in the IFNG-vaccine network (Figure 5), it is possible that more genes act in the vaccine-specific gene network through their interactions with both MAPK8 and IFNG. It is also likely that many genes shown in the IFNG-MAPK8 network but not in the IFNG-vaccine network may contribute to vaccine-induced protective immunity.
Future work includes development of a web server to store the analyzed data and provide a user-friendly web interface to query and visualize the analyzed data. We expect to provide such a user-friendly web interface for the analyses of IFNG and IFNG-vaccine gene networks in 2010. It is noted that the interactions shown in our networks may be specific for certain conditions. The interactions may not be true when experimental conditions change. One future research is to link individual interactions to specific conditions. It will provide us a more comprehensive view of the IFNG and vaccine networks. Our literature mining method will also be applied to analyze other IFNG and vaccine-related interaction networks in other animal species (e.g., mouse, rat, and cattle).
Supplementary Material
Supplementary Figure 1. The graph of the generic IFNG network extracted from the literature. The network consists of 1060 nodes (genes) and 26,313 edges (interactions). The purple nodes are the genes that are central in both the generic and the IFNG-vaccine networks. The green nodes are the genes that are central in only the generic IFNG network and the red nodes are the genes that are central in only the IFNG-vaccine network. The rest of the nodes are shown in yellow.
Supplementary File 1. The rankings of all the genes in the generic IFNG network by degree, eigenvector, betweenness, and closeness centrality metrics.
Supplementary File 2. The rankings of all the genes in the IFNG-vaccine network by degree, eigenvector, betweenness, and closeness centrality metrics.
Acknowledgments
This research is supported by NIH Grants R01AI081062 and U54-DA-021519. The authors appreciate Alex Ade's support for their access to the BioNLP database in the National Center for Integrative Biomedical Informatics (NCIBI).
References
- 1.Billiau A, Matthys P. Interferon-γ: a historical perspective. Cytokine and Growth Factor Reviews. 2009;20(2):97–113. doi: 10.1016/j.cytogfr.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 2.Wieder T, Braumuller H, Kneilling M, Pichler B, Rocken M. T cell-mediated help against tumors. Cell Cycle. 2008;7(19):2974–2977. doi: 10.4161/cc.7.19.6798. [DOI] [PubMed] [Google Scholar]
- 3.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-γ: an overview of signals, mechanisms and functions. Journal of Leukocyte Biology. 2004;75(2):163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 4.Gough DJ, Levy DE, Johnstone RW, Clarke CJ. IFNγ signaling—does it mean JAK-STAT? Cytokine and Growth Factor Reviews. 2008;19(5-6):383–394. doi: 10.1016/j.cytogfr.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Takayanagi H, Sato K, Takaoka A, Taniguchi T. Interplay between interferon and other cytokine systems in bone metabolism. Immunological Reviews. 2005;208:181–193. doi: 10.1111/j.0105-2896.2005.00337.x. [DOI] [PubMed] [Google Scholar]
- 6.Streeck H, Frahm N, Walker BD. The role of IFN-γ Elispot assay in HIV vaccine research. Nature Protocols. 2009;4(4):461–469. doi: 10.1038/nprot.2009.7. [DOI] [PubMed] [Google Scholar]
- 7.Kedzierska K, Crowe SM. Cytokines and HIV-1: interactions and clinical implications. Antiviral Chemistry and Chemotherapy. 2001;12(3):133–150. doi: 10.1177/095632020101200301. [DOI] [PubMed] [Google Scholar]
- 8.Fletcher HA. Correlates of immune protection from tuberculosis. Current Molecular Medicine. 2007;7(3):319–325. doi: 10.2174/156652407780598520. [DOI] [PubMed] [Google Scholar]
- 9.Flynn JL. Immunology of tuberculosis and implications in vaccine development. Tuberculosis. 2004;84(1-2):93–101. doi: 10.1016/j.tube.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 10.Rook GAW, Seah G, Ustianowski A. M. tuberculosis: immunology and vaccination. European Respiratory Journal. 2001;17(3):537–557. doi: 10.1183/09031936.01.17305370. [DOI] [PubMed] [Google Scholar]
- 11.Mansueto P, Vitale G, Di Lorenzo G, Rini GB, Mansueto S, Cillari E. Immunopathology of leishmaniasis: an update. International Journal of Immunopathology and Pharmacology. 2007;20(3):435–445. doi: 10.1177/039463200702000302. [DOI] [PubMed] [Google Scholar]
- 12.Roberts MTM. Current understandings on the immunology of leishmaniasis and recent developments in prevention and treatment. British Medical Bulletin. 2005;75-76(1):115–130. doi: 10.1093/bmb/ldl003. [DOI] [PubMed] [Google Scholar]
- 13.He Y, Vemulapalli R, Schurig GG. Recombinant Ochrobactrum anthropi expressing Brucella abortus Cu,Zn superoxide dismutase protects mice against B. abortus infection only after switching of immune responses to Th1 type. Infection and Immunity. 2002;70(5):2535–2543. doi: 10.1128/IAI.70.5.2535-2543.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He Y, Vemulapalli R, Zeytun A, Schurig GG. Induction of specific cytotoxic lymphocytes in mice vaccinated with Brucella abortus RB51. Infection and Immunity. 2001;69(9):5502–5508. doi: 10.1128/IAI.69.9.5502-5508.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallis RS, Doherty TM, Onyebujoh P, et al. Biomarkers for tuberculosis disease activity, cure, and relapse. The Lancet Infectious Diseases. 2009;9(3):162–172. doi: 10.1016/S1473-3099(09)70042-8. [DOI] [PubMed] [Google Scholar]
- 16.Jouanguy E, Altare F, Lamhamedi S, et al. Interferon-γ-receptor deficiency in an infant with fatal bacille Calmette-Guerin infection. New England Journal of Medicine. 1996;335(26):1956–1961. doi: 10.1056/NEJM199612263352604. [DOI] [PubMed] [Google Scholar]
- 17.Doffinger R, Helbert MR, Barcenas-Morales G, et al. Autoantibodies to interferon-gamma in a patient with selective susceptibility to mycobacterial infection and organ-specific autoimmunity. Clinical Infectious Diseases. 2004;38(1):e10–e14. doi: 10.1086/380453. [DOI] [PubMed] [Google Scholar]
- 18.Cohen BK, Hunter L. Artificial Intelligence Methods and Tools for Systems Biology. Berlin, Germany: Springer; 2004. Natural language processing and systems biology; pp. 147–173. [Google Scholar]
- 19.Hunter L, Cohen KB. Biomedical language processing: what’s beyond PubMed? Molecular Cell. 2006;21(5):589–594. doi: 10.1016/j.molcel.2006.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zanzoni A, Montecchi-Palazzi L, Quondam M, Ausiello G, Helmer-Citterich M, Cesareni G. MINT: a molecular INTeraction database. FEBS Letters. 2002;513(1):135–140. doi: 10.1016/s0014-5793(01)03293-8. [DOI] [PubMed] [Google Scholar]
- 21.Bader GD, Betel D, Hogue CWV. BIND: the biomolecular interaction network database. Nucleic Acids. 2003;31(1):248–250. doi: 10.1093/nar/gkg056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keshava Prasad TS, Goel R, Kandasamy K, et al. Human protein reference database—2009 update. Nucleic Acids Research. 2009;37, database issue(supplement 1):D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Online Mendelian Inheritance in Man (OMIM) http://www.ncbi.nlm.nih.gov/omim/.
- 24.Xiang Z, Zheng W, He Y. BBP: Brucella genome annotation with literature mining and curation. BMC Bioinformatics. 2006;7(1, article 347) doi: 10.1186/1471-2105-7-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiang Z, Tian Y, He Y. PHIDIAS: a pathogen-host interaction data integration and analysis system. Genome Biology. 2007;8(7, article R150) doi: 10.1186/gb-2007-8-7-r150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Özgür A, Vu T, Erkan G, Radev DR. Identifying gene-disease associations using centrality on a literature mined gene-interaction network. Bioinformatics. 2008;24(13):i277–i285. doi: 10.1093/bioinformatics/btn182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reynar JC, Ratnaparkhi A. A maximum entropy approach to identifying sentence boundaries. In: Proceedings of the 5th Conference on Applied Natural Language Processing; March-April 1997; Washington, DC, USA. [Google Scholar]
- 28.Tsuruoka Y, Tateishi Y, Kim J-D, et al. Developing a robust part-of-speech tagger for biomedical text. Advances in informatics. In: Proceedings of the 10th Panhellenic Conference on Informatics, vol. 3746; 2005; pp. 382–392. Lecture Notes in Computer Science. [Google Scholar]
- 29.Fan YY, Wu CY. The role of cytokines in the production of IL-17 and IFN-gamma via the induction of normal human peripheral blood mononuclear cells and CD4(+) T cells. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2007;23(10):914–916. [PubMed] [Google Scholar]
- 30.Eyre TA, Ducluzeau F, Sneddon TP, Povey S, Bruford EA, Lush MJ. The HUGO gene nomenclature database, 2006 updates. Nucleic Acids Research. 2006;34, database issue:D319–D321. doi: 10.1093/nar/gkj147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joachims T. Making large-scale support vector machine learning practical. In: Schölkopf B, Smola AJ, editors. Advances in Kernel Methods: Support Vector Learning. Cambridge, Mass, USA: MIT Press; 1999. pp. 169–184. [Google Scholar]
- 32.de Marneffe MC, Maccartney B, Manning CD. Generating typed dependency parses from phrase structure parses. In: Proceedings of the 5th International Conference on Language Resources and Evaluation (LREC ’06); 2006. [Google Scholar]
- 33.Erkan G, Özgür A, Radev DR. Semi-supervised classification for extracting protein interaction sentences using dependency parsing. In: Proceedings of the 2007 Joint Conference on Empirical Methods in Natural Language Processing and Computational Natural Language Learning (EMNLP-CoNLL ’07); 2007; pp. 228–237. [Google Scholar]
- 34.Leitner F, Krallinger M, Rodriguez-Penagos C, et al. Introducing meta-services for biomedical information extraction. Genome Biology. 2008;9(supplement 2, article S6) doi: 10.1186/gb-2008-9-s2-s6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwikowski B, Uetz P, Fields S. A network of protein-protein interactions in yeast. Nature Biotechnology. 2000;18(12):1257–1261. doi: 10.1038/82360. [DOI] [PubMed] [Google Scholar]
- 36.Spirin V, Mirny LA. Protein complexes and functional modules in molecular networks. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(21):12123–12128. doi: 10.1073/pnas.2032324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Freeman LC. Centrality in social networks: conceptual clarification. Social Networks. 1978;1(3):215–239. [Google Scholar]
- 38.Newman MEJ. The structure and function of complex networks. SIAM Review. 2003;45(2):167–256. [Google Scholar]
- 39.Page L, Brin S, Motwani R, Winograd T. Stanford InfoLab; 1999. The pagerank citation ranking: bringing order to the web. Tech. Rep. [Google Scholar]
- 40.Jeong H, Mason SP, Barabasi A-L, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411(6833):41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 41.Joy MP, Brock A, Ingber DE, Huang S. High-betweenness proteins in the yeast protein interaction network. Journal of Biomedical Biotechnology. 2005;2005(2):96–103. doi: 10.1155/JBB.2005.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hahn MW, Kern AD. Comparative genomics of centrality and essentiality in three eukaryotic protein-interaction networks. Molecular Biology and Evolution. 2005;22(4):803–806. doi: 10.1093/molbev/msi072. [DOI] [PubMed] [Google Scholar]
- 43.Li L-C, Zhao H, Shiina H, Kane CJ, Dahiya R. PGDB: a curated and integrated database of genes related to the prostate. Nucleic Acids Research. 2003;31(1):291–293. doi: 10.1093/nar/gkg008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 45.Watts DJ, Strogatz SH. Collective dynamics of ‘small-world’ networks. Nature. 1998;393(6684):440–442. doi: 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- 46.Albert R, Barabasi A-L. Statistical mechanics of complex networks. Reviews of Modern Physics. 2002;74(1):47–97. [Google Scholar]
- 47.Chen H, Sharp BM. Content-rich biological network constructed by mining PubMed abstracts. BMC Bioinformatics. 2004;5, article 147 doi: 10.1186/1471-2105-5-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoffmann R, Valencia A. Implementing the iHOP concept for navigation of biomedical literature. Bioinformatics. 2005;21(2):ii252–ii258. doi: 10.1093/bioinformatics/bti1142. [DOI] [PubMed] [Google Scholar]
- 49.Yook S-H, Jeong H, Barabasi A-L. Modeling the internet’s large-scale topology. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(21):13382–13386. doi: 10.1073/pnas.172501399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bommhardt U, Chang KC, Swanson PE, et al. Akt decreases lymphocyte apoptosis and improves survival in sepsis. Journal of Immunology. 2004;172(12):7583–7591. doi: 10.4049/jimmunol.172.12.7583. [DOI] [PubMed] [Google Scholar]
- 51.Avitzur Y, Galindo-Mata E, Jones NL. Oral vaccination against Helicobacter pylori infection is not effective in mice with fas ligand deficiency. Digestive Diseases and Sciences. 2005;50(12):2300–2306. doi: 10.1007/s10620-005-3051-5. [DOI] [PubMed] [Google Scholar]
- 52.Langhans B, Schweitzer S, Braunschweiger I, Schulz M, Sauerbruch T, Spengler U. Cytotoxic capacity of hepatitis C virus (HCV)–specific lymphocytes after in vitro immunization with HCV-derived lipopeptides. Cytometry A. 2005;65(1):59–68. doi: 10.1002/cyto.a.20136. [DOI] [PubMed] [Google Scholar]
- 53.Shi G, Mao J, Yu G, Zhang J, Wu J. Tumor vaccine based on cell surface expression of DcR3/TR6. Journal of Immunology. 2005;174(8):4727–4735. doi: 10.4049/jimmunol.174.8.4727. [DOI] [PubMed] [Google Scholar]
- 54.Dinarello CA. Interleukin-18. Methods. 1999;19(1):121–132. doi: 10.1006/meth.1999.0837. [DOI] [PubMed] [Google Scholar]
- 55.Tarcea VG, Weymouth T, Ade A. Michigan molecular interactions r2: from interacting proteins to pathways. Nucleic Acids Research. 2009;37, database issue:D642–D646. doi: 10.1093/nar/gkn722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Park H-S, Huh S-H, Kim M-S, Lee SH, Choi E-J. Nitric oxide negatively regulates c-Jun N-terminal kinase/stress-activated protein kinase by means of S-nitrosylation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(26):14382–14387. doi: 10.1073/pnas.97.26.14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pearl-Yafe M, Halperin D, Scheuerman O, Fabian I. The p38 pathway partially mediates caspase-3 activation induced by reactive oxygen species in Fanconi anemia C cells. Biochemical Pharmacology. 2004;67(3):539–546. doi: 10.1016/j.bcp.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 58.Lu B, Yu H, Chow C-W, et al. GADD45γ mediates the activation of the p38 and JNK MAP kinase pathways and cytokine production in effector TH1 cells. Immunity. 2001;14(5):583–590. doi: 10.1016/s1074-7613(01)00141-8. [DOI] [PubMed] [Google Scholar]
- 59.Hayashi K, Ishizuka S, Yokoyama C, Hatae T. Attenuation of interferon-γ mRNA expression in activated Jurkat T cells by exogenous zinc via down-regulation of the calcium-independent PKC-AP-1 signaling pathway. Life Sciences. 2008;83(1-2):6–11. doi: 10.1016/j.lfs.2008.04.022. [DOI] [PubMed] [Google Scholar]
- 60.Xue L, Firestone GL, Bjeldanes LF. DIM stimulates IFNγ gene expression in human breast cancer cells via the specific activation of JNK and p38 pathways. Oncogene. 2005;24(14):2343–2353. doi: 10.1038/sj.onc.1208434. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. The graph of the generic IFNG network extracted from the literature. The network consists of 1060 nodes (genes) and 26,313 edges (interactions). The purple nodes are the genes that are central in both the generic and the IFNG-vaccine networks. The green nodes are the genes that are central in only the generic IFNG network and the red nodes are the genes that are central in only the IFNG-vaccine network. The rest of the nodes are shown in yellow.
Supplementary File 1. The rankings of all the genes in the generic IFNG network by degree, eigenvector, betweenness, and closeness centrality metrics.
Supplementary File 2. The rankings of all the genes in the IFNG-vaccine network by degree, eigenvector, betweenness, and closeness centrality metrics.