

Abstract
X-linked cone and cone-rod dystrophies (XLCOD and XLCORD) are a heterogeneous group of progressive disorders that solely or primarily affect cone photoreceptors. Mutations in exon ORF15 of the RPGR gene are the most common underlying cause. In a previous study, we excluded RPGR exon ORF15 in some families with XLCOD. Here, we report genetic mapping of XLCOD to Xq26.1-qter. A significant LOD score was detected with marker DXS8045 (Zmax = 2.41 [θ = 0.0]). The disease locus encompasses the cone opsin gene array on Xq28. Analysis of the array revealed a missense mutation (c. 529T>C [p. W177R]) in exon 3 of both the long-wavelength-sensitive (LW, red) and medium-wavelength-sensitive (MW, green) cone opsin genes that segregated with disease. Both exon 3 sequences were identical and were derived from the MW gene as a result of gene conversion. The amino acid W177 is highly conserved in visual and nonvisual opsins across species. We show that W177R in MW opsin and the equivalent W161R mutation in rod opsin result in protein misfolding and retention in the endoplasmic reticulum. We also demonstrate that W177R misfolding, unlike the P23H mutation in rod opsin that causes retinitis pigmentosa, is not rescued by treatment with the pharmacological chaperone 9-cis-retinal. Mutations in the LW/MW cone opsin gene array can, therefore, lead to a spectrum of disease, ranging from color blindness to progressive cone dystrophy (XLCOD5).
Introduction
X-linked cone dystrophy (XLCOD) and X-linked cone-rod dystrophy (XLCORD) are a heterogeneous group of disorders in which there is dysfunction and degeneration of cone photoreceptors, often followed by later rod dysfunction.1 Affected males present in childhood or early adult life with a variable degree of photophobia, reduced central vision, and color-vision disturbance. Later in the disease there may be nyctalopia and peripheral-field loss.1 Mutations in the RPGR gene (MIM 312610) on Xp21 are the most common known cause of XLCOD/XLCORD (MIM 304020; COD1/ CORDX1), and all of the reported mutations are found in exon ORF15.2–5 XLCORD (COD3/COD4/CORDX3 [MIM 300476]) has also been described in one family from Finland with a mutation in the CACNA1F gene (MIM 300110) on Xp11.23, and an additional locus has been reported to map within the interval bounded by markers DXS292 and DXS1113 on Xq27 (MIM 300085; COD2/CORDX2).6,7
Here, we describe mapping of the disease interval in an XLCOD family to Xq26.1-qter. We subsequently identified the causative gene mutation as a missense mutation (c. 529T>C [p.W177R]) in both the long-wavelength-sensitive (LW) and the medium-wavelength-sensitive (MW) cone opsin genes, and we investigated the functional consequence of this mutation.
Subjects and Methods
Patients and Clinical Assessment
The protocol of the study adhered to the provisions of the Declaration of Helsinki and was approved by the local ethics committee at Moorfields Eye Hospital. A three-generation British family, consisting of six affected male subjects and three obligate females, was ascertained as having XLCOD (Figure 1). Four affected individuals and two obligate carrier females were available for detailed assessment. Clinical notes and fundus images were reviewed for the two remaining affected males. After informed consent was obtained, blood samples were taken from family members and genomic DNA was extracted by standard techniques. A full medical history was taken and an ophthalmological examination was performed. Affected male subjects and obligate carrier females underwent color fundus photography, fundus autofluorescence imaging (HRA2, Heidelberg Engineering, Heidelberg, Germany), color-vision testing, and electrophysiological assessment, which included a full-field electroretinogram (ERG) and pattern ERG (PERG), incorporating the standards of the International Society for Clinical Electrophysiology of Vision (ISCEV).8,9 A dark-adapted “bright flash” ERG was additionally recorded to a flash strength of 11.5 cd.s.m−2, better to demonstrate the photoreceptor-mediated a-wave. Long-duration ON-OFF ERGs used an orange stimulus (560 cd.m−2, duration 200 ms) superimposed on a green background (150 cd.m−2). Short-wavelength flash ERGs used a blue stimulus (5 ms in duration, 445 nm, 80 cd.m−2) on an orange background (620 nm, 560 cd.m−2).10 One affected male was unable to tolerate corneal electrodes, and full-field ERGs were recorded with the use of lower-eyelid-skin electrodes (Table 1).11 Color-vision tests were also carried out with the use of Ishihara pseudoisochromatic plates, Hardy, Rand, and Rittler (HRR) plates (American Optical Company, NY), the City University color vision test, the Farnsworth-Munsell (FM) D-15 and FM-100-hue tests, and Rayleigh and Moreland anomaloscope matches. The FM 100-hue test and other plate tests were performed under CIE Standard Illuminant C from a MacBeth Easel lamp or Illuminant D50 (daylight) from a daylight lighting booth. Observer II:1 was unable to perform these tests because of his very poor visual function.
Figure 1.
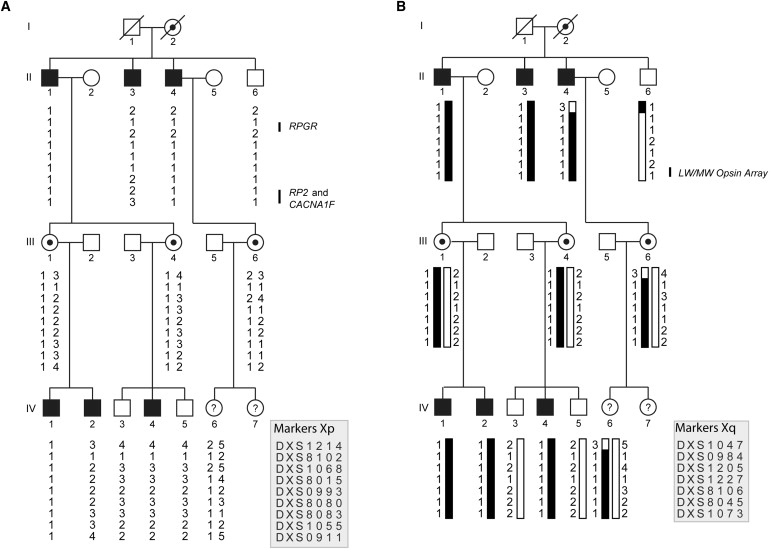
Pedigree and Haplotype Analysis of the XLCOD Family, Defining the Critical Interval as Xq26.1-qter
(A) Haplotypes for markers on Xp show that the disease does not segregate with known disease genes (RPGR, RP2, and CACNA1F).
(B) XLCOD segregates with markers on Xq26.1-qter. The affected haplotype is shaded black. Individuals II:4, II:6, and III:6 have a recombination between markers DXS1047 and DXS984, defining the proximal boundary of disease. No distal recombinations were identified; thus, the critical interval is defined as DXS1047-Xqter. The location of the LW/MW opsin array is indicated. Haplotypes are shown according to marker order on the X chromosome, as indicated in the key. Filled symbol, affected; empty symbol, unaffected; circles with dots, obligate carrier females; ?, affectation status unknown.
Table 1.
Summary of Clinical Findings
| Case | Sex | Age | VA: OD, OS | Refraction: OD, OS | Fundus | Psychophysical Testing | PERG | ERGs |
|---|---|---|---|---|---|---|---|---|
| IV:1 | M | 27 | 6/24, 6/36 | −5.5/−1.75 × 45, −4.5/−1.5 × 135 | mild bilateral macular RPE disturbance with peripheral pigmentary and atrophic changes | minimal evidence for M- or L-cone function, good S-cone function | undetectable: severe macular dysfunction | severe bilateral generalized cone dysfunction, undetectable S-cone ERG |
| IV:2 | M | 26 | 6/36, 6/36 | −5.0/−1.75 × 100, −4.5/−1.75 × 90 | bilateral macular RPE disturbance | no evidence for M- or L-cone function, good S-cone function | undetectable: severe macular dysfunction (skin electrodes) | severe generalized cone dysfunction, undetectable S-cone ERG (skin electrodes) |
| IV:4 | M | 14 | 6/18, 6/18 | −5.75/−2.5 × 180, −7.0/−1.75 × 180 | mild bilateral macular RPE disturbance | no evidence for M- or L-cone function, good S-cone function | undetectable: severe macular dysfunction | severe generalized cone dysfunction with preserved low amplitude S-cone ERG |
| III:1 | F | 56 | 6/9, 6/9 | −3.0/−0.5 × 180, −3.0/−1.0 × 90 | bilateral macular RPE changes and drusen | reduced L-cone sensitivity, normal on color vision tests | reduced: moderate macular dysfunction | mild generalized cone dysfunction with undetectable S-cone ERG |
| III:4 | F | 43 | 6/6, 6/5 | −5.0/−0.75 × 175, −4.0/−2.25 × 35 | mild bilateral macular RPE changes | reduced L-cone sensitivity, normal on color vision tests | reduced multifocal ERGs: mild macular dysfunction | mild generalized cone dysfunction, with relatively preserved S-cone ERGs |
| II:1 | M | 82 | CF, CF | −3.0/−1.5 × 80, −3.0/−2.25 × 90 | bilateral, well-demarcated macular atrophy | no evidence for M- or L-cone function, minimal residual S-cone function | undetectable: severe macular dysfunction | severe generalized cone dysfunction with relatively preserved S-cone ERG |
| II:3 | M | 80 | 6/60, CF | −4.0/−1.0 × 65, −3.5/−1.5 × 90 | bilateral, well-demarcated macular atrophy | NP | NP | NP |
| II:4 | M | 72 | 6/60, 6/60 | −7.0/−1.0 × 180, −7.0/−1.0 × 180 | bilateral, well-demarcated macular atrophy | NP | NP | NP |
Abbreviations are as follows: CF, count fingers; ERG, electroretinogram; NP, not performed; PERG, pattern ERG; RPE, retinal pigment epithelium; VA, visual acuity.
Psychophysical Experiments
The psychophysical measurements were made with the use of a Maxwellian-view optical system, as described previously.12–14 In all experiments, a flickering target of 4° of visual angle in diameter was presented in the center of a 9° diameter background field. Fixation was central. Each data point for the individual measurements represents the average of at least three settings. For L-cone critical flicker fusion (cff) measurements, the wavelength of the target was fixed at 650 nm, and its radiance varied from 6.5 to 11.0 log10 quanta s−1 deg−2 in steps of about 0.3 log10 units. The target was superimposed in the center of a 481 nm background that delivered 8.29 log10 quanta s−1 deg−2 at the cornea. This background served mainly to suppress rods, but also selectively desensitized M-cones at lower target radiances. For S-cone cff measurements, the wavelength of the target was fixed at 440 nm, and its radiance was varied from 6.5 to 11.0 log10 quanta s−1 deg−2. The target was superimposed in the center of a 620 nm background of 11.35 log10 quanta s−1 deg−2. The 620 nm background field selectively desensitized the M- and L-cones, but it had comparatively little direct effect on the S-cones. For normal observers, this field isolates the S-cone response up to a 440 nm target radiance of about 10.0 log10 quanta s−1 deg−2, as also evidenced by the change in slope of the normal S-cone cff near that radiance, above which M-cones contribute to flicker detection.15–17 For cff measurements, the observers adjusted the flicker frequency to find the frequency at which the flicker just disappeared (this frequency is known as the “critical flicker fusion” frequency). For spectral-sensitivity measurements, the wavelength of the target was varied in approximately 30 nm steps. The target was sinusoidally flickered at 5 Hz and was superimposed in the center of the same 481 nm background used in the L-cone cff measurements. At each target wavelength, the observer adjusted the target radiance to find the radiance at which the flicker just disappeared (this radiance is known as the “flicker threshold”).
Haplotype and Linkage Analysis
Genotyping was performed with the use of microsatellite markers across the X chromosome, according to the manufacturer's protocols (ABI prism linkage mapping set version 2.5 and additional microsatellite markers; primers and conditions available upon request). Two-point linkage analysis for XLCOD and informative markers was performed for calculation of LOD scores with the use of MLINK version 5.1 (Columbia University, NY). The XLCOD disease-allele frequency was set at 0.0001. The penetrance value for carrier females was set at 0.0000. Alleles at marker loci were assumed to have equal frequency. Genetic distance and marker order used for haplotype analysis and two-point linkage analysis were assigned according to genetic and physical maps obtained from NCBI and UCSC.
PCR and Sequence Analysis of the Cone Opsin Array
The cone opsin gene array is composed of a locus control region (LCR) that controls transcription of one long-wavelength-sensitive (red) opsin gene (OPN1LW [MIM 303900], herein referred to as LW) and one medium-wavelength-sensitive (green) opsin gene (OPN1MW [MIM 303800], herein referred to as MW). The LW and MW genes in the genomic array are organized in a head-to-tail tandem arrangement with a single LW opsin gene in a 5′ position, followed by one or more MW opsin genes.18,19 The number of MW genes in the array is polymorphic. However, only the first MW gene is expressed.20–22 Genomic DNA from an affected male (subject IV:2) was screened for mutations in the LCR and exons 1 to 6 of the LW/MW opsin gene array by PCR amplification and direct sequencing, as described previously.23 In the initial screen, common primer pairs designed to coamplify both LW and MW genes were used. Sequences were compared with reference sequences. Sequence electropherograms of these amplicons revealed a dual peak at sites of known nucleotide variation between the LW and MW genes. The coding sequence of exons 1 and 6 of LW and MW are identical; however, sequence upstream of the start codon of exon 1 harbors nucleotide differences that facilitated identification of both MW and LW exon 1. After the preliminary screen of the cone opsin array, exons 2, 3, and 4 of both genes were further studied with the use of LW- and MW-gene-specific primers (primer sequences and conditions available on request). These specific primers were used to confirm the presence or absence of an exon in the array. The specific primers were then paired with the coamplification primers in a series of long-range PCR amplifications to determine the structure of the array and the relationship of each LW or MW exon to its 5′ and 3′ neighbor (primer sequences and conditions available on request). High-fidelity PCR master mix (Abgene, Thermo Fisher Scientific) was used for PCR reactions according to the manufacturer's protocols. PCR products were then purified enzymatically with ExoSAP-IT (GE Healthcare) and bidirectionally sequenced (Applied Biosystems) according to the manufacturer's protocols. The sequence was examined with Lasergene DNA Star software (DNA Star). Identified sequence changes were tested for segregation with disease in the family. PCR products of exon 3 (coamplification of both LW and MW) for over 200 control chromosomes were analyzed for the mutation with the use of an MspI (New England Biolabs) restriction digest assay that was diagnostic for the presence of the mutation.
Constructs, Antibodies, and Reagents
To create cone opsin constructs with a 1D4 epitope for transient transfection, we subcloned 1D4-tagged green (MW) opsin with a C203R mutation from pGrnC203R24 into pBK-CMV. Using site-directed mutagenesis (Quickchange, Stratagene), we produced a wild-type (WT) green opsin construct and subsequently created a construct with the W177R mutation (primers available on request). Untagged and green fluorescent protein (GFP)-tagged WT rod opsin and untagged and GFP-tagged P23H mutant rod opsin were generated as previously described.25 Site-directed mutagenesis was used to introduce the W161R mutation in untagged and GFP-tagged rod opsin (primers available on request). The primary antibodies used were 1D4 and anti-calnexin (C4731, Sigma). The secondary antibodies used were HRP-conjugated goat anti-mouse (Pierce) and Alexa Fluor 488 anti-mouse or Alexa Fluor 594 anti-rabbit (Invitrogen). 9-cis-retinal was from Sigma and Endo H from New England Biolabs.
Immunoblotting and Immunocytochemistry
SK-N-SH cells were maintained and transfected essentially as previously described.26,27 In brief, in the rescue experiments, 10 μM 9-cis-retinal was added after transfection and the cells were maintained in the dark. For immunoblotting, cells were lysed 24 hr after transfection with 0.1% n-dodecyl-β-D-maltoside (DM) buffer with a protease-inhibitor cocktail (Sigma) in PBS, and cleared lysates were resolved on 10% SDS-PAGE gels. Immunodetection of both cone opsin and rod opsin was carried out with the use of the 1D4 antibody. For deglycosylation experiments, lysates were treated with Endo H for 2 hr at 37°C prior to SDS-PAGE. For immunocytochemistry, cells were seeded on 8-well permanox chamber slides and transfected with GFP-tagged rod opsin constructs or 1D4 epitope-tagged cone opsin constructs. Twenty-four hours after transfection, cells were fixed with 4% paraformaldehyde and permeabilized in 0.1% Triton X-100. Cells transfected with cone opsins were incubated with 1D4 (0.5 μg/ml) and anti-calnexin (1:600), followed by secondary antibodies Alexa Fluor 488 anti-mouse and Alexa Fluor 594 anti-rabbit (1:2000). Images were taken with a Zeiss LSM 700 laser scanning confocal microscope. The images were exported from Zen 2009 Light Edition, and figures were prepared with Adobe Photoshop and Illustrator CS2.
Results
Assessment of Phenotype
Affected subjects were aged between 14 and 82 yrs and complained of reduced central vision starting in the first decade, with subsequent gradual deterioration of visual acuity (VA) and color vision. Clinical findings are summarized in Table 1. VA ranged from 6/18 to counting fingers. All affected males were myopic. One subject had nystagmus (26 yrs old), and the remaining subjects did not report a history of nystagmus. A range of macular appearances was seen, varying from mild retinal pigment epithelial (RPE) changes in the younger subjects to extensive macular atrophy in the older generation (Figure 2A). Autofluorescence imaging was within normal limits in the three youngest affected subjects. From the older generation, subject II:1 was available for imaging and had evidence of bilateral perimacular rings of increased autofluorescence. Both obligate carriers were asymptomatic and had mild bilateral macular RPE changes; although in one (III:1) these were associated with drusen and may therefore be age-related. Autofluorescence imaging was unremarkable in both carriers. An asymptomatic subject, IV:6, was also reviewed and had a normal ocular examination.
Figure 2.
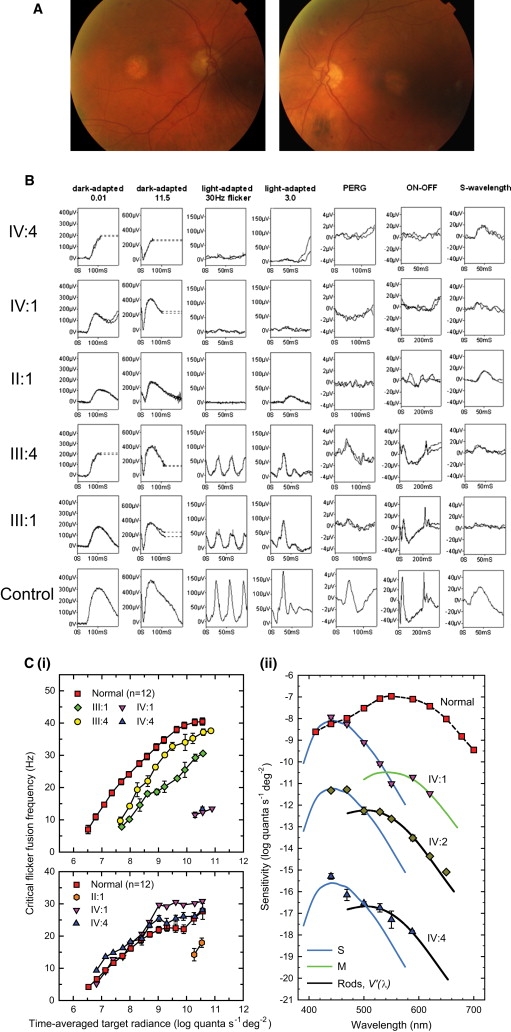
Phenotype of the XLCOD5 Family
(A) Color fundus photographs of affected male patient II:3 (80 yrs old), showing symmetrical bilateral macular atrophy.
(B) PERG and full-field ERGs for affected males IV:4, IV:1, and II:1 and obligate carrier females III:1 and III:4, as indicated, compared with those from a representative normal subject (bottom row). Recordings were performed with gold-foil corneal electrodes. Broken lines replace eye-movement artifacts.
(C) L-cone cff measurements (i, upper panel) for two affected males (IV:1 and IV:4) and two obligate carriers (III:1 and III:4), showing significant loss of L- and M-cone sensitivity in affected males and reduced L-cone sensitivity in both carriers as compared to controls. S-cone critical flicker fusion measurements (i, lower panel) for three affected males (IV:1, IV:4, and II:1), showing significant S-cone sensitivity loss in the eldest affected male (II:1) as compared to controls. Spectral-sensitivity measurements (ii) for the three youngest affected males (IV:1, IV:2, and IV:4), showing substantial losses of flicker sensitivity at middle and long wavelengths, but sensitivities at short wavelengths that are nearly normal.
Error bars represent ±1 SEM.
All four affected subjects (IV:1, IV:2, IV:4, and II:1) had severely abnormal photopic full-field ERGs consistent with severe generalized cone system dysfunction (Figure 2B). Dark-adapted ERGs to a dim flash were normal (seven eyes) or borderline subnormal (one eye; IV:2), in keeping with preserved or relatively preserved rod system function. The scotopic bright flash ERG (ISCEV “suggested”; mixed rod-cone response) for II:1 and IV:4 showed mild a-wave reduction. All four had undetectable PERGs consistent with severe macular dysfunction. ON-OFF ERGs were severely subnormal in all cases. Short-wavelength flash ERGs, normally characterized by early (L-cone and M-cone system) and late (S-cone system) components, were subnormal in two subjects (IV:1 and IV:2) and were preserved in subjects II:1 and IV:4 (Figure 2B). Both obligate carriers (III:1 and III:4) had electrophysiological evidence of mild generalized cone system dysfunction with moderate macular involvement (PERG in III:1 and multifocal ERG in III:4) (Figure 2B).
Color vision testing performed in the three youngest affected patients (IV:1, IV:2, and IV:4) revealed evidence of good tritan discrimination with no measurable discrimination along protan or deutan axes. No discernable color discrimination was possible in the eldest affected male (II:1) because of poor VA; however, psychophysical experiments detailed below established that he had only minimal residual S-cone function at the macula. The obligate carrier females performed normally on all standard color vision tests.
Psychophysical Data
L-cone cff measurements28,29 for two affected males (IV:1 and IV:4) showed a devastating loss of L- and M-cone sensitivity (Figure 2C). The target had to be set 104 times brighter than the setting for normal observers in order for the affected males to first see flicker. Control experiments carried out on subject IV:1 suggested that detection at these high levels may be partially mediated by rods. Consistent with a partial loss of L-cone function, both of the carrier females (III:1 and III:4) show some loss in cff as compared to normal. The loss in III:1 was more than that of her younger sister, III:4, averaging at about 10 Hz across levels. S-cone cff15–17 showed that the two younger affected males (IV:1 and IV:4) have normal S-cone function. By contrast, the older affected male (II:1) showed a considerable S-cone sensitivity loss, with flicker seen only at the highest radiances (Figure 2C).
Spectral-sensitivity measurements were made in the three youngest affected males. The data for IV:2 and IV:4 have been shifted down for clarity by 3.5 and 7 log10 units, respectively (Figure 2C). The S- and M-cone and rod [or V'(λ)] spectral-sensitivity curves fitted to the data for the affected males are standard functions.30,31 Compared to the normal, all three subjects showed a substantial loss of flicker sensitivity at middle and long wavelengths, but their sensitivities at short wavelengths were nearly normal (Figure 2C). Detection at short wavelengths was consistent with detection by the S-cones. Detection at longer wavelengths is mediated by rods in IV:2 and IV:4. The data for IV:1, however, suggest that his rods are more insensitive. These psychophysical data cannot entirely exclude the possibility that some residual L- or M-cone function remains in the affected younger males, particularly in the case of observer IV:1. If there is any residual function, however, it must be attenuated by more than 4 log10 units compared to normal.
Defining the Disease Interval for XLCOD
Screening of RPGR in an affected male from this family did not reveal a mutation.4 We then performed X chromosome haplotype analysis in order to define the genetic interval segregating with cone dystrophy in the family. Haplotype analysis demonstrated that markers on Xp did not segregate with disease (Figure 1A) and excluded RPGR, RP2 (MIM 300757), and CACNA1F as candidate genes. A common haplotype that segregated with disease was identified in affected males and obligate carrier females on Xq (Figure 1B). The proximal boundary of the genetic interval was defined by a recombination between markers DXS1047 and DXS984, which mapped the disease locus in this family to Xq26.1-qter, spanning approximately 26 Mb and containing 388 genes. Two-point LOD scores were calculated for markers on Xq (Table 2), and a significant LOD score was generated for marker DXS8045, Zmax = 2.41 (θ = 0), on Xq27.3. The cone opsin gene array on Xq28 was thereby a positional candidate for disease.
Table 2.
Two-Point Linkage Analysis between XLCOD and Microsatellites on the X Chromosome
|
LOD Score at Recombination Fraction (θ) of: |
|||||||
|---|---|---|---|---|---|---|---|
| Locus | Position ChrX | 0.00 | 0.05 | 0.10 | 0.20 | 0.30 | 0.40 |
| Xp | |||||||
| DXS1214 | 31160748 | −∞ | −2.88 | −1.77 | −0.77 | −0.30 | −0.07 |
| DXS8102 | 38118315 | −0.05 | −0.03 | −0.02 | −0.01 | −0.00 | −0.00 |
| DXS1068 | 38807989 | −∞ | −2.88 | −1.77 | −0.77 | −0.3 | −0.07 |
| DXS8015 | 38987415 | −∞ | −1.53 | −0.94 | −0.40 | −0.15 | −0.04 |
| DXS0993 | 41047637 | −∞ | −1.47 | −0.91 | −0.39 | −0.15 | −0.03 |
| DXS8080 | 44143324 | −∞ | −1.5 | −0.92 | −0.40 | −0.15 | −0.04 |
| DXS8083 | 45141390 | −∞ | −2.88 | −1.77 | −0.77 | −0.3 | −0.07 |
| DXS1055 | 46326364 | −∞ | −2.88 | −1.77 | −0.77 | −0.3 | −0.07 |
| DXS991 | 55419039 | −∞ | −2.88 | −1.77 | −0.77 | −0.3 | −0.07 |
| Xq | |||||||
| DXS1205 | 140162266 | 1.37 | 1.31 | 1.22 | 1.00 | 0.73 | 0.40 |
| DXS1227 | 140702381 | 0.90 | 0.81 | 0.72 | 0.52 | 0.30 | 0.09 |
| DXS8106 | 142084035 | 1.41 | 1.33 | 1.24 | 1.01 | 0.73 | 0.40 |
| DXS8045 | 145412040 | 2.41 | 2.21 | 2.00 | 1.54 | 1.03 | 0.49 |
| DXS1073 | 153728848 | 1.45 | 1.36 | 1.26 | 1.04 | 0.73 | 0.40 |
Identification of a Missense Mutation in Exon 3 of the MW Cone Opsin Gene
In the preliminary screen of the opsin array, the LCR and exons 1–6 of the LW/MW opsins of an affected individual (IV:2) were amplified and sequenced. Sequence analysis demonstrated the presence of an intact LCR and the presence of exons 1, 2, 4, 5, and 6 of the LW and MW genes (Figure S1, available online). At nucleotide c.706 in exon 4, a single peak was observed (Figure S1), representing an A>G transition in the LW gene resulting in the known SNP p.M236V. Interestingly, sequence analysis of exon 3 revealed a single MW-derived sequence (Figure S1 and Figure 3B). The exon was identified as MW, because single peaks representing MW gene common alleles were present at four positions (c.A453, c.A457 [p.M153], c.C465, c.G538 [p.A180]) out of a possible six nucleotide differences between MW and LW genes in exon 3. The allele c.G538 encodes the MW gene spectral-tuning amino acid p.A180 (Figure 3B).32 Two additional known nonsynonymous SNPs, c.521C>T (p.A174V) and c.532A>G (p.I178V), were also identified in MW exon 3 (Figure 3B). Importantly, a missense mutation, c.529T>C, was also detected in MW exon 3, creating a Trp-to-Arg change in a highly conserved residue at p.177 (p.W177R) (Figure 3B). This mutation, and the SNPs, segregated with disease in the family.
Figure 3.
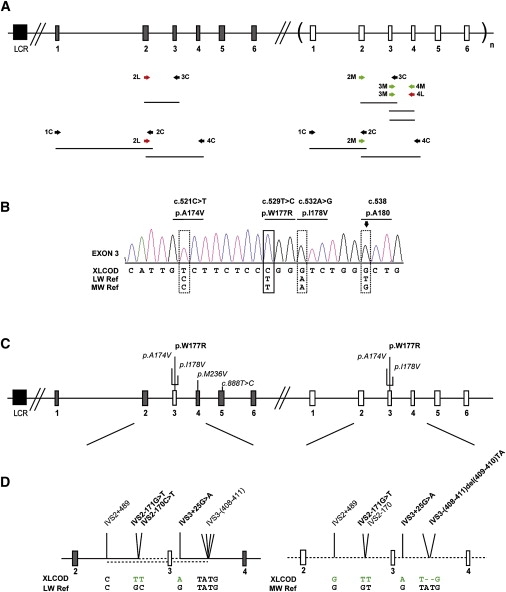
Molecular Analysis of the Cone Opsin Array and Identification of a c.529T>C Mutation in Exon 3 of the LW and MW Cone Opsin Genes
(A) The cone opsin array comprises an upstream LCR, an LW opsin gene, and one or more MW opsin genes lying in a 5′ to 3′ position in tandem on Xq28. Grey boxes represent LW opsin exons, and white boxes represent MW opsin exons. Exons are numbered below the figure. A subscript “n” represents one or more MW opsin genes. The black box represents the upstream LCR. After an initial screen of the LCR and coamplification of exons of the LW/MW genes, a strategy was developed to determine the precise opsin array structure in the XLCOD5 family. A combination of gene-specific primers (labeled “L” or “M”) and coamplification primers (labeled “C”) was used, depicted beneath the diagrammatical representation of the opsin array.
(B) Electropherogram of exon 3 in an affected male (IV:2). Reference sequences of both LW and MW genes are shown beneath the patient electropherogram (XLCOD). The dotted boxes represent known polymorphic nucleotide variants, with the resulting amino acid variant shown above. The lined box highlights the location of a c.529T>C missense mutation in exon 3 resulting in p.W177R. The arrow over a dotted box highlights a single G peak representing sequence derived from exon 3 of an MW gene only (p. A180). This indicates that the sequence changes shown are derived from an MW exon 3 only. Electropherograms for other exons (1, 2, 4, 5, and 6) in the affected male (IV:2) are shown in Figure S1.
(C) Diagrammatic representation of the cone opsin array in the XLCOD family. Both LW and MW opsin genes contain identical copies of mutant MW exon 3 with the c.529T>C missense mutation (p.W177R). Polymorphic and nonsynonymous substitutions are also indicated (italic).
(D) Detailed expansion of sequence between exons 2 and 4 of the LW and MW opsin genes in an affected male (XLCOD) compared to reference sequences. Identified nucleotide substitutions facilitated further characterization of the gene-conversion event leading to a mutated MW exon 3 in both the LW and MW genes in the opsin array. Dotted line indicates MW-gene-derived sequence, solid line indicates LW-gene-derived sequence. The upper and lower dotted lines in the LW opsin gene represent the minimal and maximal gene-conversion tracts, respectively. IVS2-171G>T and IVS3+25G>A present in the LW gene are derived from the MW gene, and they define the minimal gene-conversion interval surrounding the mutated MW exon 3.
Both the LW and MW Genes Harbor the Mutation
We developed a strategy to determine the organization of the cone opsin genes in this family. To test whether the LW copy of exon 3 was deleted, we designed LW-specific exon 3 primers. No PCR product was obtained for affected males. However, control subjects amplified successfully (data not shown), indicating that LW exon 3 was likely to be deleted. Because several array arrangements were possible (including LW/MW hybrid genes), long-range PCR was used (Figure 3A) to establish the genomic arrangement of the genes in the array. Specific amplification of MW exon 3 to MW exon 4 produced the expected 2 Kb fragment in controls and in affected and unaffected members of the XLCOD family. Importantly, specific amplification of MW exon 3 to LW exon 4 resulted in amplification of a 2 Kb fragment in affected members of the family, but not in controls (data not shown). This suggested that the affected individuals all had a gene in which MW exon 3 was attached to LW exon 4, in addition to a WT MW gene. Sequencing of both amplicons in affected family members confirmed the presence of an MW exon 3 to LW exon 4 hybrid gene and revealed that both the hybrid 3MW/4LW gene and the MW gene had the p.W177R mutation together with p.A174V and p.I178V SNPs (Figure 3C). The order of exons 1, 2, 3, and 4 in the array was established by direct sequencing of amplicons generated by pairing of LW and MW common and specific primers (Figure 3B). The length of intron 1 in the LW gene is ∼1282 bp larger than the MW gene. Thus, PCR generated two fragments of different sizes that were sequenced. This confirmed that LW exons 1 and 2 were attached to LW exon 4 but contained the mutant MW exon 3, and it also confirmed the presence of an MW gene containing the mutant MW exon 3 (Figure 3C). The cone opsin gene array in affected members of the XLCOD family therefore consists of an LW gene containing a W177R mutant MW exon 3, followed by an MW gene containing an identical W177R mutant MW exon 3 (Figure 3C). Analysis of the intervening sequence flanking the mutant MW exon 3 of both genes showed that the two genes shared a minimum identical region (or minimal converted tract) of MW-derived sequence of 492 bp, from IVS2-170 to IVS3+25 (Figure 3D). This indicates that W177R was transferred in a block of exon 3 sequence from the MW gene into the LW gene by gene conversion. This mutation was not detected in a population sample of over 200 chromosomes, equivalent to over 400 LW or MW genes.
Cellular and Biochemical Consequence of the W177R Opsin Mutation
W177 is located in transmembrane helix IV of red and green opsins (Figure 4A). This residue is highly conserved in all visual and nonvisual opsins (Figure 4B).33 Because of the small number of pathogenic mutations identified, there have been relatively few studies investigating the effect of amino acid substitutions on the function of cone opsins. Over 100 pathogenic mutations have been described in the rhodopsin gene (MIM 180380) as a cause of the degenerative retinal disease retinitis pigmentosa (RP); however, the equivalent residue in rhodopsin (W161) has not been associated with disease to date. We therefore investigated the effect of the tryptophan-to-arginine mutation in both cone opsin and rod opsin in transiently transfected SK-N-SH cells. A 1D4 epitope-tagged MW opsin construct was used to study the fate of the W177R mutation. We compared the MW W177R mutation to the previously characterized C203R mutation, known to cause blue cone monochromacy (BCM [MIM 303700]) and shown to result in protein misfolding and endoplasmic reticulum (ER) retention.24 We also studied the W161R rod opsin mutation in comparison to the common RP-causing P23H misfolding mutation.25,34
Figure 4.
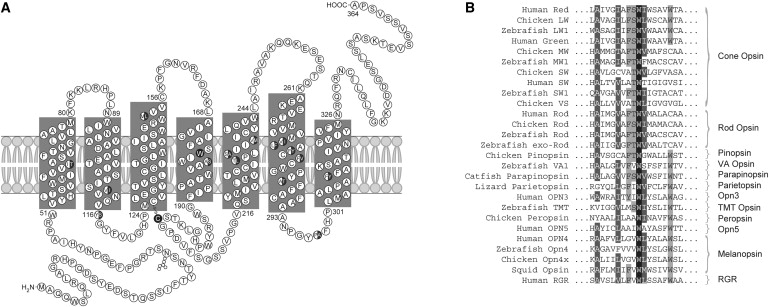
Secondary Structure of the Cone Opsins and Conservation of W177
(A) The secondary structure of the red (LW) and green (MW) cone opsins. Amino acid differences between the LW and MW opsins are shown as half-closed circles. W177 in transmembrane domain 4 and C203 are shown as a closed circle.
(B) Sequence alignment encompassing W177 of LW and MW opsins with known visual and nonvisual opsins in a variety of species, as named on the left. This tryptophan is 100% conserved in all opsins.
Immunoblot analysis of the WT MW opsin revealed a major band of approximately 40–50 kDa, with a 35 kDa species representing other glycosylated forms of the opsin and higher molecular weight aggregates near the top of the resolving gel (Figure 5A). The C203R and W177R MW proteins were expressed at lower levels in comparison to WT. Treatment with MG132 increased the levels of both mutant proteins, suggesting that the mutant proteins are degraded by the proteasome (Figure S2). The C203R and W177R MW proteins also had a different band pattern than that of WT protein (Figure 5A), with both resolving to a major species of approximately 35 kDa. WT MW opsin was resistant to the glycosidase EndoH, whereas the C203R and W177R 35 kDa species were sensitive to Endo H digestion. EndoH removes mannose-rich oligosaccharides that are added to glycoproteins in the ER, but not the complex oligosaccharides that are added in the Golgi, suggesting that the mutant proteins are retained within the ER. Immunocytochemistry of the C203R and W177R mutants confirmed that these proteins were retained within the ER, shown by colocalization with calnexin. Both mutant MW proteins were also prone to aggregation and formed intracellular inclusions (Figure S3). In contrast, WT MW opsin trafficked to the plasma membrane, with only some retention in the ER (Figure 5A).
Figure 5.
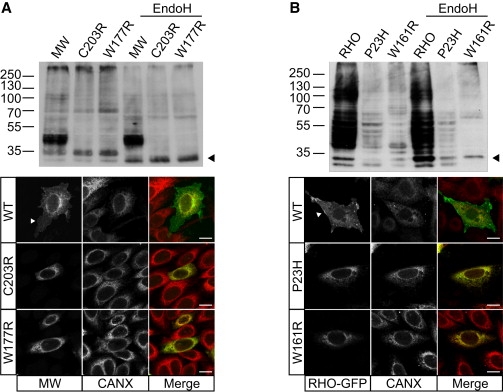
The W177R MW Mutation Causes a Cone Opsin Misfolding Defect Resulting in ER Retention
(A) Immunoblot analysis of SK-N-SH cell lysates (10 ug DM-soluble lysate) transfected with either WT MW opsin (MW), C203R MW mutant or W177R MW mutant, with Endo H treatment as indicated, probed with 1D4 opsin antibody. Both W177R and C203R show fewer glycosylated species as compared to WT and a shift in glycosylated species following Endo H treatment (arrowhead). Expression of WT MW opsin (top), C203R (middle), or W177R (bottom) in SK-N-SH cells detected with 1D4 (green in merged panel) and counterstained for the ER marker calnexin (Cnx, red in merged panel). WT cone opsin (MW) is processed in the ER and targeted to the plasma membrane (arrowhead), reflecting normal biogenesis and traffic of MW opsin, but both mutants were retained in the ER and colocalized with calnexin.
(B) Immunoblot analysis of SK-N-SH cell lysates transfected with WT rod opsin (RHO) and the P23H and W161R RHO mutants, with Endo H treatment as indicated, probed with 1D4 antibody. Endo H treatment results in a shift in glycosylated species of the W161R mutant (arrowhead). Expression of GFP-tagged RHO (top), P23H (middle), and W161R (bottom) (green) counterstained for calnexin (red). RHO-GFP is detected at the plasma membrane, but both mutant proteins are retained in the ER.
WT rod opsin protein migrated as multiple bands by immunoblot analysis, with the major species detected as a smear between 40 and 55 kDa, representing multiple glycosylated forms of the protein (Figure 5B). The P23H and W161R rod opsin mutants were expressed at lower levels than the WT protein and were sensitive to EndoH. GFP-tagged WT rod opsin localized to the plasma membrane, with some staining in the ER, whereas both P23H and W161R localized exclusively to the ER, confirming that W161R causes rod opsin misfolding (Figure 5B). W161R was also more prone to aggregation in comparison to P23H, as judged by increased intracellular inclusion formation (Figure S3).
It has previously been shown that the addition of 11-cis-retinal or 9-cis-retinal to cells expressing P23H rod opsin can improve folding and traffic.25,35 Therefore, we investigated the effect of 9-cis-retinal on cells expressing the tryptophan-to-arginine substitution in MW opsin and rod opsin. 9-cis-retinal treatment had no effect on W177R or C203R, because no change in banding pattern was detected by immunoblot analysis, indicating that the chromophore did not alleviate the mutant protein-folding defect. This was confirmed by immunocytochemistry, as both mutants remained in the ER (Figure 6A). However, 9-cis-retinal treatment of cells expressing P23H rhodopsin increased the amount of P23H, as detected by immunoblotting, and restored traffic of P23H to the plasma membrane (Figure 6B). Treatment with 9-cis-retinal had no effect on W161R rhodopsin expression or localization.
Figure 6.
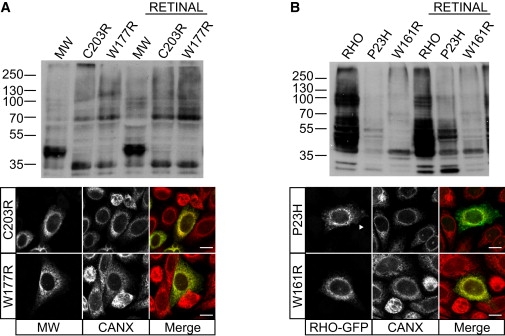
The W177R MW Mutation Is Not Rescued with 9-cis-Retinal
(A) Immunoblot analysis of SK-N-SH cell lysates transfected with WT MW opsin, W177R MW mutant and C203R MW mutant opsins, with and without 9-cis-retinal treatment, as indicated. 9-cis-retinal treatment had no effect on the level or SDS-PAGE mobility of either of the MW mutants. Immunofluorescence confirmed no retinoid-associated changes in the intracellular traffic of the mutants.
(B) Immunoblot analysis of SK-N-SH cell lysates transfected with RHO and the RHO mutants P23H and W161R, with and without 9-cis-retinal treatment resulted in increased expression of P23H, but not W161R. 9-cis-retinal treatment led to an increase in P23H traffic to the plasma membrane (arrowhead), whereas W161R was retained in the ER, where it colocalized with calnexin.
Discussion
The XLCOD5 Locus
The recent exclusion of RPGR exon ORF15 mutations as the cause of XLCOD in a number of British families led us to search for other disease-associated genes.4 In the family reported here, we mapped the disease locus to Xq26.1-qter. The proximal part of this disease region overlaps the distal part of the XLCOD2 region on Xq27; however, in the XLCOD2 family, recombination excluded the opsin array on Xq28.7 The locus for XLCOD described here caused by mutations in the cone opsin genes has therefore been assigned XLCOD5.
Gene Conversion as a Mechanism for the Cone Opsin Array Disease Haplotype
We have identified a missense mutation of a highly conserved residue, p.W177, in the LW and MW cone opsin genes that segregated with disease. The spontaneous occurrence of this inactivating point mutation in both genes is unlikely. It is more likely that the W177R mutation originally occurred in an ancestral MW gene and was then transferred to the LW gene by a gene-conversion event. This is supported by the fact that the W177R mutation in the LW gene was embedded in a block of MW sequence. The presence of a dinucleotide TT at position IVS-170-171 in both the converted tract and the donor sequence suggests a possible mechanism for gene conversion. We postulate that the ancestral LW opsin sequence in our family had originally contained a TT SNP variant at this locus, and in combination with the TT variant present in the MW gene, this could have provided an initiation site for a gene-conversion event to take place after a double-stranded break in the LW opsin gene.
The high sequence similarity (96% amino acid identity) and close linkage of the LW and MW genes predisposes the array to interlocus gene-conversion events in a manner similar to those occurring in other gene families with high homology; for example, the human fetal γ-globin gene cluster, the DRB1 loci of the major histocompatibility complex class II, and the Rh blood group antigen genes RHD and RHCE.36–39 Population genetic and statistical analyses of polymorphism at the opsin array have shown how gene conversion has contributed to sequence diversity within the array40 and, more recently, how polymorphism in the LW gene is maintained through a combination of both gene conversion and natural selection.41 Gene conversion in the opsin array has also been implicated in the pathogenesis of two cases of BCM caused by a C203R missense mutation in both LW and MW genes.42,43 Interestingly, studies of haplotype diversity have shown that sequence exchange at the opsin array locus is particularly frequent in exon 3, possibly mediated by the presence of a recombination hotspot (Chi (χ) sequence element) in the 5′ part of the exon.40,41 This high frequency of gene conversion and recombination is believed to be partly responsible for the high number of shared exon 3 polymorphisms (Figure S1) in the LW and MW genes.43 Some polymorphisms shift the λmax of the LW opsin into the “red-orange” portion of the visible spectrum, potentially enhancing color discrimination in heterozygotes.44,45 It has thus been suggested that purifying selection may be an additional factor for maintenance of exonic polymorphism between the LW and MW genes, whereas historically their introns have become homogenized.41
In this XLCOD family, the exon 3 haplotype block in the LW gene carried by affected individuals contains the mutation (W177R) in linkage disequilibrium with several SNPs (M153, V171, V174, V178, A180) (Figure S1B). The frequency of MW gene variants that contain a V174, V178, and A180 haplotype (Val:Val:Ala), as occurs in our family, is reportedly greater in individuals with only one MW opsin gene in the array (0.54 versus 0.16).43 This suggests that there may be nonrandom distribution (linkage equilibrium) of the haplotypes in the MW gene with respect to position in the array, with the proximal position (immediately downstream of the LW gene) being preferentially occupied by the Val:Val:Ala MW combination. Our findings support the suggested high frequency of gene conversion events involving exon 3 and the hypothesis of nonrandom distribution of haplotypes.44
Phenotypes Associated with LW/MW Cone Opsin Mutations
To date, the ocular phenotypes seen in association with LW and/or MW cone opsin mutations have been restricted to the various forms of red-green color deficiency and stationary cone dysfunction syndromes, including BCM.46–48 In trichromats, normal color vision requires a neural comparison between the light-absorption spectra of the three (SW, LW, and MW) different cone visual pigments. Among males, variations in red-green color vision are common and estimated to be approximately 8% for individuals of European descent.20,46 Polymorphisms and structural rearrangements within the LW/MW cone opsin genes are common, and these can alter the spectral characteristics of the encoded proteins, giving rise to a range of both normal and defective color-vision phenotypes within a population.48 Normal variants are believed to have been generated between the highly homologous LW and MW genes through ancestral gene conversions (as has been proposed for the S180A polymorphism in exon 3 that results in two spectrally different LW pigments) or by unequal recombination (as occurs in the visual pigment hybrids). Individuals with BCM have no functional LW and MW cones, vision is derived from the remaining SW cones and rod photoreceptors, and color discrimination is severely impaired from birth, with poor central vision, marked photophobia, and pendular nystagmus.47,48 The molecular genetics underlying BCM was originally reported by Nathans and colleagues.47 Three mechanisms leading to BCM are now recognized. The first pathway (60% of all cases) involves a heterogeneous group of two-step mechanisms in which nonhomologous recombination reduces the number of genes in the LW/MW array to one and, in the second step, a mutation inactivates the residual gene. Inactivating point mutations identified to date are C203R, P307L, and R247X.42,43,47 The second pathway consists of a one-step inactivation of both LW and MW genes by deletion of the LCR sequence (40% of all cases).43,47,49–52 A third, more rare, mechanism is deletion of an exon.23,53
Interestingly, the mechanism leading to the XLCOD phenotype described here is a divergent two-step pathway; inactivation mutation followed by gene conversion, leading to two nonfunctional genes in the array, which represents a fourth class of opsin array inactivation.
Lack of Cone Function versus Cone Photoreceptor Degeneration
Although in the majority of subjects BCM is a stationary condition, progression has been reported in a small number of families.23,47,49,54,55 This suggests that instead of rendering the cones nonfunctional, particular cone opsin mutation types or other modifying factors could result in cone photoreceptor loss. Molecular genetic analysis of the LW/MW opsin array in these cases has shown deletion of the LCR in two families,47,50 as well as a single 5′-LW/MW-3′ hybrid gene inactivated by a C203R mutation and a 5′-LW/MW-3′ gene with deletion of exon 2, each in a single family.23,54 No molecular diagnosis has been reported in the remaining family.55 Therefore, there is currently no clear genotype-phenotype correlation. We report a misfolding mutation, W177R, in both the LW and MW opsin genes as a cause of progressive cone dystrophy. Interestingly, mutation of the MW and LW cone opsins in this family results in an early-onset cone dystrophy, with both clinical and psychophysical evidence of cone cell death (Figure 2). There is psychophysical evidence consistent with loss of SW cone photoreceptors over time, with the eldest affected male having minimal residual SW cone function compared to the younger affected males. VA was significantly worse in the older generation of males as compared to that of the younger subjects (Table 1). Further evidence of progression was seen when comparing the mild macular RPE changes in the younger subjects with the marked macular atrophy in all three older males (Figure 2, Table 1). The clinical and psychophysical evidence of progression and gradual loss of SW cones seen in this family would not be typical of BCM. Unlike BCM, all subjects also described a definite history of better VA and color discrimination in early life that deteriorated over time. Furthermore, the VA in the youngest individual (IV:4) is better than would be expected in the vast majority of cases of BCM.23,48,54 In addition, in BCM the fundus appearance is normal in infancy and early life and may well remain normal throughout life, whereas the younger affected subjects in the XLCOD5 family all had macular RPE disturbance. Nystagmus, photophobia, and poor vision is evident from either birth or very early infancy in BCM, whereas onset in the XLCOD5 family was either several years after birth or toward the end of the first decade, with only one subject (IV:2) having nystagmus from an early age, and no individuals complaining of photophobia. Taken together, the phenotype observed in this family is more consistent with progressive COD than with BCM.
On the basis of the crystal structure of rhodopsin and comparative modeling with cone opsins,56,57 mutation of this conserved tryptophan residue would be predicted to cause a major conformational change in the structure of the encoded protein, such that mutant W177R LW and MW cone opsins would be nonfunctional in their respective photoreceptors. Misfolding class II mutations in rod opsin cause ER stress, aggregate, and exert a dominant-negative effect on the WT protein.58,59 Therefore, in addition to the loss of functional photopigment, a similar buildup of misfolded protein in both LW and MW photoreceptors could result in the cone dystrophy phenotype. We explored the biochemical and cellular consequence of this mutation in both cone opsin (MW) and rod opsin, and we compared this mutant with the most common point mutation associated with BCM, C203R, and the RP-associated rod opsin class II misfolding mutant P23H. The W177R MW mutant and the equivalent mutation in rod opsin (W161R) were retained in the ER, formed inclusions, and were targeted for degradation by the proteasome. Similarly, the C203R MW mutant, which results in misfolded protein due to disruption of a disulphide bond with C126, was retained in the ER, as reported previously.24 We then tested the ability of 9-cis-retinal treatment to rescue the protein-processing defect of all four mutants. Although P23H traffic was partially rescued after treatment, the mutants MW C203R, W177R, and rod opsin W161R were not affected by 9-cis-retinal. Although the W177R mutation results in protein misfolding and retention in the ER in a manner similar to that of the C203R mutation, the physiological difference between the resulting severity in phenotype of the C203R in stationary disease and W177R in XLCOD5 is currently not clear, and it may involve other genetic or environmental modifying factors. We have demonstrated that the W177R mutation results in misfolding of cone opsin, such that cone degeneration may result from the loss of functional photopigment, ER stress, and the accumulation of structurally defective opsin, similar to mutations in rod opsin that cause autosomal-dominant RP.58,59 The equivalent amino acid in rhodopsin (W161) is thought to be involved in packing interactions between transmembrane helices III and IV.56,57 It has also been suggested that W161 may have a role in signal transfer following light activation of the chromophore;60–62 therefore, it is possible that this mutation might affect other cone opsin properties, such as activation of the phototransduction cascade, as a potential mechanism affecting cone cell survival. Interestingly, substitution in rod opsin to W161L or W161F had no effect on rod opsin folding.60,63 In a mutagenesis study of all the tryptophan residues in LW opsin, substitution to W177F and W177Y caused misfolding of the LW opsin protein and accumulation in the ER, similar to the W177R mutant described here.64
Mutations in the LW/MW cone opsin array can cause a wider range of retinal conditions than previously recognized, ranging from the stationary congenital disorders of cone function, to BCM with progression in later life, to early-onset retinal degeneration (XLCOD5). There are now other examples of mutations in genes leading to both stationary and progressive retinal disorders, including the genes CNGA3 (MIM 600053), CNGB3 (MIM 605080), and PDE6C (MIM 600827), which are known to underlie the stationary cone-dysfunction syndrome achromatopsia.48 Mutations in these genes have been reported to also cause progressive COD and CORD.65–67 A new technique using adaptive optics has been developed to image the cone photoreceptor mosaic in vivo. Adaptive optics imaging in dichromatic individuals with a C203R MW cone opsin mutation has shown that this mutation results in disruption of the cone photoreceptor mosaic, a reduction in cone density, and thinning of the outer nuclear layer.68 The application of this technique to the study of individuals with cone dysfunction and cone dystrophy, and correlation of genotypes to cone mosaic and cone density, is likely to lead to improved understanding of the functional and visual consequence of cone opsin mutations.
Successful gene therapy for color vision deficiency in primates has been reported, highlighting the fact that this approach could be an important treatment strategy for patients with disorders affecting cone photoreceptors, such as progressive cone dystrophy resulting from LW/MW mutations.69 One important consideration for such an approach will be the evaluation of a potential toxic gain-of-function or dominant-negative effect of the mutant protein on the WT opsin introduced to photoreceptors harboring a preexisting misfolding cone opsin mutation.
Acknowledgments
This research was supported by funding from Fight for Sight UK, Moorfields Special Trustees and the National Institute for Health Research UK to the Biomedical Research Centre for Ophthalmology, based at Moorfields Eye Hospital NHS Foundation Trust and the UCL Institute of Ophthalmology (to A.J.H. and M.M.), The Wellcome Trust (065454/Z/01/Z to A.J.H., M.C., and A.T.M.), The British Retinitis Pigmentosa Society (to A.J.H.), Fight for Sight UK (to AS), and The Foundation Fighting Blindness USA (to A.T.M., G.E.H., and A.G.R.). We wish to thank the family for participating in this study. MW cone opsin 1D4 epitope-tagged construct was a kind gift from Harry Ostrer of New York University Medical School. 1D4 antibody was a kind gift from Bob Molday of the University of British Columbia.
Contributor Information
Michel Michaelides, Email: michel.michaelides@ucl.ac.uk.
Alison J. Hardcastle, Email: a.hardcastle@ucl.ac.uk.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
UCSC Genome Browser, July 2009 build, http://genome.cse.ucsc.edu/
References
- 1.Michaelides M., Hardcastle A.J., Hunt D.M., Moore A.T. Progressive cone and cone-rod dystrophies: phenotypes and underlying molecular genetic basis. Surv. Ophthalmol. 2006;51:232–258. doi: 10.1016/j.survophthal.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 2.Demirci F.Y., Rigatti B.W., Wen G., Radak A.L., Mah T.S., Baic C.L., Traboulsi E.I., Alitalo T., Ramser J., Gorin M.B. X-linked cone-rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. Am. J. Hum. Genet. 2002;70:1049–1053. doi: 10.1086/339620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Z., Peachey N.S., Moshfeghi D.M., Thirumalaichary S., Chorich L., Shugart Y.Y., Fan K., Zhang K. Mutations in the RPGR gene cause X-linked cone dystrophy. Hum. Mol. Genet. 2002;11:605–611. doi: 10.1093/hmg/11.5.605. [DOI] [PubMed] [Google Scholar]
- 4.Ebenezer N.D., Michaelides M., Jenkins S.A., Audo I., Webster A.R., Cheetham M.E., Stockman A., Maher E.R., Ainsworth J.R., Yates J.R. Identification of novel RPGR ORF15 mutations in X-linked progressive cone-rod dystrophy (XLCORD) families. Invest. Ophthalmol. Vis. Sci. 2005;46:1891–1898. doi: 10.1167/iovs.04-1482. [DOI] [PubMed] [Google Scholar]
- 5.Ruddle J.B., Ebenezer N.D., Kearns L.S., Mulhall L.E., Mackey D.A., Hardcastle A.J. RPGR ORF15 genotype and clinical variability of retinal degeneration in an Australian population. Br. J. Ophthalmol. 2009;93:1151–1154. doi: 10.1136/bjo.2008.153908. [DOI] [PubMed] [Google Scholar]
- 6.Jalkanen R., Mäntyjärvi M., Tobias R., Isosomppi J., Sankila E.M., Alitalo T., Bech-Hansen N.T. X linked cone-rod dystrophy, CORDX3, is caused by a mutation in the CACNA1F gene. J. Med. Genet. 2006;43:699–704. doi: 10.1136/jmg.2006.040741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bergen A.A.B., Pinckers A.J. Localization of a novel X-linked progressive cone dystrophy gene to Xq27: evidence for genetic heterogeneity. Am. J. Hum. Genet. 1997;60:1468–1473. doi: 10.1086/515458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marmor M.F., Fulton A.B., Holder G.E., Miyake Y., Brigell M., Bach M., International Society for Clinical Electrophysiology of Vision ISCEV Standard for full-field clinical electroretinography (2008 update) Doc. Ophthalmol. 2009;118:69–77. doi: 10.1007/s10633-008-9155-4. [DOI] [PubMed] [Google Scholar]
- 9.Holder G.E., Brigell M.G., Hawlina M., Meigen T., Vaegan, Bach M., International Society for Clinical Electrophysiology of Vision ISCEV standard for clinical pattern electroretinography—2007 update. Doc. Ophthalmol. 2007;114:111–116. doi: 10.1007/s10633-007-9053-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arden G., Wolf J., Berninger T., Hogg C.R., Tzekov R., Holder G.E. S-cone ERGs elicited by a simple technique in normals and in tritanopes. Vision Res. 1999;39:641–650. doi: 10.1016/s0042-6989(98)00182-5. [DOI] [PubMed] [Google Scholar]
- 11.Holder G.E., Robson A.G. Paediatric electrophysiology: a practical approach. In: Lorenz B., Moore A.T., editors. Essentials in Ophthalmology. Springer-Verlag; Berlin: 2006. pp. 133–155. [Google Scholar]
- 12.Stockman A., Smithson H.E., Michaelides M., Moore A.T., Webster A.R., Sharpe L.T. Residual cone vision without α-transducin. J. Vis. 2007;7:8. doi: 10.1167/7.4.8. [DOI] [PubMed] [Google Scholar]
- 13.Stockman A., Sharpe L.T., Tufail A., Kell P.D., Ripamonti C., Jeffery G. The effect of sildenafil citrate (Viagra) on visual sensitivity. J. Vis. 2007;7:4. doi: 10.1167/7.8.4. [DOI] [PubMed] [Google Scholar]
- 14.Stockman A., Smithson H.E., Webster A.R., Holder G.E., Rana N.A., Ripamonti C., Sharpe L.T. The loss of the PDE6 deactivating enzyme, RGS9, results in precocious light adaptation at low light levels. J. Vision. 2008;8:10. doi: 10.1167/8.1.10. [DOI] [PubMed] [Google Scholar]
- 15.Stockman A., MacLeod D.I.A., DePriest D.D. The temporal properties of the human short-wave photoreceptors and their associated pathways. Vision Res. 1991;31:189–208. doi: 10.1016/0042-6989(91)90111-h. [DOI] [PubMed] [Google Scholar]
- 16.Stockman A., MacLeod D.I.A., Lebrun S.J. Faster than the eye can see: blue cones respond to rapid flicker. J. Opt. Soc. Am. A. 1993;10:1396–1402. doi: 10.1364/josaa.10.001396. [DOI] [PubMed] [Google Scholar]
- 17.Stockman A., Plummer D.J. Color from invisible flicker: a failure of the Talbot-Plateau law caused by an early ‘hard’ saturating nonlinearity used to partition the human short-wave cone pathway. Vision Res. 1998;38:3703–3728. doi: 10.1016/s0042-6989(98)00049-2. [DOI] [PubMed] [Google Scholar]
- 18.Nathans J., Thomas D., Hogness D.S. Molecular genetics of human color vision: the genes encoding blue, green, and red pigments. Science. 1986;232:193–202. doi: 10.1126/science.2937147. [DOI] [PubMed] [Google Scholar]
- 19.Vollrath D., Nathans J., Davis R.W. Tandem array of human visual pigment genes at Xq28. Science. 1988;240:1669–1672. doi: 10.1126/science.2837827. [DOI] [PubMed] [Google Scholar]
- 20.Drummond-Borg M., Deeb S.S., Motulsky A.G. Molecular patterns of X chromosome-linked color vision genes among 134 men of European ancestry. Proc. Natl. Acad. Sci. USA. 1989;86:983–987. doi: 10.1073/pnas.86.3.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winderickx J., Battisti L., Motulsky A.G., Deeb S.S. Selective expression of human X chromosome-linked green opsin genes. Proc. Natl. Acad. Sci. USA. 1992;89:9710–9714. doi: 10.1073/pnas.89.20.9710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hayashi T., Motulsky A.G., Deeb S.S. Position of a ‘green-red’ hybrid gene in the visual pigment array determines colour-vision phenotype. Nat. Genet. 1999;22:90–93. doi: 10.1038/8798. [DOI] [PubMed] [Google Scholar]
- 23.Gardner J.C., Michaelides M., Holder G.E., Kanuga N., Webb T.R., Mollon J.D., Moore A.T., Hardcastle A.J. Blue cone monochromacy: causative mutations and associated phenotypes. Mol. Vis. 2009;15:876–884. [PMC free article] [PubMed] [Google Scholar]
- 24.Kazmi M.A., Sakmar T.P., Ostrer H. Mutation of a conserved cysteine in the X-linked cone opsins causes color vision deficiencies by disrupting protein folding and stability. Invest. Ophthalmol. Vis. Sci. 1997;38:1074–1081. [PubMed] [Google Scholar]
- 25.Saliba R.S., Munro P.M.G., Luthert P.J., Cheetham M.E. The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J. Cell Sci. 2002;115:2907–2918. doi: 10.1242/jcs.115.14.2907. [DOI] [PubMed] [Google Scholar]
- 26.Mendes H.F., Cheetham M.E. Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 2008;17:3043–3054. doi: 10.1093/hmg/ddn202. [DOI] [PubMed] [Google Scholar]
- 27.Kosmaoglou M., Kanuga N., Aguilà M., Garriga P., Cheetham M.E. A dual role for EDEM1 in the processing of rod opsin. J. Cell Sci. 2009;122:4465–4472. doi: 10.1242/jcs.055228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hecht S., Verrijp C.D. Intermittent stimulation by light. IV. A theoretical interpretation of the quantatitive data of flicker. J. Gen. Physiol. 1933;17:266–286. doi: 10.1085/jgp.17.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hecht S., Shlaer S. Intermittent stimulation by light. V. The relation between intensity and critical frequency for different parts of the spectrum. J. Gen. Physiol. 1936;19:965–977. doi: 10.1085/jgp.19.6.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stockman A., Sharpe L.T. The spectral sensitivities of the middle- and long-wavelength-sensitive cones derived from measurements in observers of known genotype. Vision Res. 2000;40:1711–1737. doi: 10.1016/s0042-6989(00)00021-3. [DOI] [PubMed] [Google Scholar]
- 31.Judd, D.B. (1951). Report of U.S. Secretariat Committee on Colorimetry and Artificial Daylight. In Proceedings of the Twelfth Session of the CIE, Stockholm. Vol. 1. (Paris: Bureau Central de la CIE), 11.
- 32.Neitz M., Neitz J., Jacobs G.H. Spectral tuning of pigments underlying red-green color vision. Science. 1991;252:971–974. doi: 10.1126/science.1903559. [DOI] [PubMed] [Google Scholar]
- 33.Yoshizawa T. The road to color vision: structure, evolution and function of chicken and gecko visual pigments. Photochem. Photobiol. 1992;56:859–867. doi: 10.1111/j.1751-1097.1992.tb09707.x. [DOI] [PubMed] [Google Scholar]
- 34.Illing M.E., Rajan R.S., Bence N.F., Kopito R.R. A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J. Biol. Chem. 2002;277:34150–34160. doi: 10.1074/jbc.M204955200. [DOI] [PubMed] [Google Scholar]
- 35.Noorwez S.M., Malhotra R., McDowell J.H., Smith K.A., Krebs M.P., Kaushal S. Retinoids assist the cellular folding of the autosomal dominant retinitis pigmentosa opsin mutant P23H. J. Biol. Chem. 2004;279:16278–16284. doi: 10.1074/jbc.M312101200. [DOI] [PubMed] [Google Scholar]
- 36.Chen J.M., Cooper D.N., Chuzhanova N., Férec C., Patrinos G.P. Gene conversion: mechanisms, evolution and human disease. Nat. Rev. Genet. 2007;8:762–775. doi: 10.1038/nrg2193. [DOI] [PubMed] [Google Scholar]
- 37.Stoeckert C.J., Jr., Collins F.S., Weissman S.M. Human fetal globin DNA sequences suggest novel conversion event. Nucleic Acids Res. 1984;12:4469–4479. doi: 10.1093/nar/12.11.4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gyllensten U.B., Sundvall M., Erlich H.A. Allelic diversity is generated by intraexon sequence exchange at the DRB1 locus of primates. Proc. Natl. Acad. Sci. USA. 1991;88:3686–3690. doi: 10.1073/pnas.88.9.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Innan H. A two-locus gene conversion model with selection and its application to the human RHCE and RHD genes. Proc. Natl. Acad. Sci. USA. 2003;100:8793–8798. doi: 10.1073/pnas.1031592100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winderickx J., Battisti L., Hibiya Y., Motulsky A.G., Deeb S.S. Haplotype diversity in the human red and green opsin genes: evidence for frequent sequence exchange in exon 3. Hum. Mol. Genet. 1993;2:1413–1421. doi: 10.1093/hmg/2.9.1413. [DOI] [PubMed] [Google Scholar]
- 41.Verrelli B.C., Tishkoff S.A. Signatures of selection and gene conversion associated with human color vision variation. Am. J. Hum. Genet. 2004;75:363–375. doi: 10.1086/423287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reyniers E., Van Thienen M.N., Meire F., De Boulle K., Devries K., Kestelijn P., Willems P.J. Gene conversion between red and defective green opsin gene in blue cone monochromacy. Genomics. 1995;29:323–328. doi: 10.1006/geno.1995.9998. [DOI] [PubMed] [Google Scholar]
- 43.Nathans J., Maumenee I.H., Zrenner E., Sadowski B., Sharpe L.T., Lewis R.A., Hansen E., Rosenberg T., Schwartz M., Heckenlively J.R. Genetic heterogeneity among blue-cone monochromats. Am. J. Hum. Genet. 1993;53:987–1000. [PMC free article] [PubMed] [Google Scholar]
- 44.Winderickx J., Lindsey D.T., Sanocki E., Teller D.Y., Motulsky A.G., Deeb S.S. Polymorphism in red photopigment underlies variation in colour matching. Nature. 1992;356:431–433. doi: 10.1038/356431a0. [DOI] [PubMed] [Google Scholar]
- 45.Carroll J., Neitz J., Neitz M. Estimates of L:M cone ratio from ERG flicker photometry and genetics. J. Vis. 2002;2:531–542. doi: 10.1167/2.8.1. [DOI] [PubMed] [Google Scholar]
- 46.Nathans J., Piantanida T.P., Eddy R.L., Shows T.B., Hogness D.S. Molecular genetics of inherited variation in human color vision. Science. 1986;232:203–210. doi: 10.1126/science.3485310. [DOI] [PubMed] [Google Scholar]
- 47.Nathans J., Davenport C.M., Maumenee I.H., Lewis R.A., Hejtmancik J.F., Litt M., Lovrien E., Weleber R., Bachynski B., Zwas F. Molecular genetics of human blue cone monochromacy. Science. 1989;245:831–838. doi: 10.1126/science.2788922. [DOI] [PubMed] [Google Scholar]
- 48.Michaelides M., Hunt D.M., Moore A.T. The cone dysfunction syndromes. Br. J. Ophthalmol. 2004;88:291–297. doi: 10.1136/bjo.2003.027102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deeb S.S. The molecular basis of variation in human color vision. Clin. Genet. 2005;67:369–377. doi: 10.1111/j.1399-0004.2004.00343.x. [DOI] [PubMed] [Google Scholar]
- 50.Ayyagari R., Kakuk L.E., Coats C.L., Bingham E.L., Toda Y., Felius J., Sieving P.A. Bilateral macular atrophy in blue cone monochromacy (BCM) with loss of the locus control region (LCR) and part of the red pigment gene. Mol. Vis. 1999;5:13. [PubMed] [Google Scholar]
- 51.Ayyagari R., Kakuk L.E., Bingham E.L., Szczesny J.J., Kemp J., Toda Y., Felius J., Sieving P.A. Spectrum of color gene deletions and phenotype in patients with blue cone monochromacy. Hum. Genet. 2000;107:75–82. doi: 10.1007/s004390000338. [DOI] [PubMed] [Google Scholar]
- 52.Kellner U., Wissinger B., Tippmann S., Kohl S., Kraus H., Foerster M.H. Blue cone monochromatism: clinical findings in patients with mutations in the red/green opsin gene cluster. Graefes Arch. Clin. Exp. Ophthalmol. 2004;242:729–735. doi: 10.1007/s00417-004-0921-z. [DOI] [PubMed] [Google Scholar]
- 53.Ladekjaer-Mikkelsen A.S., Rosenberg T., Jørgensen A.L. A new mechanism in blue cone monochromatism. Hum. Genet. 1996;98:403–408. doi: 10.1007/s004390050229. [DOI] [PubMed] [Google Scholar]
- 54.Michaelides M., Johnson S., Simunovic M.P., Bradshaw K., Holder G., Mollon J.D., Moore A.T., Hunt D.M. Blue cone monochromatism: a phenotype and genotype assessment with evidence of progressive loss of cone function in older individuals. Eye (Lond) 2005;19:2–10. doi: 10.1038/sj.eye.6701391. [DOI] [PubMed] [Google Scholar]
- 55.Fleischman J.A., O'Donnell F.E., Jr. Congenital X-linked incomplete achromatopsia. Evidence for slow progression, carrier fundus findings, and possible genetic linkage with glucose-6-phosphate dehydrogenase locus. Arch. Ophthalmol. 1981;99:468–472. doi: 10.1001/archopht.1981.03930010470016. [DOI] [PubMed] [Google Scholar]
- 56.Palczewski K., Kumasaka T., Hori T., Behnke C.A., Motoshima H., Fox B.A., Le Trong I., Teller D.C., Okada T., Stenkamp R.E. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 57.Stenkamp R.E., Filipek S., Driessen C.A.G.G., Teller D.C., Palczewski K. Crystal structure of rhodopsin: a template for cone visual pigments and other G protein-coupled receptors. Biochim. Biophys. Acta. 2002;1565:168–182. doi: 10.1016/s0005-2736(02)00567-9. [DOI] [PubMed] [Google Scholar]
- 58.Mendes H.F., van der Spuy J., Chapple J.P., Cheetham M.E. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol. Med. 2005;11:177–185. doi: 10.1016/j.molmed.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 59.Lin J.H., Li H., Yasumura D., Cohen H.R., Zhang C., Panning B., Shokat K.M., Lavail M.M., Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin S.W., Sakmar T.P. Specific tryptophan UV-absorbance changes are probes of the transition of rhodopsin to its active state. Biochemistry. 1996;35:11149–11159. doi: 10.1021/bi960858u. [DOI] [PubMed] [Google Scholar]
- 61.Borhan B., Souto M.L., Imai H., Shichida Y., Nakanishi K. Movement of retinal along the visual transduction path. Science. 2000;288:2209–2212. doi: 10.1126/science.288.5474.2209. [DOI] [PubMed] [Google Scholar]
- 62.Klein-Seetharaman J., Yanamala N.V.K., Javeed F., Reeves P.J., Getmanova E.V., Loewen M.C., Schwalbe H., Khorana H.G. Differential dynamics in the G protein-coupled receptor rhodopsin revealed by solution NMR. Proc. Natl. Acad. Sci. USA. 2004;101:3409–3413. doi: 10.1073/pnas.0308713101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakayama T.A., Khorana H.G. Mapping of the amino acids in membrane-embedded helices that interact with the retinal chromophore in bovine rhodopsin. J. Biol. Chem. 1991;266:4269–4275. [PubMed] [Google Scholar]
- 64.Nakayama T.A., Zhang W., Cowan A., Kung M. Mutagenesis studies of human red opsin: trp-281 is essential for proper folding and protein-retinal interactions. Biochemistry. 1998;37:17487–17494. doi: 10.1021/bi982077u. [DOI] [PubMed] [Google Scholar]
- 65.Michaelides M., Aligianis I.A., Ainsworth J.R., Good P., Mollon J.D., Maher E.R., Moore A.T., Hunt D.M. Progressive cone dystrophy associated with mutation in CNGB3. Invest. Ophthalmol. Vis. Sci. 2004;45:1975–1982. doi: 10.1167/iovs.03-0898. [DOI] [PubMed] [Google Scholar]
- 66.Thiadens A.A., Roosing S., Collin R.W., van Moll-Ramirez N., van Lith-Verhoeven J.J., van Schooneveld M.J., den Hollander A.I., van den Born L.I., Hoyng C.B., Cremers F.P., Klaver C.C. Comprehensive analysis of the achromatopsia genes CNGA3 and CNGB3 in progressive cone dystrophy. Ophthalmology. 2010;117 doi: 10.1016/j.ophtha.2009.09.008. 825–830, e1. [DOI] [PubMed] [Google Scholar]
- 67.Thiadens A.A., den Hollander A.I., Roosing S., Nabuurs S.B., Zekveld-Vroon R.C., Collin R.W., De Baere E., Koenekoop R.K., van Schooneveld M.J., Strom T.M. Homozygosity mapping reveals PDE6C mutations in patients with early-onset cone photoreceptor disorders. Am. J. Hum. Genet. 2009;85:240–247. doi: 10.1016/j.ajhg.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carroll J., Baraas R.C., Wagner-Schuman M., Rha J., Siebe C.A., Sloan C., Tait D.M., Thompson S., Morgan J.I., Neitz J. Cone photoreceptor mosaic disruption associated with Cys203Arg mutation in the M-cone opsin. Proc. Natl. Acad. Sci. USA. 2009 doi: 10.1073/pnas.0910128106. Published online November 23, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mancuso K., Hauswirth W.W., Li Q., Connor T.B., Kuchenbecker J.A., Mauck M.C., Neitz J., Neitz M. Gene therapy for red-green colour blindness in adult primates. Nature. 2009;461:784–787. doi: 10.1038/nature08401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.