

Abstract
We investigated the genetic basis for a global and uniform decrease in mitochondrial translation in fibroblasts from patients in two unrelated pedigrees who developed Leigh syndrome, optic atrophy, and ophthalmoplegia. Analysis of the assembly of the oxidative phosphorylation complexes showed severe decreases of complexes I, IV, and V and a smaller decrease in complex III. The steady-state levels of mitochondrial mRNAs, tRNAs, and rRNAs were not reduced, nor were those of the mitochondrial translation elongation factors or the protein components of the mitochondrial ribosome. Using homozygosity mapping, we identified a 1 bp deletion in C12orf65 in one patient, and DNA sequence analysis showed a different 1 bp deletion in the second patient. Both mutations predict the same premature stop codon. C12orf65 belongs to a family of four mitochondrial class I peptide release factors, which also includes mtRF1a, mtRF1, and Ict1, all characterized by the presence of a GGQ motif at the active site. However, C12orf65 does not exhibit peptidyl-tRNA hydrolase activity in an in vitro assay with bacterial ribosomes. We suggest that it might play a role in recycling abortive peptidyl-tRNA species, released from the ribosome during the elongation phase of translation.
Main Text
Combined deficiencies in the enzyme complexes of the oxidative phosphorylation (OXPHOS) system are a common cause of mitochondrial disease. They are associated with a wide variety of distinct clinical phenotypes and can be caused by mutations in either the mitochondrial (mtDNA) or nuclear genome. Mutations in mitochondrial tRNAs and rRNAs, as well as large-scale mtDNA deletions, have been known for over 20 years,1 and several gene defects, many of which are implicated in mitochondrial translation, have been identified in patients with autosomal recessive combined OXPHOS deficiencies (reviewed in 2).
The synthesis of the 13 mtDNA-encoded OXPHOS subunits is carried out on dedicated mitochondrial ribosomes with translation machinery that resembles that of prokaryotes; however, the regulation of this system is poorly understood. Although the basic components required for initiation, elongation, and termination of mitochondrial translation have been identified and cloned,3 much of our knowledge has been inferred from prokaryotic systems, and it is clear that many essential components of mammalian mitochondrial translation remain to be discovered.4 The precise roles of all the proteins involved in translation termination are particularly unclear.5
During the termination of protein synthesis, translation termination (release) factors catalyze the release of the nascent polypeptide chain when a stop codon on the translated mRNA appears in the A site of the ribosome. In prokaryotes, four proteins are involved in translation termination: two class I release factors, RF1 and RF2, which recognize the three stop codons present in prokaryotes (UAA, UAG, and UGA) and facilitate hydrolysis of the ester bond between the tRNA and nascent polypeptide in the peptidyl transferase center of the ribosome; RF3, a class II release factor with GTPase activity that stimulates the removal of RF1 and RF2 from the ribosome after the release of the polypeptide chain; and RRF, ribosomal recycling factor, which is essential for the release of ribosomes from mRNA (reviewed in 6).
The mitochondrial translation machinery utilizes a modified set of codons in which the conventional stop codon UGA has been transformed to code for tryptophan. In vertebrate mitochondria, only two stop codons (UAG and UAA) are used for termination, and these are recognized by a single class I release factor mtRF1a/mtRF1L, as demonstrated by in vitro translation termination assays that use E. coli 70S ribosomes and by in vivo restoration of respiratory deficiency resulting from deletion of the endogenous mitochondrial RF in yeast.7–9 Two ribosome recycling factors, mtRRF10,11 and mtRRF2 (EFG2), have also been described,12 but no class II release factor has been identified in mitochondria.
All known class I release factors have a GGQ motif at the active site of the protein that catalyzes the hydrolysis of the ester bond, but there are no clear sequence similarities at the stop codon recognition site. Based on this sequence homology, three additional putative mitochondrial class I release family members containing the GGQ motif can be identified: mtRF1, Ict1, and C12orf65.5 Ict1 has recently been shown to be an integral component of the large mitochondrial ribosomal subunit (mt-LSU) and to possess ribosome-dependent but codon-independent peptidyl-tRNA hydrolase activity.5 Human Ict1 is 29% identical to the bacterial protein YaeJ, which is thought to mediate stop codon independent polypeptide release activity from stalled 50S ribosomes, in which the unfinished protein remains threaded through the exit tunnel, suggesting that Ict1 might have a similar function.5,13 The precise functions of mtRF1 and C12orf65 remain unknown.
Here we have investigated patients in two unrelated pedigrees with combined OXPHOS deficiencies associated with a decrease in the synthesis of all mitochondrially encoded polypeptides. Informed consent was obtained and research studies were approved by the Helsinki Committee of the Radboud University Nijmegen Medical Centre and the Montreal Neurological Institute Institutional Review Boards.
Patient 1, a girl, was the third child born to healthy first-cousin parents of Turkish descent. The two older siblings were healthy. Birth weight was 3300 g, birth length was 51 cm. Pregnancy, birth, and the first year of life were unremarkable. From age 1, the patient had failure to thrive with swallowing problems and regression of psychomotor development. At 18 months, psychomotor retardation, ataxia, and ptosis were found. An ophthalmological examination showed bilateral paresis of N. abducens. At age 3 years, nystagmus was found, and at age 5 years, severe optic atrophy and decreased vision were found. A cerebral MRI at age 2 years showed bilateral lesions in the thalami, brain stem, and medulla spinalis, and a diagnosis of Leigh syndrome (MIM #256000) was made. At 8 years, her height and weight were normal for her age. She was hypermobile with ataxia and used a wheelchair. She had severely reduced vision with optic atrophy and ophthalmoplegia. Plasma lactate was on one occasion elevated to 2.7 mmol/l. Urine organic acids, urine amino acids, and plasma amino acids were normal. The patient died at age 8.
Patient 2, a boy, was a second child of healthy unrelated Dutch parents. His older brother was also affected. Pregnancy and delivery were uneventful. His initial development was normal. At 15 months, he developed nystagmus. MRI of the brain was normal at age 2. From the age of 3–4 years, slow deterioration of motor and cognitive function occurred. At the age of 8 years, he became wheelchair dependent. Electromyogram at 8 years of age showed signs of an axonal neuropathy. At ages 14 and 17, he had episodes of paralytic ileus. At age 14, he developed a respiratory insufficiency and from then on remained dependent on ventilatory support. He also received a percutaneous gastrostomy tube. Neurological examination at the age of 20 years revealed optic atrophy with diminished vision. A divergent squint, irregular nystagmus, and incomplete external ophthalmoplegia with limited eye movements in all directions were present. There were signs of bulbar paresis with negative masseter reflex, facial diplegia, difficulty chewing and swallowing, and a mild dysarthria. Examination of the arms and legs revealed global muscle atrophy and hypotonia. The deep tendon reflexes were negative. There was a paresis of the proximal muscles and a paralysis of distal muscles, accompanied by distal decrease of sensory functions, consistent with a polyneuropathy.
An older brother of patient 2 presented with similar development. Pregnancy, delivery, and initial development were normal. Nystagmus was noted at birth. At age 3 years, a divergent squint arose. Ophthalmological examination revealed optic atrophy. MRI of the brain was normal at that time. From the age of 7 years, slow deterioration of vision and motor and cognitive function occurred. He also developed urinary stress incontinence. Neurological examination at the age of 21 years revealed optic atrophy with diminished vision, divergent squint, irregular nystagmus, incomplete external ophthalmoplegia with limited eye movements, and bulbar paresis. Examination of the arms and legs revealed atrophy and weakness of the distal muscles, as well as distal decrease in sensory function. There were no evident signs of a cerebellar ataxia. At the age of 22 years, he developed a respiratory tract infection, respiratory insufficiency, and paralytic ileus. MRI of the brain revealed extensive signal abnormalities in the brain stem, especially the midbrain and medulla, with restricted diffusion and enhancement after contrast. There were also signal abnormalities in the cerebellar white matter and thalamus. Magnetic resonance spectroscopy of the brain stem revealed highly elevated lactate. He died a few months later.
The disease progression in all of these patients is much slower than that seen in patients with classical Leigh syndrome, caused by, for instance, SURF1 (MIM 185620) mutations,14 which is usually fatal in the first few years of life, but it is reminiscent of patients with mutations in the recently described mitochondrial translation activator TACO1.4
Primary cultured human skin fibroblasts obtained from patient biopsies were immortalized with a retrovirus expressing the E7 gene of type 16 human papilloma virus and a retroviral vector expressing the protein component (htert) of human telomerase.15 Control fibroblast lines were obtained from the cell bank of the Montreal Children's Hospital, and at least two such lines were used in each experiment. Patient 1 was shown to have decreased cytochrome c oxidase (COX, complex IV) activity in fibroblasts (COX/citrate synthase 0.27 versus control mean 0.68, control range 0.4–1.1),14 and BN-PAGE analysis16,17 revealed severe assembly defects in complexes I, IV, and V, with a milder defect in the assembly of complex III (Figure 1A). Subcomplexes could not be detected by this analysis with antibodies directed against subunits of complex I or complex IV. Synthesis of the mtDNA-encoded polypeptides18 was also severely and uniformly affected (∼30% of control levels; Figure 1B). This pattern of mitochondrial translation deficiency is distinct from that found in other genetically defined patients with mitochondrial translation defects in whom specific decreases in the synthesis of individual polypeptides have been reported.19,20 The levels of all tested mitochondrial transcripts, rRNAs, and tRNAs were unchanged or were higher when compared to controls (Figure 2). Western blot analysis, carried out on fibroblasts solubilized in 1.5% lauryl maltoside with 10–20 μg of protein, showed normal levels of mitochondrial ribosomal subunits, as well as mitochondrial translation elongation factors (Figure 1C), suggesting that the translation defect was not due to an impaired assembly of the mitochondrial ribosome.
Figure 1.
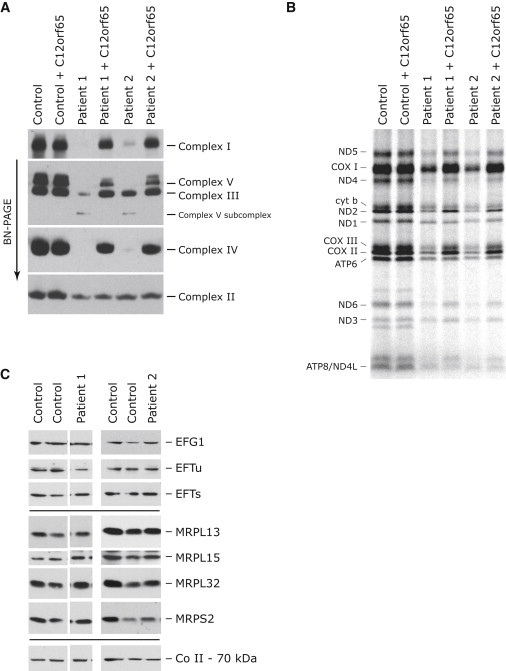
Characterization of the Molecular Defect in Patients with a Decrease in Mitochondrial Translation and Rescue of the Biochemical Phenotype by Expression of C12orf65
(A and B) Control and patient fibroblasts both alone and overexpressing C12orf65 were analyzed by BN-PAGE (A) and by pulse labeling of mitochondrial polypeptides (B). In (A), each of the five OXPHOS complexes (I–V) was visualized with a subunit-specific antibody. In (B), the seven subunits of complex I (ND), one subunit of complex III (cyt b), three subunits of complex IV (COX), and two subunits of complex V (ATP) are indicated at the left of the figure.
(C) Control and patient fibroblasts were analyzed by immunoblotting with antibodies against mitochondrial translation elongation factors (EFG1, EFTu, and EFTs) and mitochondrial ribosomal proteins (MRPL13, MRPL15, MRPL32 [a kind gift of T. Langer], and MRPS2 [a kind gift of L. Spremulli, UNC Chapel Hill]). The 70 kDa subunit of complex II was used as a loading control.
Figure 2.
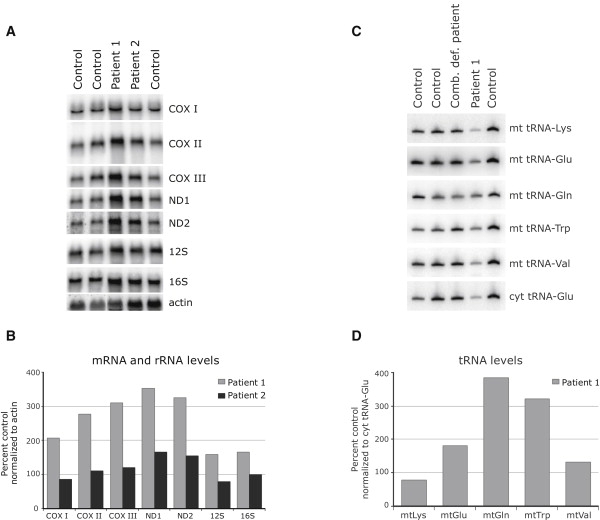
Normal Levels of Mitochondrial mRNAs, rRNAs, and tRNAs in Patient Fibroblasts
(A) RNA blot analysis was carried out with total RNA extracted from control and patient fibroblasts. Hybridization was performed with probes specific for the mitochondrial mRNAs encoding the three COX subunits, two of the complex I subunits (ND), 12S and 16S mitochondrial ribosomal RNA, and, as a loading control, a probe for beta-actin.
(B) Quantification of mitochondrial mRNA and rRNA levels in fibroblasts normalized to actin levels.
(C) Total RNA was extracted from control and patient cells, and 5 μg of RNA was run on a 10% polyacrylamide gel containing 7 M urea. After transfer to membrane, hybridization was performed with oligonucleotide probes complementary to the mitochondrial tRNAs for Lys, Glu, Gln, Trp, and Val, and, as a loading control, the cytosolic tRNA for Glu (cyt Glu), as indicated at the right of the figure.
(D) Quantification of mitochondrial tRNA levels in patient 1 fibroblasts, normalized to levels of cytosolic tRNA for Glu.
DNA extracted from peripheral blood was used for a genome-wide search for homozygosity with the Affymetrix GeneChip 50K Xba array. The SNP analysis showed a large number of homozygous regions, altogether comprising around 200 Mb and containing 1473 genes (see Table S1 available online). In order to prioritize candidate genes, we used the Maestro database to identify genes with a significant score (>4.0) for encoding a mitochondrial protein.21 The database search identified 69 genes with a score of over 4. Two of the genes, EFG1 (MIM 606639) and MRPS22 (MIM 605810), encode proteins that are part of the mitochondrial translation machinery, but no mutations were found by sequence analysis. Sequence analysis of cDNA and genomic DNA from C12orf65, a putative class I release factor, revealed a homozygous 1 bp deletion (248delT), which resulted in a premature stop codon (Figures 3A and 3B). Although we cannot rule out hemizygosity in this patient, both parents (and an older sister) were heterozygous carriers of the mutation, making it unlikely. An older brother was homozygous for the wild-type allele.
Figure 3.
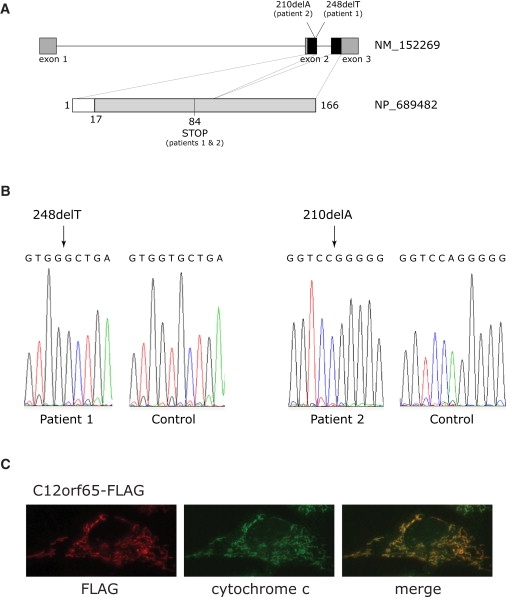
Mutational Analysis of C12orf65 and Mitochondrial Localization of the Protein
(A) Schematic diagram of the C12orf65 gene and protein. The position of the mutation in the patient DNA and the position of the resulting premature stop codon are indicated. Dark boxes in the C12orf65 gene denote the coding regions; gray boxes denote 5′UTRs and 3′UTRs. The white box in C12orf65 protein indicates the predicted mitochondrial leader sequence.
(B) Sequencing analysis of C12orf65 cDNA indicating the position of the homozygous 248delT and 210delA mutations in patient 1 and patient 2, respectively.
(C) Control fibroblasts expressing c12orf65-FLAG were grown on coverslips and incubated with antibodies against FLAG and against cytochrome c, as indicated, followed by incubation with secondary antibodies coupled to red and green fluorescent dyes. Merging the two images demonstrates the mitochondrial localization of C12orf65.
In order to confirm the pathogenicity of this mutation, we used a retroviral vector20 to express the wild-type cDNA for C12orf65 in fibroblasts from patient 1, from controls, and from a group of genetically uncharacterized combined OXPHOS deficiency patients. Retroviral vectors were created with the Gateway cloning system (Invitrogen) as previously described,20 and the cDNA from the C12orf65 gene was amplified by OneStep RT-PCR (QIAGEN) with specific primers. Retroviral constructs were used to transiently transfect a Phoenix packaging cell line via the HBS/Ca3(PO4)2 method (see Web Resources). Fibroblasts were infected 48 hr later by exposure to virus-containing medium in the presence of 4 μg/ml of polybrene.14
This approach enabled us to identify another, unrelated patient with a mutation in C12orf65. Patient 2, a Leigh-like patient, showed a virtually identical pattern of combined OXPHOS deficiency as in patient 1 (Figure 1; Figure 2; COX/citrate synthase activity of 40% of control). Expression of the wild-type C12orf65 cDNA rescued the mitochondrial protein synthesis defect in both patients (Figure 1B), which resulted in near control levels of all OXPHOS complexes in patient fibroblasts (Figure 1A). Overexpression of the mutated C12orf65, a protein with a stop codon at position 84 (Figure 3), which still contains the highly conserved class I release factor motif, did not rescue the defect in fibroblasts from patient 1; however, we do not know whether this protein is expressed, because antibodies against C12orf65 are not available. Sequence analysis of patient 2 cDNA revealed a homozygous 1 bp deletion at position 210 (210delA; Figure 3), which also resulted in a premature stop codon at position 84 in C12orf65 protein, as in patient 1. DNA from the parents of patient 2 was not available for study. The level of the C12orf65 mRNA was not significantly reduced on RT-PCR in either patient, suggesting that nonsense-mediated mRNA decay does not contribute to the loss of function of the C12orf65 gene product. Immunocytochemistry experiments with FLAG-tagged versions of C12orf65 protein revealed that it localizes exclusively to mitochondria (Figure 3C).
The molecular defects produced by knockdown of Ict1 with Stealth (Invitrogen) RNAis were very similar to those found in patients with C12orf65 mutation (Figures 4A–4C); however, expression of Ict1 from a retroviral vector failed to rescue the biochemical defect in our bank of patients with combined OXPHOS deficiencies. Strikingly, no defects in overall mitochondrial translation or the assembly and function of the OXPHOS complexes were observed after knockdown of mtRF1a/mtRF1L, the release factor that is thought to be sufficient to recognize both stop codons.8
Figure 4.
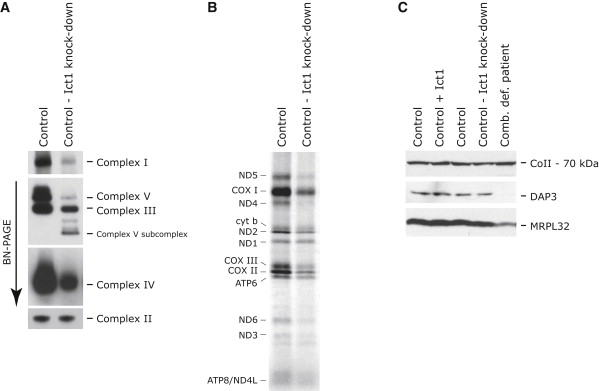
Knockdown of Ict1 in Control Fibroblasts Results in a Mitochondrial Translation Defect Similar to that in Patients with C12orf65 Mutations
(A and B) Control fibroblasts were transiently transfected with small interfering RNA constructs specific to Ict1 and analyzed by BN-PAGE (A) and [35S] Met/Cys-labeling (B) of mitochondrial polypeptides.
(C) Control fibroblasts that were untreated, overexpressing, or depleted for Ict1 were analyzed by immunoblotting for the presence of mitochondrial ribosomal proteins (DAP3 and MRPL32). A patient with a mitochondrial ribosome assembly defect (Comb. def. patient) was used as a positive control.
In order to investigate whether any of the members of the RF1 family were able to suppress the defect found in C12orf65 patients, we overexpressed the other three members (mtRF1, mtRF1a/mtRF1L, and Ict1) in control and patient fibroblasts. The overexpression of these factors in control cells did not have any effect on OXPHOS biogenesis (Figure 5A); however, overexpression of Ict1, but not mtRF1 or mtRF1a/mtRF1L, resulted in a partial (50%) increase in COX activity over that in patient fibroblasts (Figure 5B). Correspondingly, the assembly of COX was increased in patient fibroblasts overexpressing Ict1, as was the assembly of complexes I, V, and III (Figure 5A), suggesting that C12orf65 and Ict1 have similar and, to some extent, overlapping functions.
Figure 5.
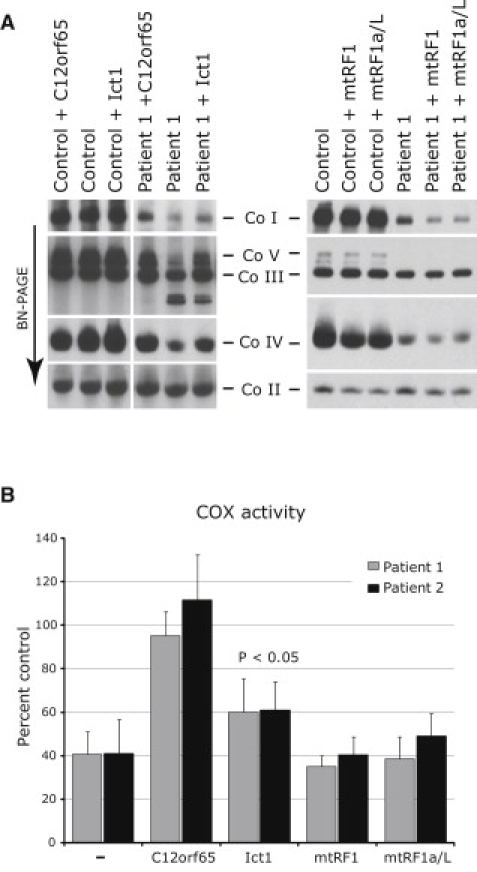
Overexpression of Ict1, but Not mtRF1 or mtRF1a/mtRF1L, Leads to a Partial Rescue of the OXPHOS Defect in C12orf65 Patients
(A) BN-PAGE analysis of untreated control and patient fibroblasts and of those overexpressing C12orf65, Ict1, mtRF1, or mtRF1a/mtRF1L (mtRF1a/L). Each of the five OXPHOS complexes (I–V) was visualized with a subunit-specific antibody.
(B) Quantification of COX activity (normalized to citrate synthase activity and expressed as a percentage of control) in fibroblasts from the patients alone and overexpressing C12orf65, Ict1, mtRF1, or mtRF1a/mtRF1L (3–9 replicates per sample).
To test whether C12orf65, like Ict1, has codon-independent peptidyl-tRNA hydrolase activity, we used a previously described assay5,8 with E. coli 70S ribosomes (Figure 6). GST-tagged proteins mtRF1a/mtRF1L and C12orf65 were prepared with E. coli strain Rosetta(DE3)pLysS (Novagen), purified and used in the in vitro translation termination assay. Although the positive control, mtRF1a, showed peptide release activity for the two termination codons (UAA and UAG), with C12orf65 no peptidyl-tRNA hydrolase activity could be detected in the presence, or in the absence, of any codon. We cannot rule out the possibility that C12orf65 does, in fact, have such an activity but that it requires 55S mitochondrial ribosomes or additional factors or that it is ribosome independent. MtRF1 similarly has no peptidyl-tRNA hydrolase activity when assayed on bacterial ribosomes.8
Figure 6.
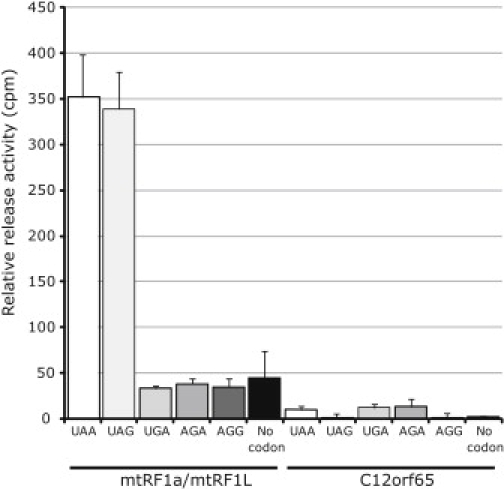
C12orf65 Does Not Have Detectable Peptidyl-tRNA Hydrolase Activity in an In Vitro Assay with Bacterial Ribosomes
E. coli ribosomes were programmed with f[3H]-Met tRNAMet at the P site and the indicated codons at the A site. Activity was measured as specific hydrolysis of f[3H]-Met from its cognate tRNAMet and expressed as cpm released. mtRF1a/mtRF1L, which recognizes the two mitochondrial termination codons UAA and UAG, was used as a positive control.
What could be the role of C12orf65? The three-dimensional (3D) structure of C12orf65, Ict1, mtRF1, and mtRF1a/mtRF1L was modeled on the crystal structure of the bacterial homolog RF26 with the I-TASSER server22–24 and was viewed with Swiss-MODEL by using the Swiss-Pdb Viewer.25
In silico 3D structure modeling of the four mitochondrial class I peptide release factor proteins predicts prokaryotic RF1 and RF2 as the best structure templates for the entire family, with mtRF1a/mtRF1L having the highest confidence score and Ict1 the lowest (Figure 7). Compared to RF2 from E. coli, C12orf 65 is missing domain 1, part of domain 2 (up to the beta3 sheet), and the last two helices of domain 4. Domain 1 interacts with the N-terminal domain of the L11 ribosomal subunit, which is thought to interact with translation factors.26 Immunoprecipitation studies suggest that C12orf65 is a soluble matrix protein that does not associate with the mitochondrial ribosome,5 so it could perhaps play a role in processing peptidyl-tRNAs that have been prematurely released during polypeptide elongation. Accumulation of such peptidyl-tRNAs would lead to depletion of aminoacylated tRNAs, resulting in diminished protein synthesis. In bacteria, recycling of these molecules is accomplished by peptidyl-tRNA hydrolase (Pth), an esterase that cleaves the ester bond between the C-terminal end of the peptide and the 2′- and 3′-hydroxyl of the ribose at the end of the tRNA.27 Two classes of Pth, which differ in substrate specificity, have also been described in eukaryotes: Pth1, an ortholog of the eubacterial enzymes, and Pth2, an ortholog of the archeal enzymes. In Saccharomyces cerevisiae, deletion of both of the Pth genes results in viable strains, but with a retarded growth on nonfermentable carbon sources, suggesting a role in mitochondrial translation.28 The human ortholog of Pth2, also called Bcl-2 inhibitor of transcription 1 (Bit1), is a mitochondrial protein with a bifunctional activity. Bit1 possesses peptidyl-tRNA hydrolase activity in vitro, and this has been presumed to be present in mitochondria; but when released into the cytoplasm, it promotes apoptosis.29 Although there are no apparent similarities of C12orf65 to the Pth domains in these proteins, this does not rule out the possibility that a class I release factor might be necessary to process some abortive translation intermediates.
Figure 7.
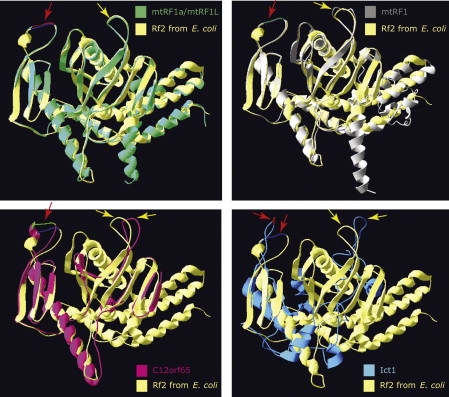
Molecular Modeling of Mitochondrial Class I Release Factor Proteins
The best fit for the predicted 3D structure of C12orf65, Ict1, mtRF1, or mtRF1a/mtRF1L is the crystal structure of translation release factor RF2 from E. coli (yellow). The red arrows indicate the conserved class I release factor motif GGQ in domain III. The yellow arrows indicate the domain II loop in which the tripeptide motif (SPF in RF2) involved in stop codon recognition is positioned.
Despite the clear structural and functional differences between C12orf65 and Ict1, overexpression of Ict1 is capable of partially suppressing the biochemical defect in C12orf65 patient cells. These data suggest that Ict1 and C12orf65 have partially overlapping functions. How might this occur? Previous experiments have shown that a large fraction of overexpressed Ict1 does not associate with the mt-LSU, but rather accumulates in the mitochondrial matrix.5 Although the peptidyl-tRNA hydrolase activity of Ict1 in the absence of E. coli ribosomes was shown to be negligible,5 it is possible that it maintains at least some activity in mammalian mitochondria when soluble and that this activity is enough to partially compensate for the loss of C12orf65. Clearly, much further work will be required to elucidate the specific function of C12orf65.
Acknowledgments
We thank the families for their participation. We thank Timothy Johns for help with cell culture and immunocytochemistry. This work was supported by grants from the Canadian Institutes of Health Research to E.A.S. and the Danish Council for Independent Research Medical Sciences to E.Ø. Z.M.C.L. would like to thank the Biotechnology and Biological Sciences Research Council (BB/F011520/1) for continuing support. E.A.S. is an International Scholar of the Howard Hughes Medical Institute.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/Omim/
Phoenix Retroviral Producer Line Protocol, http://www.stanford.edu/group/nolan/protocols/pro_helper_dep.html
GenBank Accession Numbers
The GenBank accession numbers are as follows: C12orf65, NM_152269; Ict1, NM_001545; mtRF1, NM_004294; mtRF1a/mtRF1L, NM_019041.
References
- 1.Taylor R.W., Turnbull D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Debray F.G., Lambert M., Mitchell G.A. Disorders of mitochondrial function. Curr. Opin. Pediatr. 2008;20:471–482. doi: 10.1097/MOP.0b013e328306ebb6. [DOI] [PubMed] [Google Scholar]
- 3.Spremulli L.L., Coursey A., Navratil T., Hunter S.E. Initiation and elongation factors in mammalian mitochondrial protein biosynthesis. Prog. Nucleic Acid Res. Mol. Biol. 2004;77:211–261. doi: 10.1016/S0079-6603(04)77006-3. [DOI] [PubMed] [Google Scholar]
- 4.Weraarpachai W., Antonicka H., Sasarman F., Seeger J., Schrank B., Kolesar J.E., Lochmüller H., Chevrette M., Kaufman B.A., Horvath R., Shoubridge E.A. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat. Genet. 2009;41:833–837. doi: 10.1038/ng.390. [DOI] [PubMed] [Google Scholar]
- 5.Richter R., Rorbach J., Pajak A., Smith P.M., Wessels H.J., Huynen M.A., Smeitink J.A., Lightowlers R.N., Chrzanowska-Lightowlers Z.M. A functional peptidyl-tRNA hydrolase, ICT1, has been recruited into the human mitochondrial ribosome. EMBO J. 2010;29:1116–1125. doi: 10.1038/emboj.2010.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petry S., Weixlbaumer A., Ramakrishnan V. The termination of translation. Curr. Opin. Struct. Biol. 2008;18:70–77. doi: 10.1016/j.sbi.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 7.Temperley R., Richter R., Dennerlein S., Lightowlers R.N., Chrzanowska-Lightowlers Z.M. Hungry codons promote frameshifting in human mitochondrial ribosomes. Science. 2010;327:301. doi: 10.1126/science.1180674. [DOI] [PubMed] [Google Scholar]
- 8.Soleimanpour-Lichaei H.R., Kühl I., Gaisne M., Passos J.F., Wydro M., Rorbach J., Temperley R., Bonnefoy N., Tate W., Lightowlers R., Chrzanowska-Lightowlers Z. mtRF1a is a human mitochondrial translation release factor decoding the major termination codons UAA and UAG. Mol. Cell. 2007;27:745–757. doi: 10.1016/j.molcel.2007.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nozaki Y., Matsunaga N., Ishizawa T., Ueda T., Takeuchi N. HMRF1L is a human mitochondrial translation release factor involved in the decoding of the termination codons UAA and UAG. Genes Cells. 2008;13:429–438. doi: 10.1111/j.1365-2443.2008.01181.x. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y., Spremulli L.L. Identification and cloning of human mitochondrial translational release factor 1 and the ribosome recycling factor. Biochim. Biophys. Acta. 1998;1443:245–250. doi: 10.1016/s0167-4781(98)00223-1. [DOI] [PubMed] [Google Scholar]
- 11.Rorbach J., Richter R., Wessels H.J., Wydro M., Pekalski M., Farhoud M., Kühl I., Gaisne M., Bonnefoy N., Smeitink J.A. The human mitochondrial ribosome recycling factor is essential for cell viability. Nucleic Acids Res. 2008;36:5787–5799. doi: 10.1093/nar/gkn576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsuboi M., Morita H., Nozaki Y., Akama K., Ueda T., Ito K., Nierhaus K.H., Takeuchi N. EF-G2mt is an exclusive recycling factor in mammalian mitochondrial protein synthesis. Mol. Cell. 2009;35:502–510. doi: 10.1016/j.molcel.2009.06.028. [DOI] [PubMed] [Google Scholar]
- 13.Jiang L., Schaffitzel C., Bingel-Erlenmeyer R., Ban N., Korber P., Koning R.I., de Geus D.C., Plaisier J.R., Abrahams J.P. Recycling of aborted ribosomal 50S subunit-nascent chain-tRNA complexes by the heat shock protein Hsp15. J. Mol. Biol. 2009;386:1357–1367. doi: 10.1016/j.jmb.2008.10.079. [DOI] [PubMed] [Google Scholar]
- 14.Zhu Z., Yao J., Johns T., Fu K., De Bie I., Macmillan C., Cuthbert A.P., Newbold R.F., Wang J., Chevrette M. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet. 1998;20:337–343. doi: 10.1038/3804. [DOI] [PubMed] [Google Scholar]
- 15.Lochmüller H., Johns T., Shoubridge E.A. Expression of the E6 and E7 genes of human papillomavirus (HPV16) extends the life span of human myoblasts. Exp. Cell Res. 1999;248:186–193. doi: 10.1006/excr.1999.4407. [DOI] [PubMed] [Google Scholar]
- 16.Schägger H., von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 1991;199:223–231. doi: 10.1016/0003-2697(91)90094-a. [DOI] [PubMed] [Google Scholar]
- 17.Klement P., Nijtmans L.G., Van den Bogert C., Houstěk J. Analysis of oxidative phosphorylation complexes in cultured human fibroblasts and amniocytes by blue-native-electrophoresis using mitoplasts isolated with the help of digitonin. Anal. Biochem. 1995;231:218–224. doi: 10.1006/abio.1995.1523. [DOI] [PubMed] [Google Scholar]
- 18.Leary S.C., Sasarman F., Nishimura T., Shoubridge E.A. Human SCO2 is required for the synthesis of CO II and as a thiol-disulphide oxidoreductase for SCO1. Hum. Mol. Genet. 2009;18:2230–2240. doi: 10.1093/hmg/ddp158. [DOI] [PubMed] [Google Scholar]
- 19.Coenen M.J., Antonicka H., Ugalde C., Sasarman F., Rossi R., Heister J.G., Newbold R.F., Trijbels F.J., van den Heuvel L.P., Shoubridge E.A., Smeitink J.A. Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N. Engl. J. Med. 2004;351:2080–2086. doi: 10.1056/NEJMoa041878. [DOI] [PubMed] [Google Scholar]
- 20.Antonicka H., Sasarman F., Kennaway N.G., Shoubridge E.A. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum. Mol. Genet. 2006;15:1835–1846. doi: 10.1093/hmg/ddl106. [DOI] [PubMed] [Google Scholar]
- 21.Calvo S., Jain M., Xie X., Sheth S.A., Chang B., Goldberger O.A., Spinazzola A., Zeviani M., Carr S.A., Mootha V.K. Systematic identification of human mitochondrial disease genes through integrative genomics. Nat. Genet. 2006;38:576–582. doi: 10.1038/ng1776. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 2007;69(Suppl 8):108–117. doi: 10.1002/prot.21702. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu S., Skolnick J., Zhang Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 2007;5:17. doi: 10.1186/1741-7007-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 26.Petry S., Brodersen D.E., Murphy F.V., 4th, Dunham C.M., Selmer M., Tarry M.J., Kelley A.C., Ramakrishnan V. Crystal structures of the ribosome in complex with release factors RF1 and RF2 bound to a cognate stop codon. Cell. 2005;123:1255–1266. doi: 10.1016/j.cell.2005.09.039. [DOI] [PubMed] [Google Scholar]
- 27.Das G., Varshney U. Peptidyl-tRNA hydrolase and its critical role in protein biosynthesis. Microbiology. 2006;152:2191–2195. doi: 10.1099/mic.0.29024-0. [DOI] [PubMed] [Google Scholar]
- 28.Rosas-Sandoval G., Ambrogelly A., Rinehart J., Wei D., Cruz-Vera L.R., Graham D.E., Stetter K.O., Guarneros G., Söll D. Orthologs of a novel archaeal and of the bacterial peptidyl-tRNA hydrolase are nonessential in yeast. Proc. Natl. Acad. Sci. USA. 2002;99:16707–16712. doi: 10.1073/pnas.222659199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Pereda J.M., Waas W.F., Jan Y., Ruoslahti E., Schimmel P., Pascual J. Crystal structure of a human peptidyl-tRNA hydrolase reveals a new fold and suggests basis for a bifunctional activity. J. Biol. Chem. 2004;279:8111–8115. doi: 10.1074/jbc.M311449200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.