

Abstract
Clubfoot is a common musculoskeletal birth defect for which few causative genes have been identified. To identify the genes responsible for isolated clubfoot, we screened for genomic copy-number variants with the Affymetrix Genome-wide Human SNP Array 6.0. A recurrent chromosome 17q23.1q23.2 microduplication was identified in 3 of 66 probands with familial isolated clubfoot. The chromosome 17q23.1q23.2 microduplication segregated with autosomal-dominant clubfoot in all three families but with reduced penetrance. Mild short stature was common and one female had developmental hip dysplasia. Subtle skeletal abnormalities consisted of broad and shortened metatarsals and calcanei, small distal tibial epiphyses, and thickened ischia. Several skeletal features were opposite to those described in the reciprocal chromosome 17q23.1q23.2 microdeletion syndrome associated with developmental delay and cardiac and limb abnormalities. Of note, during our study, we also identified a microdeletion at the locus in a sibling pair with isolated clubfoot. The chromosome 17q23.1q23.2 region contains the T-box transcription factor TBX4, a likely target of the bicoid-related transcription factor PITX1 previously implicated in clubfoot etiology. Our result suggests that this chromosome 17q23.1q23.2 microduplication is a relatively common cause of familial isolated clubfoot and provides strong evidence linking clubfoot etiology to abnormal early limb development.
Main Text
Congenital clubfoot is one of the most common serious congenital birth defects, with an estimated birth prevalence of 1 per 1000 live births.1 Clubfoot consists of malalignment of the bones and joints of the foot and is distinguishable from positional foot anomalies because it is rigid and not passively correctable. Twenty percent of all cases of clubfoot are associated with distal arthrogryposis, congenital myotonic dystrophy, myelomeningocele, amniotic band sequence, or other genetic syndromes such as trisomy 18 or chromosome 22q11 deletion syndrome,2,3 and in the remaining patients the etiology is unknown.4
Approximately 25% of all patients with isolated clubfoot report a family history of clubfoot.5 Twin studies also support a role for genetic factors in clubfoot given that the concordance rate is higher for identical twins than for fraternal twins (33% versus 3%).4 Additional evidence for a genetic basis is provided by differences in clubfoot prevalence across ethnic populations, with the lowest prevalence in Chinese (0.39 cases per 1000 live births) and the highest in Hawaiians and Maoris (7 per 1000).6,7 The ratio of idiopathic clubfoot among males to females is 2:1 and is consistent across ethnic groups.6
Despite the high incidence of clubfoot, few causative genes are known. We recently identified a mutation in the bicoid-related homeodomain transcription factor gene PITX1 (MIM 602149), in a multigenerational family with predominantly isolated clubfoot.8 However, additional mutations were not identified in 100 patients with idiopathic clubfoot, suggesting genetic heterogeneity. Common polymorphisms near HOX homeobox genes, insulin-like growth factor binding protein 3 (IGFBP3) (MIM 146732),9 and caspase genes10 have also been reported to be associated with idiopathic clubfoot. Currently there is no genetic testing available for patients with isolated clubfoot, and much remains to be identified regarding both the genetic and mechanistic aspects of this condition.
To identify the genes responsible for isolated clubfoot, we identified 66 isolated idiopathic clubfoot probands with at least one affected first-degree relative. Individuals were considered to have isolated idiopathic clubfoot only in the absence of additional congenital anomalies (e.g., heart defect, hypospadias, developmental delay) or known underlying etiology (e.g., arthrogryposis, myelomeningocele, myopathy). The study was approved by the local human ethics review committees. Consent and DNA samples were obtained from study subjects.
Forty familial isolated clubfoot probands were screened for genomic copy-number variants (CNVs) with the Affymetrix Genome-wide Human SNP Array 6.0, consisting of more than 1.8 million genomic DNA markers, including > 900,000 probes for the detection of copy-number variation and > 900,000 SNPs. Nearly identical 2.2 Mb chromosome 17 duplications involving 1120 markers were identified on copy-number analysis performed with the Affymetrix Genotyping Console (Figure 1A) in two unrelated probands (5103-01 and 5377-01) (Figures 2A and 2B). The duplicated region containing 17 RefSeq genes is flanked by segmental repeats and corresponds exactly to the location of the chromosome 17q23.1q23.2 microdeletion (MIM 613355) recently identified in individuals with developmental delay, heart defects, and limb anomalies.11 The duplication was verified with TaqMan copy-number assays (Hs01196629_cn and Hs02252963_cn). The chromosome 17q23.1q23.2 duplication was not present in 700 controls12,13 evaluated with the same platform (Affymetrix 6.0) and is not reported in the Database of Genomic Variants.
Figure 1.
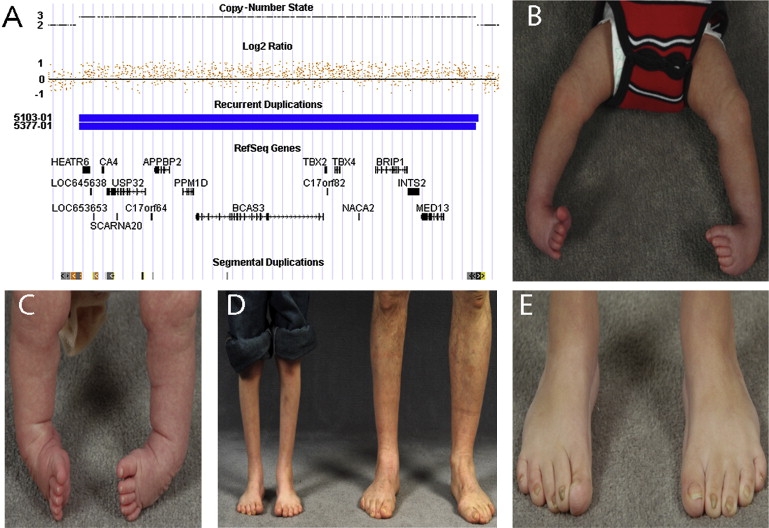
Clinical Findings in Patients with Chromosome 17q23.1q23.2 Microduplications
(A) Chromosome 17q21.1q23.2 region showing 2.2 Mb duplication at chromosome 17: 55457520–57693617 (hg18 build of the UCSC genome browser) with increased log2 ratio and copy-number state in two patients (5103-01 and 5377-01) involving 1119 and 1120 markers. Seventeen RefSeq genes are located within the interval, including TBX2 and TBX4. Paired segmental duplications flank the duplicated region (shown in orange).
(B and C) Untreated bilateral clubfoot: patient 5103-01 in (B) and patient 5377-01 in (C).
(D) Limb hypoplasia in proband with bilateral clubfoot (5103-01) and hypoplasia of the right leg in his affected father with unilateral right-sided clubfoot (5103-03). Both had been treated surgically after failing conservative therapy.
(E) Broad, short, overlapping toes with nail hypoplasia in proband (5103-01).
Figure 2.
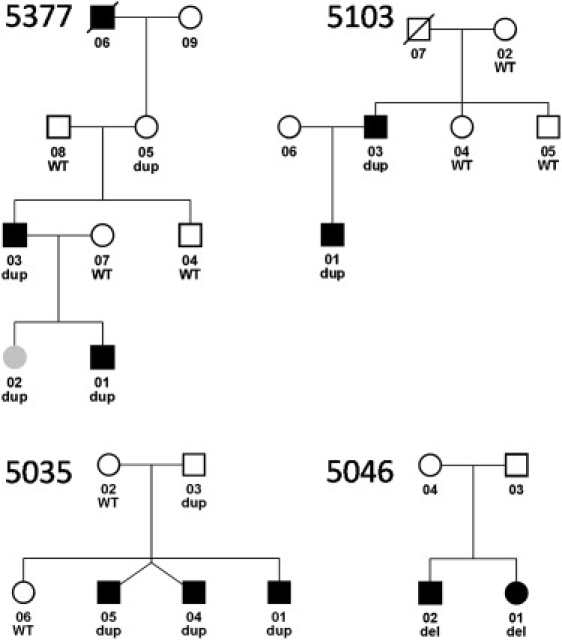
Pedigrees of Four Isolated Clubfoot Families with Chromosome 17q23.1q23.2 CNVs
Microduplications segregate with clubfoot with incomplete penetrance in three families (5377, 5103, and 5035), and microdeletions are present in two siblings with clubfoot (5046). Black affection status indicates clubfoot, and gray indicates developmental dysplasia of the hip. Dup indicates duplication, del indicates deletion, and WT indicates normal copy number. The twins in family 5035 are not identical.
To confirm the frequency of the chromosome 17q23.1q23.2 microduplication, we then screened DNA from 26 additional probands with familial isolated clubfoot for CNVs in this region by using the TaqMan assays. One chromosome 17q23.1q23.2 microduplication was identified in a proband from a multiplex family with idiopathic clubfoot (5035-01) (Figure 2C), and, surprisingly, one microdeletion was identified in a sibling pair with idiopathic clubfoot (5046-01) (Figure 2D).
To determine the inheritance of the chromosome 17q23.1q23.2 CNVs, we then obtained DNA from additional affected and unaffected family members from all three families with the chromosome 17q23.1q23.2 microduplication and one family with the reciprocal microdeletion. The chromosome 17q23.1q23.2 microduplication segregated with idiopathic clubfoot in all three families (Figure 2) but with incomplete penetrance. The parents of the siblings with the microdeletion were unavailable for testing.
Clubfoot was present in seven patients with the microduplication and absent in three patients (penetrance of 70%) (Figures 1B and 1C). One female with the duplication had bilateral hip dysplasia (5377-02), and two parents with the duplication denied any problems with their lower extremities or hips (5377-05, 5035-03) and had normal physical examinations. All cases with clubfoot were male, and clubfoot was bilateral in all except one case (Figure 1D) (Table 1). The hands and upper extremities were unaffected in all cases. Clinically, the feet were short, with broad, overlapping toes (Figure 1E), a marked contrast to the long, thin digits of individuals with the previously described chromosome 17q23 microdeletion.11 Mild nail hypoplasia was present in two affected individuals (Figure 1E). Mild short stature was common (Table 1). None of the affected individuals had the heart defects, facial dysmorphisms, or cognitive and behavioral abnormalities that are commonly associated with the chromosome 17q23.1q23.2 microdeletion.11 Notably, the two siblings with the chromosome 17q23.1q23.2 microdeletion are developmentally and cognitively normal on our examination, but have not been specifically tested for learning disabilities because of their young age (<3 years) at the time of the study.
Table 1.
Clinical Features of Subjects with Chromosome 17q23.2 Microduplication
| Patient | 5377-01 | 5377-02 | 5377-03 | 5377-06 | 5103-01 | 5103-03 | 5035-01 | 5035-04 | 5035-05 |
|---|---|---|---|---|---|---|---|---|---|
| Age | 3 | 5 | 35 | D/C | 10 | 44 | 54 | 47 | 47 |
| Sex | M | F | M | M | M | M | M | M | M |
| Height | 4% | 10% | 5% | 10% | 4% | 10% | 5% | 50% | 50% |
| Clubfoot | Bilateral Grade III | - | Bilateral Grade III | Bilateral (unknown grade) | Bilateral Grade IV | Right Grade IV | Bilateral (unknown grade) | Bilateral (unknown grade) | Bilateral (unknown grade) |
| Clubfoot treatment | Conservative | - | Conservative | Conservative | Surgery | Surgery | Surgery | Surgery | Surgery |
| Hip dysplasia | - | + | - | - | - | - | - | - | - |
| Nail hypoplasia | - | - | - | N/A | + | + | - | - | - |
| Radiographic Abnormalities of Pelvis | |||||||||
| Acetabular dysplasia | + | + | - | N/A | - | + | N/A | N/A | N/A |
| Coxa valga | + | + | + | N/A | + | - | N/A | N/A | N/A |
| Genu valgum | + | - | - | N/A | - | - | N/A | N/A | N/A |
| Tall, narrow ilium | - | + | + | N/A | + | + | N/A | N/A | N/A |
| Thickened inferior pubic ramus (ischium) | + | + | + | N/A | + | + | N/A | N/A | N/A |
| Infra-acetabular axe-cut notches | + | - | + | N/A | - | + | N/A | N/A | N/A |
| Lack of normal iliac flare | + | + | + | N/A | + | + | N/A | N/A | N/A |
| Radiographic Abnormalities of Feet | |||||||||
| Short and thickened first and/or second metatarsals | + | N/A | + | N/A | + | + | - | + | + |
| Short calcaneus | + | N/A | + | N/A | + | + | + | + | - |
| Hypoplastic distal tibial epiphysis | + | N/A | + | N/A | + | + | N/A | + | N/A |
| Tufted distal phalanx of the first toe | + | N/A | + | N/A | + | + | + | + | + |
N/A indicates not available; D/C indicates deceased.
In the general population, clubfoot ranges in severity from mild to severe,14 and few factors other than initial severity and etiology are able to predict treatment outcomes.2,15–17 For this reason, we sought to determine whether the clubfeet associated with chromosome 17q23.1q23.2 microduplications were more severe than typical or respond poorly to the conservative, predominantly nonsurgical treatment methods that are currently the standard of care.18 Three patients from one family were managed conservatively without surgery, including the proband (5377-01), who was successfully treated with the nonsurgical Ponseti method consisting of three months of casting followed by four years of brace wear.18 However, the other affected individuals were all noted to have a severe form of clubfoot (Dimeglio grade IV)19 that required multiple surgeries to correct.
A variety of subtle skeletal abnormalities were seen frequently with the chromosome 17q23.1q23.2 microduplication, including enlargement of the distal fibular head (lateral malleolus) (Figure 3A); broad, shortened first and second metatarsals and shortened calcaneus (Figures 3A and 3C); tufted, triangular distal phalanx of the first toe (Figure 3C); and hypoplastic lateral distal tibial epiphysis (Figure 3D) (Table 1). None of these findings was noted by radiologists or treating orthopedic surgeons until the radiographs were evaluated carefully as a cohort with same genetic diagnosis. In the pelvis, the ischium was thickened in patients with microduplications (Figure 3E), in contrast to the hypoplastic ossification of the ischiopubic junction seen with the reciprocal microdeletion (Figure 3F).11 The ilium was often elongated and narrow with an absent iliac flare, giving it a moth-eaten appearance (Figure 3E). Coxa valga and genu valgum were present in many cases. The patella was unaffected, in contrast to the patellar hypoplasia that is common in patients with either the chromosome 17q23.1q23.2 microdeletion11 or small patella syndrome (MIM 147891) due to TBX4 gene (MIM 601719) mutations20 (Figure 3B). Infra-acetabular axe-cut notches were occasionally present and are similar to those seen in small patella syndrome (Figures 3E and 3F).21
Figure 3.
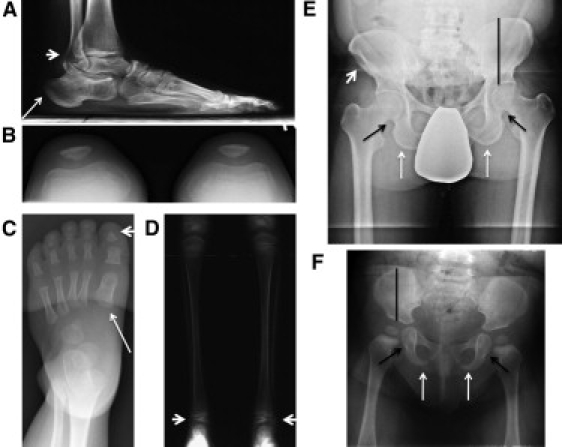
Skeletal Abnormalities Associated with Chromosome 17q23.1q23.2 CNVs in Individuals with Isolated Familial Clubfoot
All radiographic images are of patients with chromosome 17q23.1q23.2 microduplication except the image in (F), which is of an individual with a microdeletion.
(A) Large distal fibular head (lateral malleolus) (arrowhead) and shortened calcaneus (arrow) in proband's father (5103-03) imaged at age 50.
(B) Normal patella in proband (5103-01) at 7 years of age.
(C) Short, broad first metacarpal (arrow) and triangular, tufted first phalanx (arrowhead) in foot of a proband (5377-01) at 3 years of age.
(D) Bilateral hypoplastic lateral distal tibial epiphysis in proband (5103-01) at 7 years of age.
(E) Pelvis of proband's father (5103-03) showing thickened ischium (white arrow), infra-acetabular axe-cut notches (black arrow), absent normal iliac flare with moth-eaten appearance (arrowhead), and tall, narrow ilium (black line).
(F) Comparison image of pelvis of proband with microdeletion (5046-01) at 9 months of age showing hypoplastic ischium (white arrow), small infra-acetabular axe-cut notches (black arrow), and short, wide iliac bone (black line).
Few recurrent CNVs have previously been associated with isolated human birth defects.22,23 In this study, we provide evidence supporting a role for a recurrent chromosome 17q23.1q23.2 microduplication in the etiology of isolated clubfoot. Because this interval is flanked by segmental duplications that are likely to mediate nonallelic homologous recombination, this CNV is likely to be recurrent in the population. Because few genes have previously been implicated in clubfoot pathogenesis, this discovery represents the most common cause of isolated clubfoot identified to date. Clinical abnormalities related to chromosome 17q23.1q23.2 microduplications are limited to the lower extremity and typically consist only of isolated clubfoot. This phenotype differs significantly from that of the reciprocal microdeletion, in which cognitive deficits and heart defects play a major role, in addition to limb abnormalities.11 Despite our identification of a chromosome 17q23.2q23.2 microdeletion in a pair of affected siblings with clubfoot, clubfoot appears to be an unusual manifestation of the chromosome 17q23.1q23.2 microdeletion because it was not reported to be present in any of the seven previously described patients.11 Notably, those cases were identified in a series of > 19,000 patients that presumably consist of individuals with multiple congenital anomalies or neurocognitive deficits, and therefore the associated phenotype may reflect ascertainment bias. Our data support a role for recurrent CNVs in the etiology of isolated birth defects and justify the inclusion of these patient populations in future research studies.
Clubfoot occurring in the context of chromosome 17q23.1q23.2 CNVs is most likely a consequence of altered gene dosage of at least one of the two T-box transcription factors, TBX2 (MIM 600747) and TBX4, located within the 2.2 Mb interval. The TBX4 gene is expressed preferentially in the hindlimb compared to the forelimb,24,25 a pattern that makes TBX4 a prime candidate gene for lower-limb birth defects such as clubfoot. Furthermore, TBX4 is involved in limb muscle and tendon patterning26 and is critical for normal hindlimb development.26,27 Because transcription factors are exquisitely sensitive to gene dosage effects (reviewed in 28), they are therefore more likely than other genes to be involved in the pathogenesis of human disorders associated with CNVs. TBX4 haploinsufficiency, either as a result of chromosome 17q23.1q23.2 microdeletions or as a consequence of TBX4-inactivating mutations in small patella syndrome, results in variable and incompletely penetrant skeletal abnormalities, including hypoplastic patella, narrow toes, pes planus, hypoplastic ossification of the ischiopubic junction, and infra-acetabular axe-cut notches.11,20 To our knowledge, clubfoot has been described only once in an individual with small patella syndrome.21 In the current study, we find that alteration of gene dosage in the opposite direction in patients with chromosome 17q23.1q23.2 microduplications yields opposite effects, including short, broad toes, thickened ischia, and distinctly different pelvic morphology. However, some of the skeletal abnormalities including the infra-acetabular axe-cut notches are similar, highlighting the critical and complex effects of gene dosage on skeletal morphology. Though TBX2 and TBX4 are the most plausible candidate genes within the chromosome 17q23.1q23.2 CNV causing these skeletal abnormalities, the effect of duplications of the other 15 genes on the phenotype must also be considered. However, none of these genes is known to be involved in limb development.
Because the etiology of isolated clubfoot is poorly understood, this study provides additional data to support our hypothesis that clubfoot is due to abnormal early limb development. We previously demonstrated that bicoid-related homeodomain transcription factor PITX1 mutations are associated with a spectrum of congenital lower-limb abnormalities including isolated clubfoot in humans.8 Because the T-box transcription factor TBX4 is believed to be a direct transcriptional target of PITX1,29,30 the current data support a role for the PITX1-TBX4 pathway in the etiology of clubfoot. Although the skeletal abnormalities associated with chromosome 17q23.1q23.2 CNVs can be visualized radiographically, none of the identified abnormalities clearly explains the development of the severe distal limb contracture. One possibility is that the clubfoot phenotype arises predominantly from effects of these CNVs on soft tissues in the limb, given that Tbx4 loss markedly affects muscle and tendon patterning in mice.26 Studies are underway to evaluate, with magnetic resonance imaging, the soft-tissue abnormalities (i.e., muscle, tendon, and vasculature) associated with chromosome 17q23.2q23.2 CNVs in human patients.
We are intrigued by the possibility that the chromosome 17q23.1q23.2 CNVs, or genes contained within this region, may explain the gender-conditional expression of idiopathic clubfoot and/or developmental hip dysplasia. All individuals with chromosome 17q23.1q23.2 microduplication and clubfoot were male, a fact that is consistent with the 2–3-fold-higher incidence of idiopathic clubfoot in males.6 Furthermore, developmental dysplasia of the hip, a disorder that is four times more common in females,31 was only present in a single female. Additional families need to be studied to determine whether the chromosome 17q23.1q23.2 CNVs or genes contained within are responsible for the overall sex bias of these disorders.
Our identification of chromosome 17q23.1q23.2 CNVs in four patients out of 66 with familial isolated clubfoot opens up the possibility of clinical diagnostic testing in this patient population. Given the large size of this duplication, the chromosome 17q23.1q23.2 CNVs would be readily detected on any of the currently available tests for genomic CNVs (i.e., chromosomal microarray). Because the clubfeet associated with the chromosome 17q23.1q23.2 microduplication may be severe and refractory to conservative therapy, a genetic diagnosis may eventually alter treatment management, including casting or bracing methods. Diagnostic testing may also be useful to direct preventive care because of the possible increased risk of developmental dysplasia of the hip in families with chromosome 17q23.1q23.2 CNVs, though further study of the relationship between developmental dysplasia of the hip and these CNV is clearly needed. A higher incidence of developmental dysplasia of the hip in children with clubfoot has been found in some series5,6,32 but not others,4,33 suggesting that there may be a common etiology in some, but not all, cases. Diagnostic testing may also be useful to predict the risk of clubfoot recurrence in families. However, interpreting the significance of chromosome 17q23.1q23.2 CNVs is complicated by incomplete penetrance and variable expressivity, characteristics that are commonly described in CNVs associated with neurocognitive disorders.34–37 Incomplete penetrance suggests that additional genetic or environmental risk factors may be involved in clubfoot pathogenesis. Though our current study demonstrates that clubfoot is the predominant abnormality associated with chromosome 17q23.1q23.2 microduplications, accurate prediction of the frequency or severity of musculoskeletal abnormalities (i.e., clubfoot and developmental hip dysplasia) will not be possible until additional families with these CNVs are studied.
In summary, we have shown that four of 66 probands with familial isolated clubfoot carry a chromosome 17q23.1q23.2 CNV, three microduplications and one microdeletion, that segregates with clubfoot in all four families. This represents one of the few CNVs to be associated with an isolated birth defect and broadens the role of recurrent CNVs in human disease etiology. Though further study is needed, this discovery opens up the possibility of clinical genetic testing for patients with familial isolated clubfoot and provides important insight into the developmental pathway responsible for human limb birth defects.
Acknowledgments
This work was supported by grants from the Shriners Hospital for Children, the Children's Discovery Institute, the St. Louis Children's Hospital Foundation, the National Institute of Health (K12 HD001459), the March of Dimes Basil O'Connor Starter Scholar Research Award, the Orthopaedic Research and Education Foundation, and the Pediatric Orthopaedic Society of North America. We kindly thank Seth Crosby and Mike Heinz at the Washington University Genome Center for processing the Affymetrix microarrays. Genotyping of control samples was provided through the Genetic Association Information Network (GAIN). The control data set used for the analysis described in this manuscript was obtained from the Database of Genotype and Phenotype (dbGaP) through dbGaP accession number phs000017.v3.p1. Data for control samples were provided by John R. Kelsoe and John Nurnberger as part of the National Institute of Mental Health Bipolar Genetics Collaborative.
Contributor Information
Matthew B. Dobbs, Email: dobbsm@wudosis.wustl.edu.
Christina A. Gurnett, Email: gurnettc@neuro.wustl.edu.
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Database of Genomic Variants, http://projects.tcag.ca/variation/
Database of Genotype and Phenotype (dbGaP), http://www.ncbi.nlm.nih.gov/gap/
USCS Genome Browser, http://genome.ucsc.edu/
References
- 1.Wynne-Davies R. Genetic and environmental factors in the etiology of talipes equinovarus. Clin. Orthop. Relat. Res. 1972;84:9–13. doi: 10.1097/00003086-197205000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Gurnett C.A., Boehm S., Connolly A., Reimschisel T., Dobbs M.B. Impact of congenital talipes equinovarus etiology on treatment outcomes. Dev. Med. Child Neurol. 2008;50:498–502. doi: 10.1111/j.1469-8749.2008.03016.x. [DOI] [PubMed] [Google Scholar]
- 3.Brewer C., Holloway S., Zawalnyski P., Schinzel A., FitzPatrick D. A chromosomal deletion map of human malformations. Am. J. Hum. Genet. 1998;63:1153–1159. doi: 10.1086/302041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wynne-Davies R. Family studies and the cause of congenital club foot. Talipes equinovarus, talipes calcaneo-valgus and metatarsus varus. J. Bone Joint Surg. Br. 1964;46:445–463. [PubMed] [Google Scholar]
- 5.Lochmiller C., Johnston D., Scott A., Risman M., Hecht J.T. Genetic epidemiology study of idiopathic talipes equinovarus. Am. J. Med. Genet. 1998;79:90–96. [PubMed] [Google Scholar]
- 6.Chung C.S., Nemechek R.W., Larsen I.J., Ching G.H. Genetic and epidemiological studies of clubfoot in Hawaii. General and medical considerations. Hum. Hered. 1969;19:321–342. doi: 10.1159/000152236. [DOI] [PubMed] [Google Scholar]
- 7.Beals R.K. Club foot in the Maori: A genetic study of 50 kindreds. N. Z. Med. J. 1978;88:144–146. [PubMed] [Google Scholar]
- 8.Gurnett C.A., Alaee F., Kruse L.M., Desruisseau D.M., Hecht J.T., Wise C.A., Bowcock A.M., Dobbs M.B. Asymmetric lower-limb malformations in individuals with homeobox PITX1 gene mutation. Am. J. Hum. Genet. 2008;83:616–622. doi: 10.1016/j.ajhg.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ester A.R., Weymouth K.S., Burt A., Wise C.A., Scott A., Gurnett C.A., Dobbs M.B., Blanton S.H., Hecht J.T. Altered transmission of HOX and apoptotic SNPs identify a potential common pathway for clubfoot. Am. J. Med. Genet. A. 2009;149A:2745–2752. doi: 10.1002/ajmg.a.33130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ester A.R., Tyerman G., Wise C.A., Blanton S.H., Hecht J.T. Apoptotic gene analysis in idiopathic talipes equinovarus (clubfoot) Clin. Orthop. Relat. Res. 2007;462:32–37. doi: 10.1097/BLO.0b013e318073c2d9. [DOI] [PubMed] [Google Scholar]
- 11.Ballif B.C., Theisen A., Rosenfeld J.A., Traylor R.N., Gastier-Foster J., Thrush D.L., Astbury C., Bartholomew D., McBride K.L., Pyatt R.E. Identification of a recurrent microdeletion at 17q23.1q23.2 flanked by segmental duplications associated with heart defects and limb abnormalities. Am. J. Hum. Genet. 2010;86:454–461. doi: 10.1016/j.ajhg.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dick D.M., Foroud T., Flury L., Bowman E.S., Miller M.J., Rau N.L., Moe P.R., Samavedy N., El-Mallakh R., Manji H. Genomewide linkage analyses of bipolar disorder: A new sample of 250 pedigrees from the National Institute of Mental Health Genetics Initiative. Am. J. Hum. Genet. 2003;73:107–114. doi: 10.1086/376562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McInnis M.G., Dick D.M., Willour V.L., Avramopoulos D., MacKinnon D.F., Simpson S.G., Potash J.B., Edenberg H.J., Bowman E.S., McMahon F.J. Genome-wide scan and conditional analysis in bipolar disorder: Evidence for genomic interaction in the National Institute of Mental Health genetics initiative bipolar pedigrees. Biol. Psychiatry. 2003;54:1265–1273. doi: 10.1016/j.biopsych.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Diméglio A., Bensahel H., Souchet P., Mazeau P., Bonnet F. Classification of clubfoot. J. Pediatr. Orthop. B. 1995;4:129–136. doi: 10.1097/01202412-199504020-00002. [DOI] [PubMed] [Google Scholar]
- 15.Dyer P.J., Davis N. The role of the Pirani scoring system in the management of club foot by the Ponseti method. J. Bone Joint Surg. Br. 2006;88:1082–1084. doi: 10.1302/0301-620X.88B8.17482. [DOI] [PubMed] [Google Scholar]
- 16.Scher D.M., Feldman D.S., van Bosse H.J., Sala D.A., Lehman W.B. Predicting the need for tenotomy in the Ponseti method for correction of clubfeet. J. Pediatr. Orthop. 2004;24:349–352. doi: 10.1097/00004694-200407000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Dobbs M.B., Rudzki J.R., Purcell D.B., Walton T., Porter K.R., Gurnett C.A. Factors predictive of outcome after use of the Ponseti method for the treatment of idiopathic clubfeet. J. Bone Joint Surg. Am. 2004;86-A:22–27. doi: 10.2106/00004623-200401000-00005. [DOI] [PubMed] [Google Scholar]
- 18.Ponseti I.V., Smoley E.N. Congenital club foot: The results of treatment. J. Bone Joint Surg. 1963;45:261–344. [Google Scholar]
- 19.Flynn J.M., Donohoe M., Mackenzie W.G. An independent assessment of two clubfoot-classification systems. J. Pediatr. Orthop. 1998;18:323–327. [PubMed] [Google Scholar]
- 20.Bongers E.M., Duijf P.H., van Beersum S.E., Schoots J., Van Kampen A., Burckhardt A., Hamel B.C., Losan F., Hoefsloot L.H., Yntema H.G. Mutations in the human TBX4 gene cause small patella syndrome. Am. J. Hum. Genet. 2004;74:1239–1248. doi: 10.1086/421331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bongers E.M., Van Bokhoven H., Van Thienen M.N., Kooyman M.A., Van Beersum S.E., Boetes C., Knoers N.V., Hamel B.C. The small patella syndrome: Description of five cases from three families and examination of possible allelism with familial patella aplasia-hypoplasia and nail-patella syndrome. J. Med. Genet. 2001;38:209–214. doi: 10.1136/jmg.38.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greenway S.C., Pereira A.C., Lin J.C., DePalma S.R., Israel S.J., Mesquita S.M., Ergul E., Conta J.H., Korn J.M., McCarroll S.A. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat. Genet. 2009;41:931–935. doi: 10.1038/ng.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mefford H.C., Clauin S., Sharp A.J., Moller R.S., Ullmann R., Kapur R., Pinkel D., Cooper G.M., Ventura M., Ropers H.H. Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am. J. Hum. Genet. 2007;81:1057–1069. doi: 10.1086/522591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodriguez-Esteban C., Tsukui T., Yonei S., Magallon J., Tamura K., Izpisua Belmonte J.C. The T-box genes Tbx4 and Tbx5 regulate limb outgrowth and identity. Nature. 1999;398:814–818. doi: 10.1038/19769. [DOI] [PubMed] [Google Scholar]
- 25.Takeuchi J.K., Koshiba-Takeuchi K., Matsumoto K., Vogel-Höpker A., Naitoh-Matsuo M., Ogura K., Takahashi N., Yasuda K., Ogura T. Tbx5 and Tbx4 genes determine the wing/leg identity of limb buds. Nature. 1999;398:810–814. doi: 10.1038/19762. [DOI] [PubMed] [Google Scholar]
- 26.Hasson P., DeLaurier A., Bennett M., Grigorieva E., Naiche L.A., Papaioannou V.E., Mohun T.J., Logan M.P. Tbx4 and tbx5 acting in connective tissue are required for limb muscle and tendon patterning. Dev. Cell. 2010;18:148–156. doi: 10.1016/j.devcel.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naiche L.A., Papaioannou V.E. Tbx4 is not required for hindlimb identity or post-bud hindlimb outgrowth. Development. 2007;134:93–103. doi: 10.1242/dev.02712. [DOI] [PubMed] [Google Scholar]
- 28.Veitia R.A., Birchler J.A. Dominance and gene dosage balance in health and disease: Why levels matter! J. Pathol. 2010;220:174–185. doi: 10.1002/path.2623. [DOI] [PubMed] [Google Scholar]
- 29.Logan M., Tabin C.J. Role of Pitx1 upstream of Tbx4 in specification of hindlimb identity. Science. 1999;283:1736–1739. doi: 10.1126/science.283.5408.1736. [DOI] [PubMed] [Google Scholar]
- 30.Menke D.B., Guenther C., Kingsley D.M. Dual hindlimb control elements in the Tbx4 gene and region-specific control of bone size in vertebrate limbs. Development. 2008;135:2543–2553. doi: 10.1242/dev.017384. [DOI] [PubMed] [Google Scholar]
- 31.Lewis K., Jones D.A., Powell N. Ultrasound and neonatal hip screening: the five-year results of a prospective study in high-risk babies. J. Pediatr. Orthop. 1999;19:760–762. [PubMed] [Google Scholar]
- 32.Carney B.T., Vanek E.A. Incidence of hip dysplasia in idiopathic clubfoot. J. Surg. Orthop. Adv. 2006;15:71–73. [PubMed] [Google Scholar]
- 33.Westberry D.E., Davids J.R., Pugh L.I. Clubfoot and developmental dysplasia of the hip: Value of screening hip radiographs in children with clubfoot. J. Pediatr. Orthop. 2003;23:503–507. [PubMed] [Google Scholar]
- 34.Stefansson H., Ophoff R.A., Steinberg S., Andreassen O.A., Cichon S., Rujescu D., Werge T., Pietiläinen O.P., Mors O., Mortensen P.B., Genetic Risk and Outcome in Psychosis (GROUP) Common variants conferring risk of schizophrenia. Nature. 2009;460:744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Bon B.W., Mefford H.C., Menten B., Koolen D.A., Sharp A.J., Nillesen W.M., Innis J.W., de Ravel T.J., Mercer C.L., Fichera M. Further delineation of the 15q13 microdeletion and duplication syndromes: A clinical spectrum varying from non-pathogenic to a severe outcome. J. Med. Genet. 2009;46:511–523. doi: 10.1136/jmg.2008.063412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharp A.J., Mefford H.C., Li K., Baker C., Skinner C., Stevenson R.E., Schroer R.J., Novara F., De Gregori M., Ciccone R. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat. Genet. 2008;40:322–328. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]