

Abstract
Mesomelia-synostoses syndrome (MSS) or mesomelic dysplasia with acral synostoses Verloes-David-Pfeiffer type is a rare autosomal-dominant disorder characterized by mesomelic limb shortening, acral synostoses, and multiple congenital malformations. So far, five patients in four unrelated families have been reported worldwide with MMS. By using whole-genome oligonucleotide array CGH, we have identified an interstitial deletion at 8q13 in all patients. The deletions vary from 582 Kb to 738 Kb in size, but invariably encompass only two genes: SULF1, encoding the heparan sulfate 6-O-endosulfatase 1, and SLCO5A1, encoding the solute carrier organic anion transporter family member 5A1. SULF1 acts as a regulator of numerous growth factors in skeletal embryonic development whereas the function of SLCO5A1 is yet unknown. Breakpoint sequence analyses performed in two families showed nonrecurrent deletions. Real-time quantitative RT-PCR analysis showed the highest levels of SULF1 transcripts in human osteoblasts and cartilage whereas SLCO5A1 was highly expressed in human fetal and adult brain and heart. Our results strongly suggest that haploinsufficiency of SULF1 contributes to this mesomelic chondrodysplasia, highlighting the critical role of endosulfatase in human skeletal development. Codeletion of SULF1 and SLCO5A1—which does not result from a low-copy repeats (LCRs)-mediated recombination event in at least two families—was found in all patients, so we suggest that haploinsufficiency of SULF1 combined with haploinsufficiency of SLCO5A1 (or the altered expression of a neighboring gene through position effect) could be necessary in the pathogenesis of MSS.
Main Text
Mesomelia-synostoses syndrome (MSS [MIM 600383])—or mesomelic dysplasia with acral synostoses Verloes-David-Pfeiffer type1—is a rare clinical entity mainly characterized by mesomelic limb shortening and acral synostoses, originally delineated by Verloes and David (1995)2 and independently by Pfeiffer et al. (1995).3 MSS is inherited as an autosomal-dominant trait.2,4 MSS stands apart from all other mesomelic dysplasias because of the coexistence with synostoses between metacarpal/metatarsal bones and carpal/tarsal bones and specific craniofacial dysmorphism (Figure 1). Five patients with MSS have been reported so far, three of whom present with extraskeletal anomalies including renal malformations and/or congenital heart defects.3,5–7 A report of long-term follow-up of MSS patients has illustrated the progressive course of deformation within the mesomelic limb anomalies.4 The main clinical and radiological features of the patients are listed in Table 1.
Figure 1.
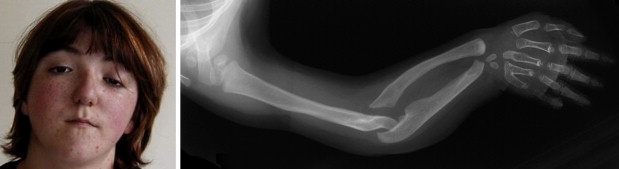
Clinical and Radiological Features of Patient II-2 in Family 1
Specific facial dysmorphy with downslanting palpebral fissures, ptosis, beaked nose, and small mouth (photograph at 15 years of age reproduced with patient's permission). Radiograph at 18 months of age of right upper limb showing severe bowing and shortening of the forearm, metacarpal, carpometacarpal fusions, and brachymetacarpy of the 3rd to 5th ray.
Table 1.
Clinical and Radiological Features in Patients with Mesomelia-Synostosis Syndrome
| Patient | F.1 I-1 | F.1 II-2 | F.2 II-3 | F.3 II-1 | F.4 II-1 |
|---|---|---|---|---|---|
| Polyhydramnios | NA | - | + | - | + |
| Craniofacial | |||||
| Palpebral fissures downslanted | + | + | + | + | + |
| Eyelid ptosis | + | + | + | + | + |
| Telecanthus | + | + | ± | + | + |
| Hypoplasia soft palate; absent uvula | + | + | + | + | + |
| Mild micrognathia | + | + | + | + | + |
| Short sublingual frenula; excess frenula | + | + | NA | NA | NA |
| Cognitive and Sensory Functions | |||||
| Developmental delay | - | - | - | - | - |
| Speech with nasal quality | + | + | + | + | + |
| Myopia | + | + | - | - | - |
| Hearing deficit | + | - | - | - | - |
| General Physical Features | |||||
| Short stature | + | + | + | + | + |
| Complex congenital heart defect | - | - | + | - | + |
| Umbilicus; short cord; excess skin; hernia | + | + | - | NA | + |
| Congenital hydronephrosis | NA | - | + | + | + |
| Limited motion in joints | + | + | + | + | + |
| Mesomelic bowing and shortening in both upper and lower limbs | + | + | + | + | + |
| Complex brachydactyly (postaxial) | + | + | + | + | + |
| Ulnar deviation of hands | + | + | + | + | + |
| Narrow, short feet | + | + | + | + | + |
| Brachydactylic toes (fibular side) | + | + | + | + | + |
| Dysfunctional ankle joints | + | + | + | + | + |
| Radiological Features | |||||
| Proximal carpometacarpal fusion | + | + | + | + | + |
| Shortened phalanges | + | + | + | + | + |
| Mild bowing of distal part of femora | + | + | + | + | - |
| Medial bowing tibial diaphyses | + | + | + | + | + |
| Shortened metatarsals | + | + | + | + | - |
| Tarsometatarsal synostoses | + | + | + | + | + |
Notation is as follows: +, present; -, absent; F., family; NA, not available.
For the last few years, array-based comparative genomic hybridization (array CGH) has led to the identification of several genes involved in monogenic disorders, such as CHD7 in CHARGE syndrome (MIM #214800),8 TCF4 in Pitt-Hopkins syndrome (MIM #610954),9 and MEF2C in mental retardation (MIM ∗600662).10 Here, we report the identification by array CGH of a 8q13 microdeletion in each one of the five patients with MSS in the four unrelated families previously reported (Figure 2).2,3,5–7
Figure 2.
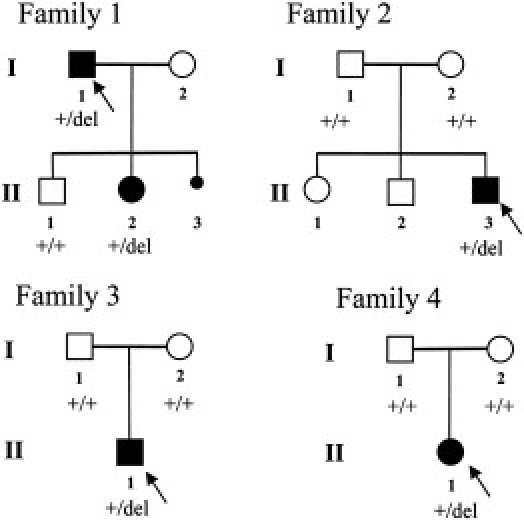
Pedigrees of the Families with Mesomelia-Synostoses Syndrome
The families 1, 2, 3, and 4 were reported by Verloes and David (1995),2 Leroy et al. (2001),5 Pfeiffer et al. (1995),3 and Day-Salvatore and McLean (1998),7 respectively. The arrows indicate the probands. +/del indicate the presence of heterozygous 8q13 deletion. +/+ indicate the presence of two copies of the 8q13 region. NA, DNA sample not available.
Informed consent for cytogenetic and molecular analyses was obtained from each family member, and research was approved by the local ethics committee of the University Hospital Center of Nantes. Karyotypes, performed via standard methods on metaphase spreads of peripheral blood from each individual, were normal. Molecular karyotyping in all patients and the available clinically normal parents was performed with Agilent Human Genome CGH 400K and 44K oligonucleotide arrays (Agilent, Santa Clara, CA) (Table S1 available online), respectively. We have identified a submicroscopic 8q13 microdeletion in all five patients. In each family, the deletion encompasses only two genes: SULF1, encoding the heparan sulfate 6-O-endosulfatase 1 (MIM ∗610012; GenBank accession number NM_015170), and SLCO5A1, encoding the solute carrier organic anion transporter family member 5A1 (GenBank accession number NM_030958). No other genomic imbalance was detected. Deletion size was different in each of the four unrelated families, ranging from 582 Kb to 738 Kb (Figure 3 and Table S2). In family 1,2 the proband (I-1) and his affected daughter (II-2) carried the same deletion at 8q13. Fluorescence in situ hybridization (FISH) performed on patient II-2 and his affected father (I-1) with two probes (the RP11-676B14 BAC clone located in the 8q13 deleted region and the RP4-585L5 BAC clone as the control probe in the subtelomeric 8p region) has confirmed the presence of the deletion in both patients (Figure 4). Normal results were obtained in the healthy brother (family 1, individual II-1). In families 2,5 3,3 and 4,7 CGH on 44K oligonucleotide arrays showed normal results for all phenotypically normal parents, demonstrating that the deletions arose de novo in these three families. Real-time quantitative PCR (qPCR) with primers (Table S3) designed inside intron 1 and exon 6 of SULF1 and introns 2 and 5 of SLCO5A1 were used for independent confirmation of the array CGH results as described with minor modifications.11 Presence of a deletion in all patients and of two normal alleles at 8q13 in all phenotypically normal parents and the normal brother in family 1 were confirmed by qPCR (Figure S1).
Figure 3.
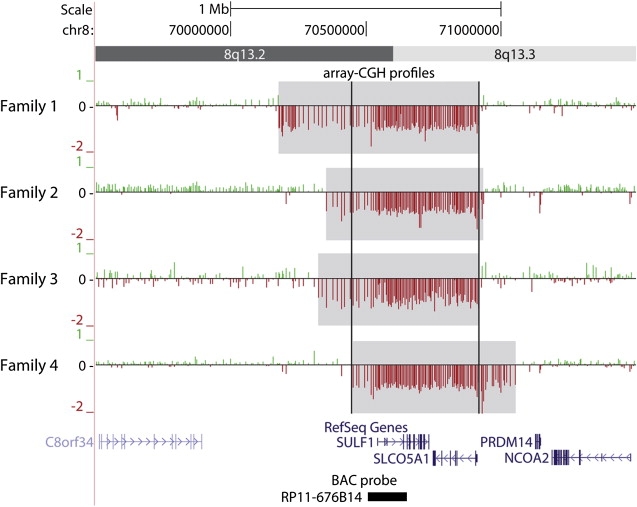
Array-CGH Profiles at 8q13
The 400K oligonucleotide arrays performed on patients from families 1, 2, 3, and 4 were analyzed with the Agilent scanner and the Feature Extraction software (v. 9.1.3). For each patient, positive and negative log2 ratio values are depicted by green and red vertical bars, respectively. Deleted segments are highlighted in gray whereas black vertical bars delimit the minimal deleted interval. RefSeq genes (including SULF1 and SLCO5A1) are indicated in blue. The black box at the bottom indicates the BAC probe RP11-676B14, which was used by FISH validation. Note that five noncoding RNA pseudogenes are predicted or present in the deleted regions.
Figure 4.
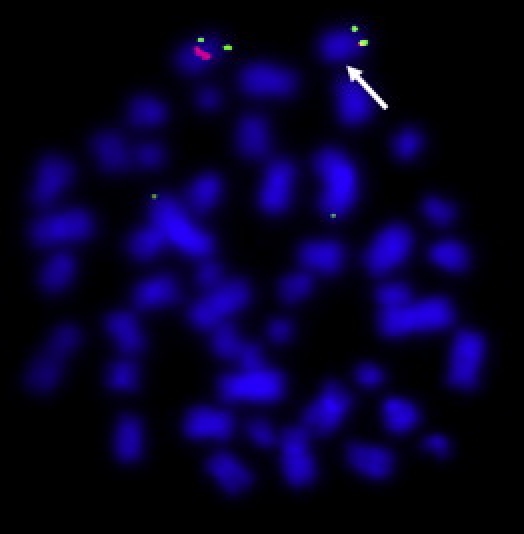
FISH Analysis
Dual-color FISH analysis was performed on metaphases of patient II-1 of family 1 via the RP11-676B14 BAC probe in the 8q13 deletion and a subtelomeric 8p probe (RP4-585L5) used as a control (labeled with SpectrumOrange and SpectrumGreen, respectively). The arrow indicates the deletion.
Genomic disorders are caused by recurrent chromosomal rearrangements involving most frequently nonallelic homologous recombination (NAHR) between highly similar duplicated sequences.12 In contrast, several mechanisms, including nonhomologous end-joining (NHEJ)13 or fork stalling and template switching (FoSTeS),14 can be responsible for nonrecurrent rearrangements without the need for a homologous template or only requiring microhomology. By long-range PCR (TaKaRa LA Taq, TAKARA) and direct sequencing (primer sequences are available upon request), we characterized the breakpoints at a molecular level in families 1 and 4 (Figure S2). Breakpoint sequence analyses confirmed that the deletions were nonrecurrent in these two families. The deletion in family 4 may have been mediated by NHEJ whereas a more complex rearrangement was observed in family 1. The variable size of the deletions and the absence of segmental duplications at the breakpoint junctions indicate that MMS is not a genomic disorder associated with deletions recurring from NAHR in at least two families. To our knowledge, this microdeletion has never been associated with any other disease. None of the deletions identified here have been reported in the Database of Genomic Variants nor in our cohort of patients with mental retardation analyzed by array CGH (n = 850; unpublished data). These observations demonstrate that the 8q13 deletions are responsible for MSS.
SULF1 contains 23 exons and shares about 64% homology with its ortholog SULF2 and 93% identity with its murine paralog Sulf1. Heparan sulfate 6-O-endosulfatases, such as SULF1, modulate the function of heparan sulfate by altering binding sites for signaling molecules. They regulate the activity of growth factors such as bone morphogenetic proteins (BMPs),15 fibroblast growth factors (FGFs),16 and Wnt ligands17 and thus control important developmental processes such as cell growth and differentiation. Sulf1 is expressed in most adult mouse tissues, highest levels being observed in bone, testis, stomach, skeletal muscle, lung, and kidney.18 During mouse embryonic development, Sulf1 is widely expressed in the forelimb, arm girdle, the condensing mesenchyme in the distal limb buds, the cartilaginous parts in digits, the brain, somites, clefts of the branchial arches, eyes, palate, tongue, and nasal pits.18,19 The expression pattern of Sulf1 during embryonic development overlaps the dysostoses and malformations observed in MSS.
The role of Sulf1 and Sulf2 during embryonic development has been studied in two sulfatase-deficient mouse models. Surprisingly, targeted disruption of Sulf1 or Sulf2 leading to a profound loss of expression of the Sulf1 or Sulf2 proteins exhibited only subtle skeletal anomalies, such as mild forms of dorsally split vertebrae in Sulf1 mutants.19 In contrast, mice deficient in both Sulf1 and Sulf2 exhibited increased perinatal lethality and developmental defects, including skeletal and renal abnormalities. Holst et al. (2007)18 showed that double mutant Sulf1 and Sulf2 embryos exhibited altered cartilage zonation in the ulna and radius and a decrease in bone volume and length in the axial and appendicular skeleton. In a second sulfatase-deficient mouse model, Ratzka et al. (2008)19 showed that double mutants for Sulf1 and Sulf2 exhibited skeletal anomalies such as reduced bone length, premature vertebrae ossification, and fusion of sternebrae and tail vertebrae. In the mouse, loss of increasing numbers of sulfatase alleles enhances severity and penetrance of the skeletal alterations and leads to malformations at additional sites. Overall, these murine phenotypes strongly suggest that SULF1 haploinsufficiency plays a key role in the skeletal abnormalities in patients with MSS. However, compositional analysis by RPIP-HPLC of heparan sulfate20 recovered from the urine of patient II-2 of family 1 revealed no abnormalities in heparan sulfate structure (Figure S3).
The role of SULF1 in human carcinogenesis is under active investigation. SULF1 inhibits the coreceptor function of heparan sulfate proteoglycans (HSPGs) in multiple receptor tyrosine kinase signaling pathways. SULF1 also promotes drug-induced apoptosis of cancer cells in vitro and inhibits tumorigenesis and angiogenesis in vivo.21 None of the patients with the SULF1 deletions presented with any type of tumor.
We have identified nonrecurrent deletions in all analyzed patients, invariably including both SULF1 and SLCO5A1. For most monogenic diseases resulting from haploinsufficiency, point mutations are identified in the majority of patients whereas genomic deletions are only rarely detected. Because nonrecurrent deletions have been found in the four known families and do not result from NAHR in at least two families, the hypothesis that the deletions were observed by chance is unlikely.
An alternative hypothesis is that haploinsufficency of SULF1 is necessary but not sufficient: expression of MSS phenotype would require an additional effect, either through the deletion of SLCO5A1 or through an alteration of the expression of neighboring gene(s) by position effect. In such case, (1) SULF1 haploinsufficiency might cause the skeletal phenotype while additional gene(s) would explain the other abnormalities such as the cardiac or renal defects or (2) the entire phenotype might be due to epistasis between SULF1 and SLCO5A1 or neighboring gene(s), as suggested by the mild phenotype observed in Sulf1 mutant mice.18,19
SLCO5A1 encodes a protein with 12 transmembrane domains. It is a member of the solute carrier organic anion transporter superfamily. Both expression pattern and function of SLCO5A1 are still unknown. Among previously reported copy number variants (CNVs) at 8q13, only one partial deletion of SLCO5A1 has been identified in a single normal individual.22 This may suggest that the deletion of this gene alone is unlikely to be responsible for MSS.
We have performed expression analyses for SULF1 and SLCO5A1 with total RNA extracted from different human tissues. Both adult and fetal RNA were obtained from brain, heart, kidney, and liver tissues (Clontech). Primary human osteoblasts were isolated from bone explants23 and human osteoclasts were differentiated from purified CD14+ cells.24 Healthy cartilage was harvested from patients referred to the Department of Orthopaedic Surgery during operations for intramedullary nail removal. Total RNA was extracted according to the previously described procedure.25 Real-time quantitative reverse transcription PCR (qRT-PCR) was performed via the ΔΔCt method11 and two housekeeping genes (Table S4). SULF1 was highly expressed in adult osteoblasts and cartilage and moderately so in fetal kidney and brain (Figure 5A). SLCO5A1 was highly expressed in fetal and adult brain and heart (Figure 5B).
Figure 5.
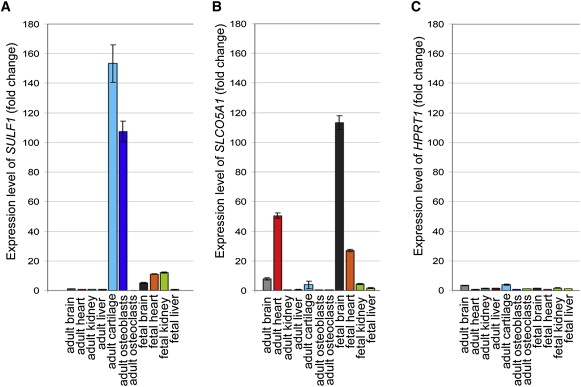
Expression Patterns of SULF1 and SLCO5A1 in a Panel of Human Tissues
cDNA were obtained via the MMLV reverse transcriptase (Invitrogen) with random primers from 1 μg of human total RNA of five adult and four fetal tissues and two cultured cells (osteoblasts and osteoclasts). Real-time PCR was performed in triplicate with Takara SYBR premix on Light Cycler 480 (Roche diagnostics). The ΔΔCt method11 was used to assess expression level of two target genes—SULF1 (A) and SLCO5A1 (B)—relative to the expression level of the GAPDH housekeeping gene. For a given target gene, the ΔCt of each tissue was compared to the median of the ΔCt of the 11 tissues analyzed. As a control, we assessed the HPRT1 housekeeping gene expression in the different tissues with the same method and showed minor differences between HPRT1 and GAPDH genes (C).
In conclusion, we have shown that MSS is caused by a 8q13 microdeletion, enclosing only the genes SULF1 and SLCO5A1. Deletion of SULF1 is the more likely culprit of at least the skeletal abnormalities in MSS. Because it was combined with SLCO5A1 deletion in all pedigrees, we hypothesize that MSS could either result from the combined haploinsufficiency of both genes or be the result of SULF1 haploinsufficiency associated with altered expression of a neighboring gene through position effect. Our observation may point to a possible role of SULF1 and/or SLCO5A1 as the cause of one or more other rare mesomelic dysplasias. The Kantaputra mesomelic dysplasia (MIM 156232) is one example of these mesomelic dysplasias with unknown molecular basis. We have directly sequenced the coding regions and surrounding exon/intron boundaries in SULF1 and SLCO5A1 (Table S5) in two patients with Kantaputra type mesomelic dysplasia26,27 and studied the genomic region by 44K oligonucleotide array. We did not identify any deleterious point mutations in these two genes nor any deletion and/or duplication at 8q13 or elsewhere in the genome. More patients with this or other less-well-delineated mesomelic dysplasias need to be investigated in order to further address the specificity of the correlation between the deletion 8q13 and MSS.
Acknowledgments
We are grateful to the patients and their families who participated in this study; to Rémi Houlgatte and Catherine Chevalier from Biogenouest de Nantes, France; to Inger Eriksson, Annaig Briand, Aude Derevier, and Hadja Eldjouzi for technical assistance; and to Martine Berreur for providing us RNA extracted from the osteoblasts, osteoclasts, and cartilage.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ArrayExpress, http://www.ebi.ac.uk/microarray-as/ae/
BACPAC Resources Center, http://bacpac.chori.org/home.htm
Database of Genomic Variants, http://projects.tcag.ca/variation/?source=hg18
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
UCSC Genome Browser, http://genome.ucsc.edu/
Accession Numbers
The ArrayExpress accession number for the genomic microarray data reported in this paper is E-MEXP-2710.
References
- 1.Superti-Furga A., Unger S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am. J. Med. Genet. 2007;143A:1–18. doi: 10.1002/ajmg.a.31483. [DOI] [PubMed] [Google Scholar]
- 2.Verloes A., David A. Dominant mesomelic shortness of stature with acral synostoses, umbilical anomalies, and soft palate agenesis. Am. J. Med. Genet. 1995;55:205–212. doi: 10.1002/ajmg.1320550211. [DOI] [PubMed] [Google Scholar]
- 3.Pfeiffer R.A., Hirschfelder H., Rott H.D. Specific acromesomelia with facial and renal anomalies: A new syndrome. Clin. Dysmorphol. 1995;4:38–43. [PubMed] [Google Scholar]
- 4.Isidor B., Hamel A., Plasschaert F., Claus L., Mercier J.M., Mortier G.R., Leroy J.G., Verloes A., David A. Mesomelic dysplasia with acral synostoses Verloes-David-Pfeiffer type: Follow-up study documents progressive clinical course. Am. J. Med. Genet. A. 2009;149A:2220–2225. doi: 10.1002/ajmg.a.32926. [DOI] [PubMed] [Google Scholar]
- 5.Leroy, J.G., Claus, L., and Mortier, G.R. (2001). Dysmorphic facies, heart defect, hydronephrosis, bilateral club hand, and club foot due to acromesomelic dysostosis: Second edition of Verloes-David syndrome? Abstracts from the 2000 DW Smith Workshop on malformations and morphogenesis, Proc. Greenwood Genetic Center 20, 149–150.
- 6.Leroy J.G., Claus L., Lee B., Mortier G.R. Mesomelic dysplasia with specific autopodal synostoses: A third observation and further delineation of the multiple congenital anomaly syndrome. Pediatr. Pathol. Mol. Med. 2003;22:23–35. doi: 10.1080/pdp.22.1.23.35. [DOI] [PubMed] [Google Scholar]
- 7.Day-Salvatore D., McLean D. Blepharophimosis, hypoplastic radius, hypoplastic left heart, telecanthus, hydronephrosis, fused metacarpals, and “prehensile” halluces: A new syndrome? Am. J. Med. Genet. 1998;80:309–313. [PubMed] [Google Scholar]
- 8.Vissers L.E., van Ravenswaaij C.M., Admiraal R., Hurst J.A., de Vries B.B., Janssen I.M., van der Vliet W.A., Huys E.H., de Jong P.J., Hamel B.C. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 9.Zweier C., Peippo M.M., Hoyer J., Sousa S., Bottani A., Clayton-Smith J., Reardon W., Saraiva J., Cabral A., Gohring I. Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome) Am. J. Hum. Genet. 2007;80:994–1001. doi: 10.1086/515583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le Meur N., Holder-Espinasse M., Jaillard S., Goldenberg A., Joriot S., Amati-Bonneau P., Guichet A., Barth M., Charollais A., Journel H. MEF2C haploinsufficiency caused either by microdeletion of the 5q14.3 region or mutation is responsible for severe mental retardation with stereotypic movements, epilepsy and/or cerebral malformations. J. Med. Genet. 2010;47:22–29. doi: 10.1136/jmg.2009.069732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 12.Stankiewicz P., Lupski J.R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 13.Lieber M.R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- 14.Lee J.A., Carvalho C.M., Lupski J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 15.Viviano B.L., Paine-Saunders S., Gasiunas N., Gallagher J., Saunders S. Domain-specific modification of heparan sulfate by Qsulf1 modulates the binding of the bone morphogenetic protein antagonist Noggin. J. Biol. Chem. 2004;279:5604–5611. doi: 10.1074/jbc.M310691200. [DOI] [PubMed] [Google Scholar]
- 16.Wang S., Ai X., Freeman S.D., Pownall M.E., Lu Q., Kessler D.S., Emerson C.P., Jr. QSulf1, a heparan sulfate 6-O-endosulfatase, inhibits fibroblast growth factor signaling in mesoderm induction and angiogenesis. Proc. Natl. Acad. Sci. USA. 2004;101:4833–4838. doi: 10.1073/pnas.0401028101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ai X., Do A.T., Lozynska O., Kusche-Gullberg M., Lindahl U., Emerson C.P., Jr. QSulf1 remodels the 6-O sulfation states of cell surface heparan sulfate proteoglycans to promote Wnt signaling. J. Cell Biol. 2003;162:341–351. doi: 10.1083/jcb.200212083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holst C.R., Bou-Reslan H., Gore B.B., Wong K., Grant D., Chalasani S., Carano R.A., Frantz G.D., Tessier-Lavigne M., Bolon B. Secreted sulfatases Sulf1 and Sulf2 have overlapping yet essential roles in mouse neonatal survival. PLoS ONE. 2007;2:e575. doi: 10.1371/journal.pone.0000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ratzka A., Kalus I., Moser M., Dierks T., Mundlos S., Vortkamp A. Redundant function of the heparan sulfate 6-O-endosulfatases Sulf1 and Sulf2 during skeletal development. Dev. Dyn. 2008;237:339–353. doi: 10.1002/dvdy.21423. [DOI] [PubMed] [Google Scholar]
- 20.Ledin J., Staatz W., Li J.P., Götte M., Selleck S., Kjellén L., Spillmann D. Heparan sulfate structure in mice with genetically modified heparan sulfate production. J. Biol. Chem. 2004;279:42732–42741. doi: 10.1074/jbc.M405382200. [DOI] [PubMed] [Google Scholar]
- 21.Lai J.P., Sandhu D.S., Shire A.M., Roberts L.R. The tumor suppressor function of human sulfatase 1 (SULF1) in carcinogenesis. J. Gastrointest. Cancer. 2008;39:149–158. doi: 10.1007/s12029-009-9058-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Smith A.J., Tsalenko A., Sampas N., Scheffer A., Yamada N.A., Tsang P., Ben-Dor A., Yakhini Z., Ellis R.J., Bruhn L. Array CGH analysis of copy number variation identifies 1284 new genes variant in healthy white males: implications for association studies of complex diseases. Hum. Mol. Genet. 2007;16:2783–2794. doi: 10.1093/hmg/ddm208. [DOI] [PubMed] [Google Scholar]
- 23.Chipoy C., Berreur M., Couillaud S., Pradal G., Vallette F., Colombeix C., Rédini F., Heymann D., Blanchard F. Down-regulation of osteoblast markers and induction of the glial fibrillary acidic protein by oncostatin M in osteosarcoma cells required PKCd and STAT3. J. Bone Miner. Res. 2004;19:1850–1861. doi: 10.1359/JBMR.040817. [DOI] [PubMed] [Google Scholar]
- 24.Duplomb L., Baud'huin M., Charrier C., Berreur M., Trichet V., Blanchard F., Heymann D. Interleukin-6 inhibits receptor activator of nuclear factor kB ligand-induced osteoclastogenesis by diverting cells into the macrophage lineage: Key role of serine727 phosphorylation of signal transducer and activator of transcription factor 3. Endocrinology. 2008;149:3688–3697. doi: 10.1210/en.2007-1719. [DOI] [PubMed] [Google Scholar]
- 25.Grimaud E., Soubigou L., Couillaud S., Coipeau P., Moreau A., Passuti N., Gouin F., Rédini F., Heymann D. Receptor Activator of Nuclear Factor kB Ligand (RANKL)/Osteoprotegerin (OPG) ratio is increased in severe osteolysis. Am. J. Pathol. 2003;163:2021–2031. doi: 10.1016/s0002-9440(10)63560-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shears D.J., Offiah A., Rutland P., Sirimanna T., Bitner-Glindzicz M., Hall C. Kantaputra mesomelic dysplasia: A second reported family. Am. J. Med. Genet. A. 2004;128A:6–11. doi: 10.1002/ajmg.a.20640. [DOI] [PubMed] [Google Scholar]
- 27.Siwicka K.A., Kitoh H., Nishiyama M., Ishiguro N. A case of mesomelic dysplasia Kantaputra type–New findings and a new diagnostic approach. J. Pediatr. Orthop. B. 2008;17:271–276. doi: 10.1097/BPB.0b013e32830cc3c8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.