

Abstract
Terminal osseous dysplasia (TOD) is an X-linked dominant male-lethal disease characterized by skeletal dysplasia of the limbs, pigmentary defects of the skin, and recurrent digital fibroma with onset in female infancy. After performing X-exome capture and sequencing, we identified a mutation at the last nucleotide of exon 31 of the FLNA gene as the most likely cause of the disease. The variant c.5217G>A was found in six unrelated cases (three families and three sporadic cases) and was not found in 400 control X chromosomes, pilot data from the 1000 Genomes Project, or the FLNA gene variant database. In the families, the variant segregated with the disease, and it was transmitted four times from a mildly affected mother to a more seriously affected daughter. We show that, because of nonrandom X chromosome inactivation, the mutant allele was not expressed in patient fibroblasts. RNA expression of the mutant allele was detected only in cultured fibroma cells obtained from 15-year-old surgically removed material. The variant activates a cryptic splice site, removing the last 48 nucleotides from exon 31. At the protein level, this results in a loss of 16 amino acids (p.Val1724_Thr1739del), predicted to remove a sequence at the surface of filamin repeat 15. Our data show that TOD is caused by this single recurrent mutation in the FLNA gene.
Main Text
Terminal osseous dysplasia (MIM 300244) is a rare condition, characterized by terminal skeletal dysplasia, pigmentary defects of the skin, and recurrent digital fibromata during infancy. It has been described as a male-lethal X-linked dominant disease in the previously reported families and cases.1 Linkage studies mapped the mutation to Xq27.3-q28.2 However, no disease-causing gene had been discovered.
In the present study, we examined terminal osseous dysplasia (TOD) in three families and three sporadic case individuals (patients 1, 2, and 3 described by Horii,3 Drut,4 and Breuning5). The Dutch family (Figure 1A, family 1) and Italian family (Figure 1A, family 2) have been described before (Breuning5 and Baroncini6). The third family (Figure 1A, family 3) has not been reported before and is nonconsanguineous and of Israeli Arab origin. All patients, a mother and her two daughters, have normal cognitive development. The mother (3I:2) suffers from chronic mild obstructive lung disease and vitamin B12 deficiency. Since her childhood, she has had multiple minor surgeries to remove small skin lesions from her hands and legs. On clinical examination at the age of 25 yrs, her head circumference was 54 cm (25%–50%), her height was 170 cm (75%–90%), and her arm span was 171 cm. Her right hand showed brachydactyly of digit III-V, a short fingernail on digitus IV, and lateral deviation of the fifth digit. On her left hand, there was lateral deviation of the fourth digit, with a small lesion on the lateral aspect of the distal phalanx, and clinodactyly of the fifth digit (Figure 1B). Her right foot showed a short and highly implanted fourth digit. There was bilateral widening of the distal portion of the second–fifth digits. She had no gingival extra frenulum and no pterygium. A skeletal X-ray survey revealed unilateral flattening of her vertebral bodies at L1-L3, secondary right scoliosis, and wedging of her L1 vertebral body. Her daughter (3II:4) underwent surgery at 2 mo of age to remove small skin lesions from her hands, feet, and gingiva. On clinical examination at the age of 3 yrs, she had a head circumference of 48 cm (25%–50%), a height of 85 cm (< 3%), and a weight of 11.1 kg (< 3%). She showed hypertelorism—interpupillary distance of 5.4cm (> 97%), a right epicanthal fold, a normal palate, an upper and lower accessory frenulum (Figure 1C), a short neck, and a short thorax. Despite earlier surgery, she had bilateral skin lesions on her second and fifth digits and bilateral clinodactyly of the fifth digit (Figure 1D). Her feet showed a lesion in her third toes and thickening of the nail of the fifth toes bilaterally. A skeletal X-ray survey revealed bilateral lytic lesions in the proximal humerus and the proximal femur, as well as multiple soft-tissue lesions in her feet and hands. The youngest daughter (3II:5) was born with multiple lesions on her hands and feet, including bilateral camptodactyly of the third digit, and bilateral overriding of the fourth toe. Echocardiogram at birth showed persistent foramen ovale. On clinical examination at the age of 6 mo, her head circumference was 42 cm (25%–50%), her height was 58.8 cm (< 3%), and her weight was 5.1 kg (< 3%). She has mild hypertelorism, three brownish pigmented spots of different sizes (3 mm to 1.5 cm) in her right temporal groove, mild retrognathia, a right upper accessory frenulum, a cleft palate, a short neck, and a short thorax. She has a bilateral axillary pterygium (Figure 1E), which is more severe on the right side. Bilaterally, there is limited extension of her elbows, with normal supination and pronation of her hands. In her right hand (Figure 1F), she had multiple skin lesions on her second–fifth digits, clinodactyly and lateral deviation of her second and third digits, and a narrow fifth digit with an absent distal crease. Her left hand showed skin lesions on her second-fourth digits. Her second digit was narrow and laterally deviated. There was camptodactyly of the third-fifth digits, brachydactyly and clinodactyly of the fifth digit, and absence of a distal crease. In her feet, she had bilateral plantar pits. The right foot has distal broadening of the second-fifth toe and brachydactyly of the second and third toe accompanied by syndactyly. There was overriding of the third and fourth toe. On her left foot, the second-fifth toes were distally broad. She had a overlapping of the second and fourth toes over her third toe, brachydactyly of the third toe that was proximally implanted. A skeletal X-ray survey revealed bilateral lytic lesions of the proximal humerus, lytic lesions of the left proximal femur, and multiple soft-tissue lesions. She had underdeveloped tarsal bones in her feet. The phenotypes from different patients are summarized in Table 1.
Figure 1.
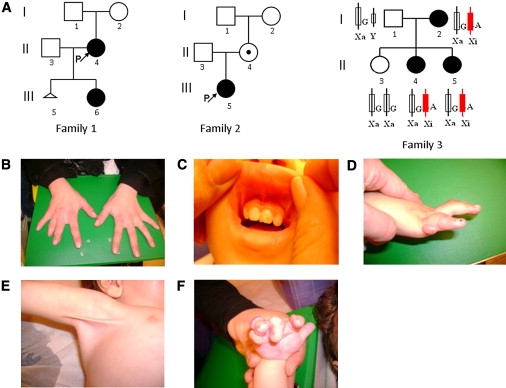
The Pedigrees and the Phenotype of Family 3
(A) The pedigrees investigated in this study. In family 3, XCI patterns show the silencing of the X chromosome that carries the mutant allele.
(B) The hands of 3I:2.
(C) Multiple frenula of 3II:4.
(D) The right hand of 3II:4. She has clinodactyly and digital fibroma.
(E) The right axillary pterygium of 3II:5.
(F) The right hand of 3II:5.
Table 1.
Clinical Features of the Patients Studied in This Report
| 1II:4 | 1III:6 | 2II:4 | 2III:5 | 3I:2 | 3II:4 | 3 II:5 | Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Origin | ||||||||||
| Dutch | Dutch | Italian | Italian | Israeli Arab | Israeli Arab | Israeli Arab | Japanese | Argentinian | Dutch | |
| Age at Onset | ||||||||||
| 1 mo | 3 mo | 7 mo | 2 mo | birth | 3 mo | 4 mo | ||||
| Pigmentary Skin Anomalies | ||||||||||
| Face | + | + | − | + | + | + | + | + | ||
| Scalp | − | − | + | − | ||||||
| Fibromatosis | ||||||||||
| Digital fibromas | + | + | − | + | + | + | + | + | + | + |
| Limbs and Skeletal System | ||||||||||
| Synadactyly | − | − | − | + | + | + | − | − | − | |
| Brachydactyly | + | − | + | + | + | + | ||||
| Clinodactyly | − | + | + | + | + | |||||
| Camptodactyly | + | + | ||||||||
| Metacarpal disorganization | + | + | − | + | + | + | + | |||
| Metatarsal disorganization | + | + | − | + | + | + | + | + | ||
| Limb long bones anomalies | − | + | − | + | − | + | + | + | + | + |
| Articular abnormalities | + | + | − | + | + | + | + | |||
| Facial Features | ||||||||||
| Cleft palate | − | − | − | − | − | + | − | − | ||
| Upslanting palpebral fissures | − | − | + | + | ||||||
| Hypertelorism/Telecanthus | + | − | − | + | + | + | ||||
| Epicanthic folds | − | − | + | + | + | |||||
| Coloboma of Iris | − | + | − | − | − | − | − | |||
| Flat/depressed nasal tip | − | + | − | − | + | − | ||||
| Thick lips/Prominent | + | − | + | |||||||
| Lower Lip | ||||||||||
| Papillomata | − | − | − | − | ||||||
| Multiple frenula | + | + | − | |||||||
| Preauricular pits and tags | + | − | ||||||||
DNA of patients and family members were extracted from peripheral blood (families 1, 2, and 3), buccal cells (patient 1), or paraffin-embedded tissue (patients 2 and 3). Two probands (1II:4 and 2III:5) of the Dutch and the Italian families were tested with the X-exome target-enrichment methodology (SureSelect, Agilent) and next-generation sequencing (Illumina Genome Analyzer II). The methods used for sequence capture, enrichment, and elution followed instructions and protocols provided by the manufacturers (SureSelect, Agilent) with a little modification. In brief, 500 ng of DNA was fragmented (Bioruptor, Diagenode) according to manufacturer's instructions to yield fragments from 200 to 300 bp. Paired-end adaptor oligonucleotides from Illumina were added to both ends. The DNA-adaptor-ligated fragments were then hybridized to 250 ng of SureSelect X chromosome oligo capture library (SureSelect, Agilent) for 14 hr. After hybridization, washing, and elution, the elute was amplified to create sufficient DNA template for downstream applications. The eluted-enriched DNA fragments were sequenced with the Illumina technology platform. We prepared the paired-end flow cell on the supplied cluster station, following the instructions of the manufacturer.
The reads were aligned to the reference human genome (hg 18, NCBI build 36.2) by Bowtie7 (Table S1, available online). Substitution-variant calling was performed by searching for positions where a variant nucleotide was present in more than 30% of the reads. After removing substitutions present with high frequency in dbSNP, the variants located in the previously identified TOD linkage interval, Xq27.3-q28, were listed in Table 2. From these variants, c.5217G>A, the only variant shared by the two patients, in the FLNA gene was selected for further study for the following reasons: (1) c.5217G>A, the last nucleotide of exon 31, was predicted to affect splicing by Human Splicing Finder.8 The score of the splicing donor site dropped from 91.2 to 80.63, indicating that the wild-type site may not function as usual. (2) Mutations in FLNA have been reported to be involved in diseases showing a partial phenotypic overlap with TOD.9
Table 2.
List of all Exonic Variants with Low Frequency in the European Population in Xq27.3-Xq28
| HGVS Name | Gene | Predicted Function | Predicted Protein Change | 1II:4 | 2III:5 | |
|---|---|---|---|---|---|---|
| NM_002025.2:c.1653A>G | AFF2 | silent | p.(=) | − | + | |
| NM_001183.4:c.∗461A>C | ATP6AP1 | 3′ UTR | p.(=) | − | + | |
| NM_001009932.1:c.364G>A | DNASE1L1 | silent | p.(=) | + | − | |
| NM_001110556.1:c.5217G>A | FLNA | silent | p.(=) | + | + | |
| NM_001110556.1:c.5814C>T | FLNA | silent | p.(=) | − | + | rs2070825, high frequency in a group of multiple populations |
| NM_001110556.1:c.5850T>C | FLNA | silent | p.(=) | + | − | does not segregate with phenotype |
| NM_004961.3:c.186G>A | GABRE | silent | p.(=) | + | − | |
| NM_005342.2:c.166G>C | HMGB3 | missense | p.(Glu56Gln) | − | + | |
| NM_005367.4:c.888A>G | MAGEA12 | silent | p.(=) | − | + | |
| NM_005362.3:c.455G>T | MAGEA6 | missense | p.(Ser152Ile) | + | − | repetitive region |
| NM_005365.4:c.92C>A | MAGEA9 | missense | p.(Pro31His) | + | − | repetitive region |
| NM_001170944.1:c.468C>T | PNMA6B | silent | p.(=) | + | − | |
| NM_005629.3:c.324A>G | SLC6A8 | silent | p.(=) | − | + | |
| NM_032539.2:c.1002T>C | SLITRK2 | silent | p.(=) | − | + | |
| NM_032539.2:c.309G>A | SLITRK2 | silent | p.(=) | − | + | |
| NM_001009615.1:c.240C>A | SPANXN2 | silent | p.(=) | + | − | |
| NM_014370.2:c.1014G>A | SRPK3 | silent | p.(=) | + | − | |
| NM_006280.1:c.430G>A | SSR4 | missense | p.(Gly144Arg) | + | − |
All of the HGVS numbers were generated with the use of the longest isoforms if multiple transcripts existed.
Sanger sequencing results confirmed the presence of c.5217G>A (Figure 2A) and c.5850T>C (Figure 2B) in all affected cases (1II:4 and 1III:6) in family 1, as well as c.5686+84A>G found in an intron but not in an unaffected individual (1I:2). Further evidence came from the analysis of the Italian family, in whom affected cases (2II:4 and 2III:5) carry exactly the same variant, c.5217G>A, together with another exonic variant, c.5814C>T. Unfortunately, we did not have access to material from both parents and therefore could not determine whether the mutations occurred de novo. Notably, families 1 and 2 had two distinct variants adjacent to the c.5217G>A mutation, making a close and common ancestor highly unlikely. Finally, upon analysis of a third TOD family and three unrelated sporadic cases, we identified exactly the same c.5217G>A variant again in all patients, but not in unaffected family members (1I:2, 3I:1, and 3II:3).
Figure 2.
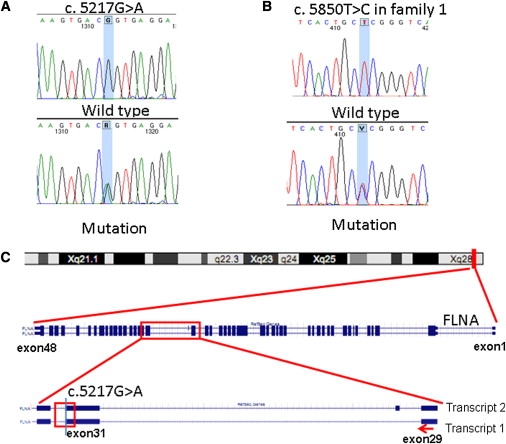
Genomic Structure and Mutation Analysis of FLNA
(A) c.5217G>A was confirmed by Sanger sequencing in all of the patients. The unaffected family members and controls carry the homozygous normal allele.
(B) The sequence of c.5850T>C in family 1.
(C) FLNA is located in Xq28, the target region of linkage analysis. c.5217G>A alters the last nucleotide of exon 31 of FLNA.
Variant c.5217G>A affects the last nucleotide of exon 31 of the FLNA gene (Figure 2C). At the protein level, it is not predicted to change the encoded amino acid, but as the last nucleotide of an exon, it may affect splicing.10–12 RNA was isolated from cultured fibroblasts of arm skin from 1III:6, removed during a recent orthopaedic procedure under general anesthesia with informed consent. Cells were cultured in standard medium for human fibroblasts (Dulbecco's modified Eagle's medium with 10% FBS, 1% penicillin/streptomycin, 1% glucose, 1% glutamax) with 5% CO2 in 37°C. RNA was extracted with the RNeasy Mini Kit (QIAGEN). cDNA was synthesized from 500 ng of total RNA by RevertAid RNaseH-M-MuLV reverse transcriptase in a total volume of 20 μl according to the protocol provided by the supplier (MBI-Fermentas). Target regions were amplified by RT-PCR with the use of the primers listed in Table S2. The products were evaluated with the Bioanalyzer 2100 DNA chip 1000 (Agilent), according to the manufacturer's instructions. RNA from patient fibroblasts showed only normal transcripts, both transcripts 1 (NM_01456) and 2 (NM_001110556) differing by insertion of the 24 bp exon 30 in transcript 2. Although transcript 1 has been reported as the predominant transcript in controls,13 we detected about equal expression levels in controls (Figure 3B, lanes 2–4 and 8) and higher expression of transcript 2 in patient fibroblasts (Figure 3B, lane 1). Both bands were isolated from the agarose gel by the Qiaquick Gel Extraction Kit (QIAGEN) and analyzed by Sanger sequencing. Interestingly, we detected no expression of the mutant allele. This could be due to nonsense-mediated decay 14 and/or skewed X chromosome inactivation (XCI). To test the first possibility, the fibroblasts were treated with cycloheximide15 for 4.5 hr followed by RNA analysis using the same procedures as those for RNA from untreated cells. The mutant allele was still absent in RNA from cycloheximide treated cells. XCI was analyzed with the Androgen Receptor (AR) assay.16 The assay showed random XCI in 1I:2 versus 100% XCI of the mutant chromosome in patient 1II:4 (patient 1III:6 was uninformative), indicating that the mutant allele was inactivated.
Figure 3.
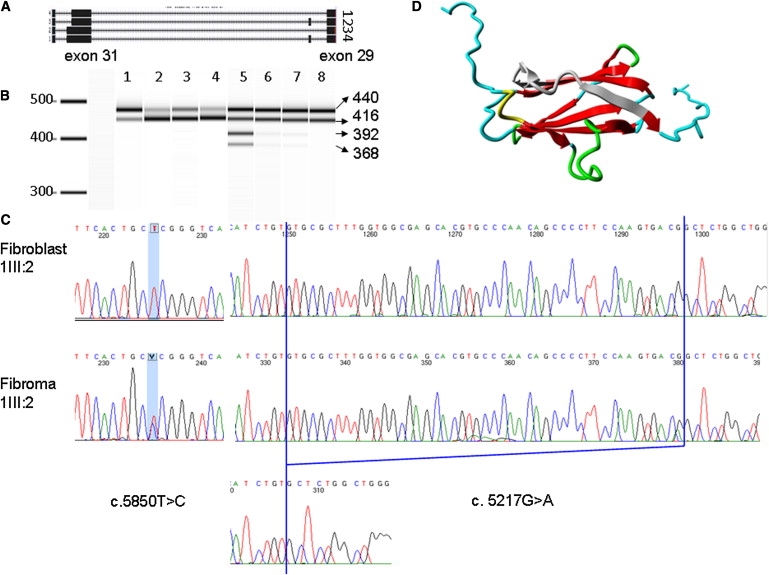
Detection of Alternative Splicing and 3D Protein Model
(A) Diagram of four FLNA transcripts in fibroma cells: transcripts 1 and 2, which carry the 48 bp deletion at the end of exon 31, as well as the normal transcripts 3 and 4.
(B) RT-PCR result from Agilent 2100 Bioanalyzer. Lane 1 is the product of the fibroblasts of 1III:6, which has a predominant longer isoform. Lanes 2–4 and 8 are four control human fibroblasts. Lanes 5–7 show RT-PCR products that were obtained from fibroma cells of 1III:6, the normal bands from two FLNA isoforms, and two extra shorter bands, which are faint in lane 6 (left fifth finger) and lane 7 (fifth toe of the left foot), whereas lane 5 (right fifth finger) shows four dark bands.
(C) Sanger sequencing results of c.5858T>C and c.5217G>A in fibroblast and fibroma cells of 1III:6.
(D) The 3D model of FLNA domain 15. The deleted 16 amino acids are marked in gray. Beta strands are marked in red. Green represents a turn. Yellow indicates a 3/10 helix. Random coils are colored in cyan.
Fifteen years ago, at the age of 1 yr, patient 1III:6 had fibroma tissue from the fifth digits of both hands and the fifth toe of the left foot surgically removed and stored in liquid nitrogen. We cultured these cells and analyzed RNA. In the fibroma cells, we observed two sets of two bands (Figure 3B, lanes 5–7), indicating altered splicing. One set had the same length as that observed in normal fibroblasts (Figure 3A, transcripts 1 and 2), and the other set was shorter (Figure 3A, transcripts 3 and 4, faint from RNA of a tumor in left fifth finger and toe; Figure 3B, lanes 6 and 7). Note that the fibroma always contains a mixture of tumor and normal stroma cells. Sequence analysis showed a deletion removing the last 48 nucleotides of exon 31 (Figure 3C), resulting in a deletion of 16 amino acids.
To facilitate clinical diagnostics of FLNA gene mutations, we have established a web-based FLNA gene variant database using the LOVD software.17 In this publicly available database, we have collected all variants reported in the literature thus far (83 in total; see FLNA mutation database), including the variants described here. The c.5217G>A variant detected in TOD patients has not been described before; it is listed neither in dbSNP nor in the pilot study 1 of the 1000 Genomes Project. Finally, over 400 chromosomes have been sequenced and the mutant allele was not found (data not shown).
Mutations in FLNA have been reported to cause a wide range of developmental malformations in the brain, bones, limbs, heart,18 and other organs19 in human,9 including periventricular heterotopia (PVNH [MIM 300049])20–24 and otopalatodigital (OPD) spectrum disorders,25 which include otopalatodigital syndrome type 1 (MIM 311300)26–28 and type 2 (MIM 304120),26,29 frontometaphyseal dysplasia (MIM 305620),26,30,31 and Melnick-Needles syndrome (OMIM 309350).26,27 Although each of the OPD spectrum disorders are characterized by specific clinical symptoms, there clearly is a clinical overlap with TOD, including a generalized bone dysplasia that includes craniofacial anomalies and anomalies in digits and long bones.9,32 Interestingly, the most conspicuous symptoms of TOD patients are skeletal dysplasia of the limbs and recurrent digital fibroma, suggesting a significant role of the FLNA mutation in the TOD phenotype.
The FLNA gene encodes a cytoskeletal protein, filamin A, which crosslinks actin filaments into an orthogonal network and links these to the cell membrane. Within the cytoskeleton, filamin A also mediates functions relating to cell signaling, transcription, and development.33 Filamin A consists of two calponin homology sequences (CH1 and CH2) at the N terminus and connects with 24 immunoglobin-like filamin repeats, divided by two hinges, one between repeats 15 and 16 and one between repeats 23 and 24. To check the stability of filamin A in patient cells, protein was extracted from both fibroblast and fibroma cells. Immunoblot was performed with the use of mouse human filamin A monoclonal antibody, MAB1680, from Millipore. No difference in molecular weight or quantity was observed. The difference of 18 amino acids was likely too small to be distinguished by immunoblot. The c.5217G>A mutation is located in a highly conserved position at the DNA level, across a wide range of vertebrate and invertebrate species except rodent, and found in all ten affected patients from six different unrelated families. In addition, the mutation introduced abnormal splicing in fibroma cells. At the protein level, c.5217G>A encodes the second-to-last amino acid of repeat 15, which is immediately adjacent to hinge 1. Recent studies demonstrated repeats 9–15 contain an F-actin binding domain necessary for high avidity F-actin binding.34 Hinge 1 plays an important part in maintaining the viscoelastic properties of actin networks.35 Moreover, this region interacts with many binding partners, such as TRAF1, TRAF2,36 CaR extracellular Ca2+ receptor,37 and FAP52.38 Because no crystal structure has yet been described for this region, the crystal structure of repeat 15 in filamin B(PDB file 2 dmb), which shows the highest identity (58%) with this region of interest, was used as a template for building a 3D model (Figure 3D). The model was built with the use of the WHAT IF and YASARA twinset.39 Repeat 15 consists of two beta sheets. The in-frame deletion causes the removal of the top of a beta strand in the middle of one beta sheet, and of two beta strands at the side of that sheet (gray part of Figure 3D). These residues are likely to form some kind of beta strand-like structure and to substantially alter the structure of the highly conserved tertiary structure of filamin repeat 15. Furthermore, this structure will affect the residues following the beta sheet and linking repeat 15 to hinge 1. Although there is no way to predict what will happen to those linking residues, we believe it will affect the overall conformation of the protein and likely influence the interaction between filamin A and other molecules.
The precise mechanism of TOD remains unclear. However, like other X-linked diseases, XCI might be a key component of how the disease develops. The developmental role of FLNA is borne out by the presence of the skeletal and skin malformations at birth. Multiple fibroma on digits begins to occur in the first years, and fibromas spontaneously stop by the age of five. Skewed XCI is known to vary in different tissues and to correlate with age under the pressure of secondary selection.40 Several mechanisms may contribute to the skewing, including stochastic effects, a selective growth advantage of the cell that carries either the mutated or the normal allele (secondary cell selection), and genetic processes yielding preferential inactivation of specific alleles. Primarily the XCI choice is random, but during cell proliferation, either in all cells or in a tissue specific manner, cells that carry an active mutated allele may have a significant disadvantage, are gradually lost or selected against, and are thus less represented in the adult female.41 Disorders caused by defects in the FLNA gene often show a skewed XCI pattern,26 suggesting that cells need normal filamin to survive. Several studies in TOD families showed that patients had skewed XCI, while unaffected individuals had random inactivation.1,6 We examined the XCI pattern in family 1 (1I:2, 1II:4, and 1III:6) and family 3 (3I:2, 3II:3, 3II:4, and 3II:5; Figure 1A) by AR assay. Apart from the uninformative patient 1III:6, all of the other patients—1II:4, 3I:2, 3II:4, and 3II:5—showed extremely skewed XCI (0/100%), whereas the normal family member 1I:2 showed random XCI (30/70%), as did 3II:3 (50/50%). Because there was no mutant allele detectable in the RNA of normal fibroblast, we deduced that 1III:6 also had 100% skewed XCI with the preferential inactivation of the mutant allele. We tested the XCI of 2II:4 and 2III:5, and both showed 100% skewing.6 2II:4 was interpreted by the authors as unaffected. However, we assume that 2II:4 is a carrier of TOD, given that she has only mild manifestations (multiple frenula in the mouth). She probably has skewed XCI at a very early stage. Local XCI patterns may influence the severity of the phenotype of carrier females and are also associated with selective female survival in male-lethal, X-linked, dominant disorders.
Taken together, these data suggest that TOD is caused by a unique variant, c.5217G>A (p.Val1724_Thr1739del), in the FLNA gene. The variant is not found in other databases, has not been seen in other patients with pathogenic FLNA variants, segregates with the disease, and is located in Xq28, where the potential mutated gene causing this disorder was mapped previously. The mutation was found in six unrelated families. It will affect splicing, and it causes a deletion of 16 amino acids at the protein level. The missing region in the filamin A protein is hypothesized to affect or prevent the interaction of filamin A with other proteins.
Acknowledgments
We would like to thank the patients and their family members for their willingness to join the project, the China Scholarship Council (CSC) scholarship for supporting Yu Sun's studies in The Netherlands, Filip Kluin for sending paraffin-embedded tissue and Hans Morreau for isolating DNA from the tissue, Tobias Messemaker for helping us with immunoblotting, and the Leiden Genome Technology Center (LGTC) and the Laboratory for Diagnostic Genome Analysis (LDGA) for help with sequencing, DNA extraction, and XCI detection. X-exome capture was implemented in collaboration with ServiceXS (Leiden, http://www.servicexs.com). The research leading to these results has received funding from the European Community's Seventh Framework Program (FP7/2007-2013) under grant agreements 223026 (NMD-chip), 223143 (TechGene), and 200754 (Gen2Phen). B.F. was funded by the Italian Telethon Foundation.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/page.php
FLNA gene variant database, http://www.lovd.nl/FLNA
Human Splicing Finder, http://www.umd.be/HSF
Online Mendelian Inheritance in Man(OMIM), http://www.ncbi.nlm.nih.gov/entrez/Omim/
SureSelect manual, http://www.genomics.agilent.com/files/Manual/G3360-90020_SureSelect_Indexing_1.0.pdf
UCSC Genome Browser, http://genome.ucsc.edu/
YASARA, http://www.yasara.org/
References
- 1.Bacino C.A., Stockton D.W., Sierra R.A., Heilstedt H.A., Lewandowski R., Van den Veyver I.B. Terminal osseous dysplasia and pigmentary defects: clinical characterization of a novel male lethal X-linked syndrome. Am. J. Med. Genet. 2000;94:102–112. doi: 10.1002/1096-8628(20000911)94:2<102::aid-ajmg2>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 2.Zhang W., Amir R., Stockton D.W., Van Den Veyver I.B., Bacino C.A., Zoghbi H.Y. Terminal osseous dysplasia with pigmentary defects maps to human chromosome Xq27.3-xqter. Am. J. Hum. Genet. 2000;66:1461–1464. doi: 10.1086/302868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horii E., Sugiura Y., Nakamura R. A syndrome of digital fibromas, facial pigmentary dysplasia, and metacarpal and metatarsal disorganization. Am. J. Med. Genet. 1998;80:1–5. doi: 10.1002/(sici)1096-8628(19981102)80:1<1::aid-ajmg1>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 4.Drut R., Pedemonte L., Rositto A. Noninclusion-body infantile digital fibromatosis: a lesion heralding terminal osseous dysplasia and pigmentary defects syndrome. Int. J. Surg. Pathol. 2005;13:181–184. doi: 10.1177/106689690501300209. [DOI] [PubMed] [Google Scholar]
- 5.Breuning M.H., Oranje A.P., Langemeijer R.A., Hovius S.E., Diepstraten A.F., den Hollander J.C., Baumgartner N., Dwek J.R., Sommer A., Toriello H. Recurrent digital fibroma, focal dermal hypoplasia, and limb malformations. Am. J. Med. Genet. 2000;94:91–101. doi: 10.1002/1096-8628(20000911)94:2<91::aid-ajmg1>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 6.Baroncini A., Castelluccio P., Morleo M., Soli F., Franco B. Terminal osseous dysplasia with pigmentary defects: clinical description of a new family. Am. J. Med. Genet. A. 2007;143:51–57. doi: 10.1002/ajmg.a.31557. [DOI] [PubMed] [Google Scholar]
- 7.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desmet F.O., Hamroun D., Lalande M., Collod-Béroud G., Claustres M., Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robertson S.P. Filamin A: phenotypic diversity. Curr. Opin. Genet. Dev. 2005;15:301–307. doi: 10.1016/j.gde.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 10.Agarwal N., Kutlar F., Mojica-Henshaw M.P., Ou C.N., Gaikwad A., Reading N.S., Bailey L., Kutlar A., Prchal J.T. Missense mutation of the last nucleotide of exon 1 (G->C) of beta globin gene not only leads to undetectable mutant peptide and transcript but also interferes with the expression of wild allele. Haematologica. 2007;92:1715–1716. doi: 10.3324/haematol.11543. [DOI] [PubMed] [Google Scholar]
- 11.Yamada K., Fukao T., Zhang G., Sakurai S., Ruiter J.P., Wanders R.J., Kondo N. Single-base substitution at the last nucleotide of exon 6 (c.671G>A), resulting in the skipping of exon 6, and exons 6 and 7 in human succinyl-CoA:3-ketoacid CoA transferase (SCOT) gene. Mol. Genet. Metab. 2007;90:291–297. doi: 10.1016/j.ymgme.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 12.Kuivaniemi H., Tromp G., Bergfeld W.F., Kay M., Helm T.N. Ehlers-Danlos syndrome type IV: a single base substitution of the last nucleotide of exon 34 in COL3A1 leads to exon skipping. J. Invest. Dermatol. 1995;105:352–356. doi: 10.1111/1523-1747.ep12320704. [DOI] [PubMed] [Google Scholar]
- 13.Maestrini E., Patrosso C., Mancini M., Rivella S., Rocchi M., Repetto M., Villa A., Frattini A., Zoppè M., Vezzoni P. Mapping of two genes encoding isoforms of the actin binding protein ABP-280, a dystrophin like protein, to Xq28 and to chromosome 7. Hum. Mol. Genet. 1993;2:761–766. doi: 10.1093/hmg/2.6.761. [DOI] [PubMed] [Google Scholar]
- 14.Holbrook J.A., Neu-Yilik G., Hentze M.W., Kulozik A.E. Nonsense-mediated decay approaches the clinic. Nat. Genet. 2004;36:801–808. doi: 10.1038/ng1403. [DOI] [PubMed] [Google Scholar]
- 15.Kim C.E., Gallagher P.M., Guttormsen A.B., Refsum H., Ueland P.M., Ose L., Folling I., Whitehead A.S., Tsai M.Y., Kruger W.D. Functional modeling of vitamin responsiveness in yeast: a common pyridoxine-responsive cystathionine beta-synthase mutation in homocystinuria. Hum. Mol. Genet. 1997;6:2213–2221. doi: 10.1093/hmg/6.13.2213. [DOI] [PubMed] [Google Scholar]
- 16.Kubota T., Nonoyama S., Tonoki H., Masuno M., Imaizumi K., Kojima M., Wakui K., Shimadzu M., Fukushima Y. A new assay for the analysis of X-chromosome inactivation based on methylation-specific PCR. Hum. Genet. 1999;104:49–55. doi: 10.1007/s004390050909. [DOI] [PubMed] [Google Scholar]
- 17.Fokkema I.F., den Dunnen J.T., Taschner P.E. LOVD: easy creation of a locus-specific sequence variation database using an “LSDB-in-a-box” approach. Hum. Mutat. 2005;26:63–68. doi: 10.1002/humu.20201. [DOI] [PubMed] [Google Scholar]
- 18.Kyndt F., Gueffet J.P., Probst V., Jaafar P., Legendre A., Le Bouffant F., Toquet C., Roy E., McGregor L., Lynch S.A. Mutations in the gene encoding filamin A as a cause for familial cardiac valvular dystrophy. Circulation. 2007;115:40–49. doi: 10.1161/CIRCULATIONAHA.106.622621. [DOI] [PubMed] [Google Scholar]
- 19.Gargiulo A., Auricchio R., Barone M.V., Cotugno G., Reardon W., Milla P.J., Ballabio A., Ciccodicola A., Auricchio A. Filamin A is mutated in X-linked chronic idiopathic intestinal pseudo-obstruction with central nervous system involvement. Am. J. Hum. Genet. 2007;80:751–758. doi: 10.1086/513321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fox J.W., Lamperti E.D., Ekşioğlu Y.Z., Hong S.E., Feng Y., Graham D.A., Scheffer I.E., Dobyns W.B., Hirsch B.A., Radtke R.A. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21:1315–1325. doi: 10.1016/s0896-6273(00)80651-0. [DOI] [PubMed] [Google Scholar]
- 21.Sheen V.L., Dixon P.H., Fox J.W., Hong S.E., Kinton L., Sisodiya S.M., Duncan J.S., Dubeau F., Scheffer I.E., Schachter S.C. Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum. Mol. Genet. 2001;10:1775–1783. doi: 10.1093/hmg/10.17.1775. [DOI] [PubMed] [Google Scholar]
- 22.Moro F., Carrozzo R., Veggiotti P., Tortorella G., Toniolo D., Volzone A., Guerrini R. Familial periventricular heterotopia: missense and distal truncating mutations of the FLN1 gene. Neurology. 2002;58:916–921. doi: 10.1212/wnl.58.6.916. [DOI] [PubMed] [Google Scholar]
- 23.Zenker M., Rauch A., Winterpacht A., Tagariello A., Kraus C., Rupprecht T., Sticht H., Reis A. A dual phenotype of periventricular nodular heterotopia and frontometaphyseal dysplasia in one patient caused by a single FLNA mutation leading to two functionally different aberrant transcripts. Am. J. Hum. Genet. 2004;74:731–737. doi: 10.1086/383094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheen V.L., Jansen A., Chen M.H., Parrini E., Morgan T., Ravenscroft R., Ganesh V., Underwood T., Wiley J., Leventer R. Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology. 2005;64:254–262. doi: 10.1212/01.WNL.0000149512.79621.DF. [DOI] [PubMed] [Google Scholar]
- 25.Robertson S.P. Otopalatodigital syndrome spectrum disorders: otopalatodigital syndrome types 1 and 2, frontometaphyseal dysplasia and Melnick-Needles syndrome. Eur. J. Hum. Genet. 2007;15:3–9. doi: 10.1038/sj.ejhg.5201654. [DOI] [PubMed] [Google Scholar]
- 26.Robertson S.P., Twigg S.R., Sutherland-Smith A.J., Biancalana V., Gorlin R.J., Horn D., Kenwrick S.J., Kim C.A., Morava E., Newbury-Ecob R., OPD-spectrum Disorders Clinical Collaborative Group Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nat. Genet. 2003;33:487–491. doi: 10.1038/ng1119. [DOI] [PubMed] [Google Scholar]
- 27.Robertson S.P., Thompson S., Morgan T., Holder-Espinasse M., Martinot-Duquenoy V., Wilkie A.O., Manouvrier-Hanu S. Postzygotic mutation and germline mosaicism in the otopalatodigital syndrome spectrum disorders. Eur. J. Hum. Genet. 2006;14:549–554. doi: 10.1038/sj.ejhg.5201586. [DOI] [PubMed] [Google Scholar]
- 28.Hidalgo-Bravo A., Pompa-Mera E.N., Kofman-Alfaro S., Gonzalez-Bonilla C.R., Zenteno J.C. A novel filamin A D203Y mutation in a female patient with otopalatodigital type 1 syndrome and extremely skewed X chromosome inactivation. Am. J. Med. Genet. A. 2005;136:190–193. doi: 10.1002/ajmg.a.30792. [DOI] [PubMed] [Google Scholar]
- 29.Mariño-Enríquez A., Lapunzina P., Robertson S.P., Rodríguez J.I. Otopalatodigital syndrome type 2 in two siblings with a novel filamin A 629G>T mutation: clinical, pathological, and molecular findings. Am. J. Med. Genet. A. 2007;143A:1120–1125. doi: 10.1002/ajmg.a.31696. [DOI] [PubMed] [Google Scholar]
- 30.Zenker M., Nährlich L., Sticht H., Reis A., Horn D. Genotype-epigenotype-phenotype correlations in females with frontometaphyseal dysplasia. Am. J. Med. Genet. A. 2006;140:1069–1073. doi: 10.1002/ajmg.a.31213. [DOI] [PubMed] [Google Scholar]
- 31.Giuliano F., Collignon P., Paquis-Flucklinger V., Bardot J., Philip N. A new three-generational family with frontometaphyseal dysplasia, male-to-female transmission, and a previously reported FLNA mutation. Am. J. Med. Genet. A. 2005;132A:222. doi: 10.1002/ajmg.a.30396. [DOI] [PubMed] [Google Scholar]
- 32.Robertson S.P. Molecular pathology of filamin A: diverse phenotypes, many functions. Clin. Dysmorphol. 2004;13:123–131. doi: 10.1097/01.mcd.0000130235.95356.40. [DOI] [PubMed] [Google Scholar]
- 33.Zhou A.X., Hartwig J.H., Akyürek L.M. Filamins in cell signaling, transcription and organ development. Trends Cell Biol. 2010;20:113–123. doi: 10.1016/j.tcb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura F., Osborn T.M., Hartemink C.A., Hartwig J.H., Stossel T.P. Structural basis of filamin A functions. J. Cell Biol. 2007;179:1011–1025. doi: 10.1083/jcb.200707073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gardel M.L., Nakamura F., Hartwig J.H., Crocker J.C., Stossel T.P., Weitz D.A. Prestressed F-actin networks cross-linked by hinged filamins replicate mechanical properties of cells. Proc. Natl. Acad. Sci. USA. 2006;103:1762–1767. doi: 10.1073/pnas.0504777103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arron J.R., Pewzner-Jung Y., Walsh M.C., Kobayashi T., Choi Y. Regulation of the subcellular localization of tumor necrosis factor receptor-associated factor (TRAF)2 by TRAF1 reveals mechanisms of TRAF2 signaling. J. Exp. Med. 2002;196:923–934. doi: 10.1084/jem.20020774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Awata H., Huang C., Handlogten M.E., Miller R.T. Interaction of the calcium-sensing receptor and filamin, a potential scaffolding protein. J. Biol. Chem. 2001;276:34871–34879. doi: 10.1074/jbc.M100775200. [DOI] [PubMed] [Google Scholar]
- 38.Nikki M., Meriläinen J., Lehto V.P. FAP52 regulates actin organization via binding to filamin. J. Biol. Chem. 2002;277:11432–11440. doi: 10.1074/jbc.M111753200. [DOI] [PubMed] [Google Scholar]
- 39.Krieger E., Koraimann G., Vriend G. Increasing the precision of comparative models with YASARA NOVA—a self-parameterizing force field. Proteins. 2002;47:393–402. doi: 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- 40.Sharp A., Robinson D., Jacobs P. Age- and tissue-specific variation of X chromosome inactivation ratios in normal women. Hum. Genet. 2000;107:343–349. doi: 10.1007/s004390000382. [DOI] [PubMed] [Google Scholar]
- 41.Orstavik K.H. X chromosome inactivation in clinical practice. Hum. Genet. 2009;126:363–373. doi: 10.1007/s00439-009-0670-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.