

Abstract
Mitochondrial respiratory chain disorders are a heterogeneous group of disorders in which the underlying genetic defect is often unknown. We have identified a pathogenic mutation (c.156C>G [p.F52L]) in YARS2, located at chromosome 12p11.21, by using genome-wide SNP-based homozygosity analysis of a family with affected members displaying myopathy, lactic acidosis, and sideroblastic anemia (MLASA). We subsequently identified the same mutation in another unrelated MLASA patient. The YARS2 gene product, mitochondrial tyrosyl-tRNA synthetase (YARS2), was present at lower levels in skeletal muscle whereas fibroblasts were relatively normal. Complex I, III, and IV were dysfunctional as indicated by enzyme analysis, immunoblotting, and immunohistochemistry. A mitochondrial protein-synthesis assay showed reduced levels of respiratory chain subunits in myotubes generated from patient cell lines. A tRNA aminoacylation assay revealed that mutant YARS2 was still active; however, enzyme kinetics were abnormal compared to the wild-type protein. We propose that the reduced aminoacylation activity of mutant YARS2 enzyme leads to decreased mitochondrial protein synthesis, resulting in mitochondrial respiratory chain dysfunction. MLASA has previously been associated with PUS1 mutations; hence, the YARS2 mutation reported here is an alternative cause of MLASA.
Introduction
Mitochondrial respiratory chain (RC) disorders are among the most common inborn errors of metabolism, with an incidence of at least 1 in 8000 births, and are one of the most challenging to diagnose, treat, or prevent.1 Diagnosis of RC disorders is usually dependent on demonstration of altered activities of one or more of the RC enzyme complexes. In up to 50% of cases, RC enzyme deficiencies can only be detected in muscle or liver biopsy samples and do not manifest in fibroblasts, making diagnosis more complex.2
For the five RC enzyme complexes to be functional, 13 mitochondrially encoded subunits and more than 75 nuclear-encoded subunits must be expressed in a coordinated manner.3 In addition, there are a number of essential nuclear-encoded proteins that are involved in regulation of mitochondrial DNA (mtDNA) transcription and translation and RC subunit assembly and stability. Mutations in any of these genes may result in a mitochondrial RC disorder.
Myopathy, lactic acidosis, and sideroblastic anemia (MLASA [MIM 600462]) is a mitochondrial RC disorder characterized by progressive exercise intolerance and sideroblastic anemia. Severity of symptoms varies between and within affected families and sometimes includes mental retardation.4,5 MLASA is associated with a mutation in pseudouridylate synthase 1 (PUS1 [MIM 610957]), resulting in decreased pseudouridylation of some cytoplasmic and mitochondrial tRNAs6; this decreased pseudouridylation may affect protein translation.
In the present study, we performed genome-wide SNP analyses of a consanguineous family with two children affected by MLASA. We identified a c.156C>G (p.F52L) mutation at chromosome 12p11.21 in YARS2 (MIM 610957) in the affected children and another MLASA patient. This tyrosyl-tRNA synthetase catalyzes covalent binding of tyrosine to its cognate tRNA. Functional studies of recombinant p.F52L YARS2 protein showed that the mutant protein had reduced catalytic efficiency.
Material and Methods
Clinical Information
The Ethics Committee of the Children's Hospital at Westmead approved this research, and, as required by that approval, written consent was obtained from adult family members.
Family 1
The proband (II:2; Figure 1), the second child of first-cousin parents of Lebanese background, was born at term after an uncomplicated pregnancy and delivery. At around 10 weeks of age, pallor and lethargy became apparent, and he was found to have sideroblastic anemia. He became transfusion dependent, initially requiring packed cell transfusions every 2–3 months and in later years every 3–4 weeks.
Figure 1.
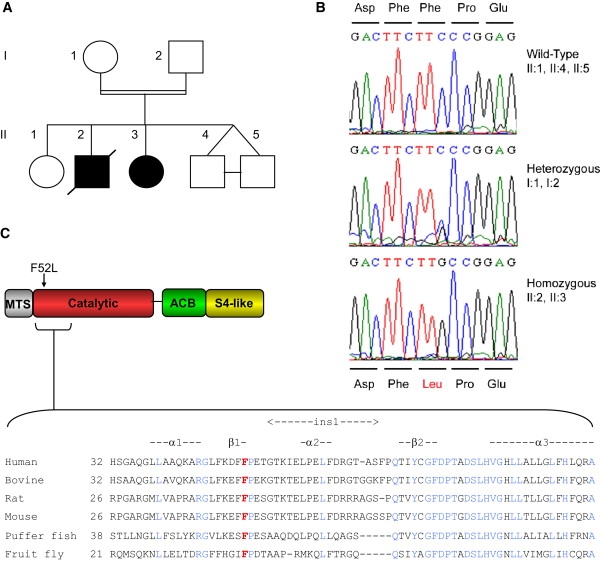
Identification of a Mutation in YARS2 Associated with MLASA
(A) Pedigree of the family used for homozygosity mapping.
(B) DNA sequence identifying the c.156C>G (p.F52L) YARS2 mutation.
(C) Schematic diagram of YARS2 domains showing location of the variant F52 residue and its conservation among species. The following abbreviations are used: MTS, mitochondrial targeting sequence; catalytic, catalytic domain; ACB, anticodon binding domain; and S4-like, S4 ribosomal protein-like domain.
He was further investigated at 3 months of age because of failure to thrive and was found to have a persistent lactic acidemia (ranging over the years from 3 to 13.7 mmol/l; normal range 0.7–2.0 mmol/l) and hypertrophic cardiomyopathy, the latter of which subsequently resolved without specific therapy. Pyridoxal phosphate and thiamine appeared to have no impact on the anemia. He was commenced on iron chelators—to prevent iron overload in infancy—which were initially delivered subcutaneously and, in later years, orally.
Early gross and fine motor development was normal, and cognition was normal, with above-average school performance. However, he suffered from chronic lethargy and progressive exercise intolerance. By the age of 7 years he could no longer walk long distances or climb stairs. Hearing and vision remained normal. At 17 years of age, cardiomyopathy was again noted, and by this time he was essentially restricted to a wheelchair, although he was able to walk short distances on a level surface. He had also developed dysphagia, with intermittent choking with meals.
At 17 years 10 months he developed acute respiratory difficulties not preceded by eating or drinking nor associated with an intercurrent illness, and he suffered an acute respiratory arrest, requiring ventilatory support. At that time his weight was 40.4 kg (<3rd percentile) and height was 160.4 cm (<3rd percentile). Further investigations found no evidence of an acute cardiac ischemic event, myocarditis, or pulmonary embolus. Over the next 3 months there were a number of attempts to wean him from mechanical ventilation, which were not successful. There was no deterioration in cognitive abilities throughout this time. He developed permanent vocal cord paresis and required enteral nutritional support. After extensive discussion with the patient and his parents, he elected to have heroic therapeutic measures withdrawn, and he succumbed shortly thereafter.
A muscle biopsy at 2 years of age showed many regions of subsarcolemmal mitochondrial aggregates and some incipient “ragged red” fibers on histology, clearly decreased cytochrome oxidase staining intensity, but a more subtle reduction in the amount of NADH-tetrazolium reductase and succinate dehydrogenase staining. Lipid and glycogen staining was normal. Muscle mitochondrial RC enzymology showed low activities of complexes I and IV (Table 1). Fibroblast RC activities were normal. Molecular testing for some of the common mtDNA mutations (m.3243A>G, m.8344A>G, m.8993T>G, and m.8993T>C) and for mtDNA deletions was normal. In addition, sequencing of the PUS1 gene was normal (kindly performed by N. Fischel-Ghodsian, Cedars-Sinai Medical Center, Los Angeles). Once he was found to have a primary RC disorder, he was subjected to trials of vitamins, cofactors, and other pharmacological agents over a number of years, including folic acid, coenzyme Q, vitamin C, vitamin E, nicotinamide, riboflavin, carnitine, and creatine, none of which were found to be of any obvious clinical benefit or to have a positive impact on blood lactate levels.
Table 1.
RC Enzyme Activity in Skeletal Muscle of Individuals with the YARS2 Mutation
|
Enzyme Activity (% of Control Mean Relative to Citrate Synthase) |
|||||
|---|---|---|---|---|---|
| Enzyme | Family 1, Patient 1 (II:2) | Family 2, Patient 3 | Normal Rangea | Family 1, Patient 2 (II:3) | Normal Rangea |
| Complex I | 9 | 22 | 32–149 | 3 | 70–149 |
| Complex II | 164 | 120 | 53–160 | 139 | 80–110 |
| Complex III | n.t.b | n.t.b | n.t.b | 3 | 31–170 |
| Complex IV | 21 | 13 | 53–283 | 2 | 50–138 |
| Citrate synthasec | 344 | 125 | 55–152 | 319 | 66–139 |
The observed range was determined on at least nine muscle biopsies obtained from children lacking evidence of RC disease. Note that patient 2 was assayed at a later time to the other patients.
n.t. denotes not tested.
Citrate synthase activity was expressed relative to protein.
His younger sister (II:3; Figure 1) also developed a transfusion-dependent sideroblastic anemia in infancy and went on to have progressive muscle weakness, following a trajectory similar to her brother's. She also developed swallowing difficulties with weakened pharyngeal peristalsis, first apparent at the age of 15 years. Because of worsening dysphagia, she had a gastrostomy inserted for enteral nutrition. When last reviewed at 16 years of age, her height was in the 6th percentile and weight was below the 3rd percentile. She was pale with a hyperdynamic circulation. She had no hepatosplenomegaly. Muscle tone was generally reduced, with mild muscle weakness and normal deep tendon reflexes. Cognition was normal. She was unable to walk more than about 20 m without having to stop to rest.
She too was found to have persistent lactic acidemia (2.5–8.4 mmol/l). A muscle biopsy at 15 years of age showed reduced complex I, III, and IV activities, with increased activities of complex II and citrate synthase (Table 1).
The affected siblings have an older sister (II:1) and younger identical twin brothers (II:4 and II:5), all of who are clinically well.
Family 2
The proband is the fourth child of first-cousin parents of Lebanese background and is not known to be related to family 1. She had delayed motor milestones (unable to walk unaided until 3 years of age) but was otherwise healthy until 7 years of age, when she developed sideroblastic anemia, with a hemoglobin of 70 g/l (normal range 115–140). She also had a raised blood lactate of 4.1 mmol/l, and her urine metabolic screen showed raised alanine and lactate. Her parents refused to allow her to have blood transfusions. They then embarked on an alternative-medicine approach to her care, which included a raw protein powder preparation, an algal-based preparation, mineral salts, lipoic acid, B group vitamins, regular intramuscular pyridoxal-5-phosphate, and oral coenzyme Q10. When diligently taking these preparations, the family believed that her hemoglobin could be maintained between 80 and 110 g/l, and when not taking these treatments her hemoglobin could drop to as low as 40 g/l and was associated with severe lethargy and weakness. In recent years she had also been involved in a regular fitness program. She has been able to participate in running events (walking only) of up to 10 km.
When last reviewed at age 24 years, she had a mild skeletal myopathy and mild peripheral muscle weakness, with normal deep tendon reflexes. The rest of her general physical examination was unremarkable.
A muscle biopsy when she was 8 years old showed lipid vacuoles. Specific stains for RC enzymes were not used. Muscle mitochondrial RC enzymology showed low activities of complex I and IV (Table 1). Molecular testing for some of the common mtDNA mutations (m.3243A>G, m.8344A>G, m.8993T>G, and m.8993T>C) and for mtDNA deletions was normal.
She has two older brothers, an older sister, two younger sisters, and a younger brother, all of whom are healthy and do not have anemia or a myopathy. A maternal aunt is said to have had transfusion-dependent sideroblastic anemia and a skeletal myopathy, but never had a muscle biopsy. She died at 26 years of age after a lower-segment Caesarean section for her fifth pregnancy (the previous four having miscarried). The details of this are unclear, but in the days before her death she developed a fever and became comatose.
RC Enzyme Activities
Enzyme activities were determined as previously described.7
Genotyping and Linkage Analysis
Genome-wide SNP analysis was performed by the Australian Genome Research Facility (Melbourne, Australia) with Affymetrix 250K NspI SNP chips. Data files for linkage analysis were generated with LINKDATAGEN8 and were analyzed with MERLIN.9 Haplotypes were generated with HaploPainter.10 A list of candidate mitochondrial genes was generated from MitoCarta.11 PCR-amplified COX6C (MIM 124090), MRPL13 (MIM 610200), MRPS28 (MIM 611990), MRPS35 (MIM 611995), TIMM13 (MIM 607383), and YARS2 cDNA or YARS2 gDNA was sequenced by Macrogen (Korea). Control genomic DNA was screened by a PCR-RFLP test utilizing a MboII restriction site abolished by the c.156C>G mutation (primers 5′-AGTAGGTGGGTGTGGTGGG-3′, 5′-CAGCAGCGCAAGTAGATGAC-3′).
Immunoblotting
Skeletal muscle and fibroblast samples were lysed as previously described12 and resolved by 10% Bis-Tris precast gels (Invitrogen). For immunoblot analysis, polyvinylidene fluoride (PVDF) membranes were probed with 1:200 anti-YARS2 (AP7838b, Abgent), 1:500 anti-OXPHOS cocktail (consisting of five antibodies, for detection of each of the five RC complexes; MitoSciences), 1:5000 anti-GAPDH (Millipore), or 1:1000 anti-mitofilin (MitoSciences) overnight at 4°C and then with either 1:2500 ECL anti-rabbit or anti-mouse IgG horseradish peroxidase (HRP; GE Healthcare) for 2 hr at room temperature (RT). Membranes were developed with enhanced chemiluminescence (ECL) reagents and exposed to Hyperfilm ECL. Films were scanned on a Microtek ScanMaker 8700 and analyzed with ImageQuant software (GE Healthcare).
Immunofluorescence
Muscle sections (0.8 μm) were fixed and dehydrated.13 Muscle sections were probed with 1:100 COI (complex IV subunit 1) antibody (Invitrogen) O/N at 4°C and then 1:250 anti-mouse AlexaFluor555 (Invitrogen) and 1:1000 TOPRO-3 (Invitrogen) for 2 hr at RT. Images were taken on a Leica SP2 scanning laser confocal microscope.
Mitochondrial Protein-Synthesis Assay
Assays were performed on myotubes and fibroblasts as previously described.14 Myotubes were generated by transdifferentiation of fibroblasts12 with an adenoviral MyoD vector. Fibroblasts were transduced at 70%–80% confluence with 500 transforming units in the presence of 0.5 μg/ml Polybrene (Sigma). Mitochondrial protein-synthesis assays were performed on myotubes after 6 days of differentiation.
Cloning and Aminoacylation Assay
Recombinant p.F52L YARS2 was generated for use in functional assays. YARS2 was PCR amplified from patient DNA and cloned into a TOPO TA vector (Invitrogen). A BamHI and BglII doubly digested fragment containing the DNA mutation was then subcloned into the wild-type YARS2 vector,15 from which the corresponding fragment had been removed. Recombinant wild-type and p.F52L YARS2 were expressed and purified from E. coli as previously described.15 Tyrosylation assays were performed according to previously described methods.15 Apparent kinetic parameters were determined from Lineweaver-Burk plots in the presence of either wild-type or mutated (p.F52L) YARS2 (diluted in 100 mM HEPES-NaOH [pH 7.4], 1 mM DTT, 5 mg/mL bovine serum albumin, and 10% glycerol) and native E. coli tRNATyr (Sigma) with concentrations ranging from 4 to 17 μM. Both wild-type and mutant enzymes were used at concentrations of 23 and 35 nM, respectively. Experimental errors on kcat and Km varied at most by 20%. Numerical values are averages of at least two independent experiments.
Results
We investigated the cause of MLASA in three patients from two apparently unrelated consanguineous families. RC enzyme analyses of muscle biopsies revealed functional defects of complexes I, III, and IV enzyme activity relative to citrate synthase activity in affected patients (Table 1; note that complex III was not tested in all patients). Enzyme activities were within normal limits in fibroblasts (data not shown).
Genome-wide SNP-based homozygosity analysis was performed in the family with two affected children (Figure 1A) after PUS1 mutation screening proved negative. Regions of homozygosity common to the affected family members were identified within five chromosomes: 2p25.1-p25.3 (10.6 cM), 8q12.1-q24.12 (16.4 cM), 12p12.1-q13.11 (13.2 cM), 18p11.31-q12.1 (32.4 cM), and 19p13.3 (5.6 cM) (Table S1, Figure S1). The candidate gene set was refined by cross-referencing to genes that expressed a mitochondrially localized protein with the MitoCarta database,11 narrowing the list to 28 candidate genes (Table S1). Six of these genes were sequenced, with no mutations found in COX6C, MRPL13, MRPS28, MRPS35, or TIMM13. YARS2, which encodes the human mitochondrial tyrosyl-tRNA synthetase, was among the first genes selected for sequencing, given the key role of aminoacyl-tRNA synthetases (ARSs) in protein synthesis, and previous reports of pathogenic mutations in the mt-ARS genes DARS2 (MIM 610956) and RARS2 (MIM 611524) causing neurological disease and lactic acidosis.16 A homozygous c.156C>G (p.F52L) mutation was identified in YARS2 in the affected children but not in the unaffected children, whereas both parents were heterozygous (Figure 1B). Phenylalanine 52 lies in the catalytic domain of YARS2 within the β1-strand and close to the “ins1” insertion domain and is conserved in vascularized metazoans17 (Figure 1C). We subsequently identified the same homozygous mutation in another MLASA patient (Table 1) from a second consanguineous family. Both families were of Lebanese origin, and haplotype mapping (data not shown) suggests that they may carry a founder mutation that arose on a relatively common haplotype defined by three SNPs that span the YARS2 gene. The haplotype consisting of rs1183094, rs17541794, and rs11539444 has an inferred haplotype frequency of 0.82 among the Lebanese population based on haplotype frequency calculations using data from 50 Lebanese control individuals with HAPLOVIEW.18 The YARS2 mutation was not found in 220 control chromosomes, including 104 of Lebanese origin.
Because myopathy was a major symptom of the YARS2 mutation, we examined YARS2 expression in patient muscle and its effect on RC complexes. Immunoblot analysis of skeletal muscle from patient II:3 revealed that mutant YARS2 was expressed in patient muscle at slightly reduced levels relative to three age- and sex-matched controls (Figure 2A), with broadly normal levels of YARS2 observed in fibroblast lines (Figure S2). Abnormal RC complex I, III, and IV enzymatic function associated with the YARS2 mutation (Table 1) was supported by immunoblotting (Figure 2A) and immunolabeling (Figure 2B) of patient skeletal muscle. Immunoblot analysis of subunits for RC enzyme complexes, dependent upon RC complex formation for protein stability, revealed a severe reduction in complex I (subunit NDUFB8), III (subunit core 2), and IV (subunit 2) in patient muscle, relative to controls (Figure 2A). Immunolabeling of skeletal muscle cryosections from patient II:3 also showed a marked reduction in levels of complex IV (subunit 1) (Figure 2B). Levels of complex II (subunit FeS) were elevated in patient muscle (Figure 2A), in agreement with RC enzymology results showing increased activity of complex II (Table 1). Complex II is the only RC complex that does not contain mitochondrially encoded subunits, suggesting that the dysfunction in complexes I, III, and IV may be related to the requirement for mitochondrially encoded subunits to form functional complexes. Complex V, which was not assayed for enzyme activity, was investigated by immunoblotting. The nuclear-encoded subunit α of complex V was elevated in patient muscle on immunoblotting (Figure 2A). Accumulation of subcomplexes containing subunit α when mitochondrial subunits are defective has been reported previously.19 Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and mitofilin were used as loading controls; however, the levels of both also differed from controls. Coomassie staining was used to confirm equivalent loadings predicted by protein assay. Patient muscle showed decreased levels of the glycolytic enzyme GAPDH and increased levels of mitofilin, an inner mitochondrial membrane protein with critical functions in mitochondrial fusion and fission (Figure 2A). These changes are presumably secondary to the mitochondrial proliferation and myopathic changes that have occurred in response to the YARS2 mutation. Immunoblotting of patient fibroblasts showed only minor, if any, decrease in levels of RC subunits relative to loading controls and no change in GAPDH and mitofilin levels (Figure S2).
Figure 2.
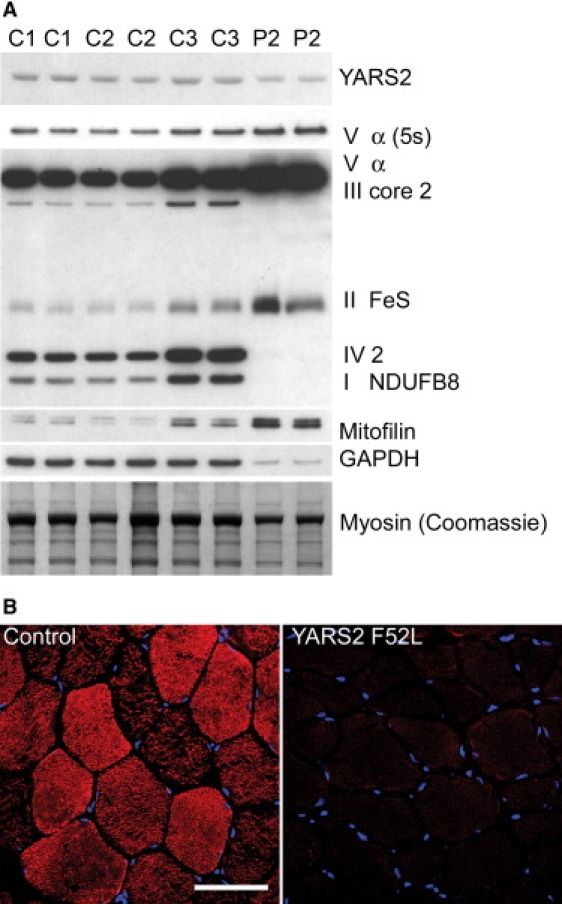
Effect of the YARS2 Mutation on Skeletal Muscle RC Protein Expression
Immunoblot analysis of YARS2 and the RC complexes in (A) patient II:3 muscle (P2) and three controls (C1, C2, C3). A 1 min exposure of specific subunits of the five RC complexes (I–V) detected by the anti-OXPHOS cocktail are shown on one blot. An additional shorter exposure (5 s) of complex V subunit α is shown for clarity. Note that these subunits are labile if the RC complex is not fully assembled. GAPDH and mitofilin were used as loading controls; however, both were affected. Five micrograms of total protein was loaded, and equivalent loadings were confirmed by Coomassie staining.
(B) Immunolabeling of patient II:3 and control muscle cryosections with CO1 (complex IV, subunit 1; red) and nuclear staining with TOPRO-3 (blue). The scale bar represents 76 μm.
Because YARS2 encodes mitochondrial tyrosyl-tRNA synthetase, we hypothesized that the p.F52L mutation may result in reduced mitochondrial protein synthesis. There are 37 genes encoded by mtDNA: 24 are involved in mt translation and 13 encode subunits of the RC.3 A mitochondrial protein-synthesis assay involving [35S]-methionine pulse-chase labeling of patient myotubes revealed decreased levels of mitochondrially encoded protein subunits of RC complexes III (cytb), IV (CO1, CO2/3), and V (ATP6) after [35S]-methionine pulse-chase labeling of patient myotubes (Figure 3A). Reduced steady-state levels of complex IV (CO1, CO2) subunits and complex I (ND1) subunits were confirmed by immunoblotting of mitochondrial extracts derived from patient myotubes (Figure 3B). In contrast, steady-state levels of a nuclear-encoded subunit of complex II (70 kDa) were unaffected. These data suggest that the p.F52L YARS2 mutation leads to a deficiency in mitochondrial protein synthesis and thus a reduction in RC complexes I, III, IV, and V, which contain seven, one, three, and two mitochondrially encoded subunits, respectively.3 No significant defect in mitochondrial protein synthesis was observed in fibroblasts (Figure S3). Because immunoblot analysis showed that mutant YARS2 levels were close to normal in patient muscle, the apparent inability to supply sufficient mitochondrially encoded subunits of the RC may be due to a functional defect in enzyme activity of mutant YARS2.
Figure 3.
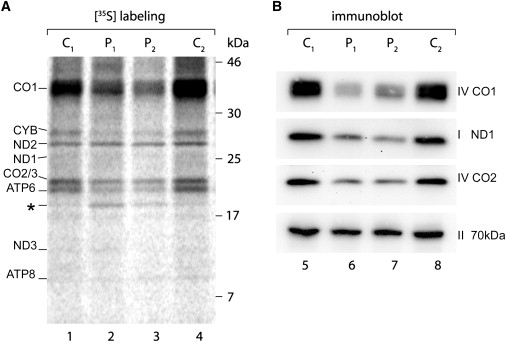
The Mutation in YARS2 Results in Reduced Mitochondrial Protein Synthesis in Patient Myotubes
(A) SDS-PAGE phosphorimaging analysis of [35S]-methionine-labeled mitochondrial proteins from patient (P1 = family 2, P2 = II:3) and control (C1, C2) myotubes. Mitochondrially encoded subunits of complex I (ND1, ND2, ND3), complex III (CYB), complex IV (CO1, CO2, CO3; note that CO2 and CO3 comigrate), and complex V (ATP6, ATP8) are shown. An additional band (∗) seen in patient samples may represent a degradation or incomplete translation product.
(B) Immunoblot of selected mitochondrial- (complex I; ND1, complex IV; CO1, CO2) and nuclear-encoded (complex II; 70 kDa) RC subunits in patient myotubes.
Functional assays were performed with purified recombinant YARS2 to examine the consequence of the p.F52L substitution upon YARS2 enzymatic activity. An aminoacylation assay, measuring incorporation of [14C]-tyrosine into E. coli native tRNATyr substrate, showed that p.F52L YARS2 was catalytically active but with a ∼2-fold reduced rate as compared to the wild-type protein (kcat = 55 versus 128 × 10−3 s−1). The affinity of p.F52L YARS2 for tRNATyr was also reduced as indicated by an increased Km (Km = 3.9 versus 1.2 μM). Overall, there was a 9-fold loss in catalytic efficiency (kcat / Km) in p.F52L YARS2.
Discussion
Here we report the identification of a YARS2 mutation as a cause of MLASA in three patients from two apparently unrelated consanguineous families. Analysis of a control cohort suggests that this may be a founder mutation in the Lebanese population.
The mutation results in reduced tRNATyr aminoacylation efficiency. Analysis of the YARS2 crystal structure shows that the p.F52L mutation lies near the catalytic center where tRNA is tyrosylated17 and suggests an effect of F52 for recognizing and/or stabilizing the amino acid-accepting helix of tRNATyr that is lost in the mutant (Figure 4). The reduction in aminoacylation efficiency results in defective mitochondrial protein synthesis. Reduced availability of mitochondrially encoded RC complex subunits is probably rate limiting for RC complex assembly, underlying complex I, III, and IV RC dysfunction, each of which contains mitochondrially encoded subunits. It appears that fibroblasts can maintain sufficient levels of mitochondrially encoded RC components in the presence of this mutation; however, RC deficiency is manifest in muscle, possibly because of the higher requirements for RC components in muscle.20–22
Figure 4.
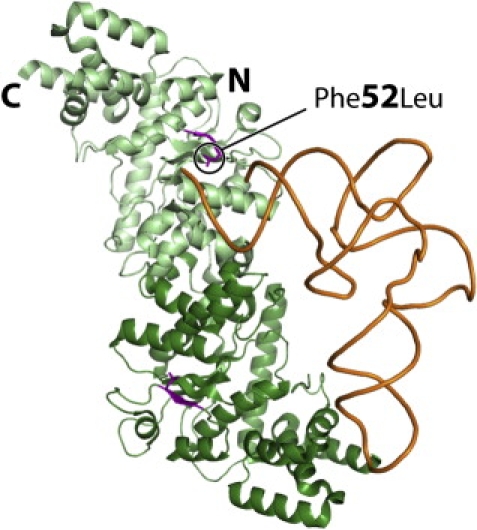
Interaction Model of Human YARS2 with tRNA
The model shows the crystal structure of the enzyme17 (without the S4-like C-terminal domain) on which one tRNATyr molecule has been docked according to its location in the crystal structure of a bacterial tryosyl-tRNA synthetase and tRNATyr complex.31 The two monomers are distinguished by dark- and light-green color with the first β strand encompassing residue 52 (p.F52L), shown as the mutant L52, highlighted in purple. Note the proximity of L52 with the accepting end of tRNA shown as orange.
The YARS2 mutation gives rise to a unique phenotype compared to other ARS mutations, despite their similar roles in protein synthesis. However, the possibility that mutations in other ARS genes could cause MLASA should not be excluded in patients with unidentified mutations. Mutations reported in both cytoplasmic and mitochondrial ARS genes have primarily affected the central nervous system16 whereas the YARS2 mutation does not appear to have any effects on brain structure or function. The reduction in oxidative phosphorylation capacity that results from the RC deficits would explain the myopathy, lactic academia,23 and sideroblastic anemia.24 In addition, YARS2 may have noncanonical functions, as is known for many ARSs,16,25 which may be affected by this mutation and thereby contribute to the unique phenotype compared to those resulting from DARS2 and RARS2 mutations.
The YARS2 phenotype more closely resembles that arising from PUS1 mutations because both are associated with MLASA. Onset of symptoms was at an earlier age (infancy–childhood) than generally reported for PUS1 mutation-positive patients (childhood–adolescence).5,26 As for PUS1 mutations,5 variability in severity of the phenotype was observed for the YARS2 mutation. The reduced severity of the disease in one family raises the possibility that an unknown modifier allows compensation for the decreased enzyme activity in vivo, or that perhaps a chaperone molecule with a stabilizing effect may be found among the alternative treatments of the less severely affected patient. It is also possible that the more severely affected family carries an additional mutation in another gene involved in mitochondrial RC homeostasis.
The pathogenesis of YARS2 and PUS1 mutations may be similar. PUS1 mutations result in loss of pseudouridine in some tRNAs, which may affect protein synthesis.6 The second anticodon position of tRNATyr is invariably uridine and is pseudouridylated in cytosolic human tRNATyr (ref. 27) and possibly in mitochondrial species. In patients with pathogenic PUS1 mutations, there is greatly reduced pseudouridylation6 that could occur at positions such as U35, where pseudouridylation has been shown to participate in tyrosine identity.28,29 This could imply a decreased tyrosylation capacity of hypomodified tRNATyr and therefore suggests that the marked clinical similarity between patients with PUS1 and YARS2 mutations is through a common mechanism triggered by altered tRNATyr charging. However, it has also been proposed that the clinical features associated with PUS1 mutations may be due to effects on non-tRNA molecules.30
In conclusion, the p.F52L YARS2 mutation results in reduced mitochondrial tRNATyr aminoacylation activity leading to a reduction in mitochondrial protein synthesis and is another cause of MLASA. It remains to be established whether mutations in YARS2 will be frequently observed in other cases of MLASA and other clinical phenotypes where sideroblastic anemia is a predominant feature.
Acknowledgments
Thanks to N. Fischel-Ghodsian for PUS1 mutation screening, L. Bonnefond for preparation of Figure 4, S. Taplin for assistance in the generation of Figure S1, L. Waddell for cutting muscle sections, and M. Menezes for preliminary haplotype analysis. This work was supported by a grant from the March of Dimes Foundation. D.T. is supported by an Australian National Health and Medical Research Council Principal Research Fellowship. M.B. is supported by an Australian National Health and Medical Research Council Career Development Award.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Skladal D., Halliday J., Thorburn D.R. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126:1905–1912. doi: 10.1093/brain/awg170. [DOI] [PubMed] [Google Scholar]
- 2.Thorburn D. Practical problems in detecting abnormal mitochondrial function and genomes. Hum. Reprod. 2000;15 (Suppl):57–67. doi: 10.1093/humrep/15.suppl_2.57. [DOI] [PubMed] [Google Scholar]
- 3.DiMauro S., Schon E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 4.Zeharia A., Fischel-Ghodsian N., Casas K., Bykhocskaya Y., Tamari H., Lev D., Mimouni M., Lerman-Sagie T. Mitochondrial myopathy, sideroblastic anemia, and lactic acidosis: An autosomal recessive syndrome in Persian Jews caused by a mutation in the PUS1 gene. J. Child Neurol. 2005;20:449–452. doi: 10.1177/08830738050200051301. [DOI] [PubMed] [Google Scholar]
- 5.Fernandez-Vizarra E., Berardinelli A., Valente L., Tiranti V., Zeviani M. Nonsense mutation in pseudouridylate synthase 1 (PUS1) in two brothers affected by myopathy, lactic acidosis and sideroblastic anaemia (MLASA) J. Med. Genet. 2007;44:173–180. doi: 10.1136/jmg.2006.045252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patton J.R., Bykhovskaya Y., Mengesha E., Bertolotto C., Fischel-Ghodsian N. Mitochondrial myopathy and sideroblastic anemia (MLASA): Missense mutation in the pseudouridine synthase 1 (PUS1) gene is associated with the loss of tRNA pseudouridylation. J. Biol. Chem. 2005;280:19823–19828. doi: 10.1074/jbc.M500216200. [DOI] [PubMed] [Google Scholar]
- 7.Kirby D., Crawford M., Cleary M., Dahl H., Dennett X., Thorburn D. Respiratory chain complex I deficiency: An underdiagnosed energy generation disorder. Neurology. 1999;52:1255–1264. doi: 10.1212/wnl.52.6.1255. [DOI] [PubMed] [Google Scholar]
- 8.Bahlo M., Bromhead C.J. Generating linkage mapping files from Affymetrix SNP chip data. Bioinformatics. 2009;25:1961–1962. doi: 10.1093/bioinformatics/btp313. [DOI] [PubMed] [Google Scholar]
- 9.Abecasis G.R., Wigginton J.E. Handling marker-marker linkage disequilibrium: Pedigree analysis with clustered markers. Am. J. Hum. Genet. 2005;77:754–767. doi: 10.1086/497345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thiele H., Nürnberg P. HaploPainter: A tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–1732. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- 11.Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.E., Walford G.A., Sugiana C., Boneh A., Chen W.K. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooper S.T., Kizana E., Yates J.D., Lo H.P., Yang N., Wu Z.H., Alexander I.E., North K.N. Dystrophinopathy carrier determination and detection of protein deficiencies in muscular dystrophy using lentiviral MyoD-forced myogenesis. Neuromuscul. Disord. 2007;17:276–284. doi: 10.1016/j.nmd.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 13.Tanji K., Bonilla E. Light microscopic methods to visualize mitochondria on tissue sections. Methods. 2008;46:274–280. doi: 10.1016/j.ymeth.2008.09.027. [DOI] [PubMed] [Google Scholar]
- 14.McKenzie M., Lazarou M., Ryan M.T. Analysis of respiratory chain complex assembly with radiolabeled nuclear- and mitochondrial-encoded subunits. Methods Enzymol. 2009;456:321–339. doi: 10.1016/S0076-6879(08)04418-2. [DOI] [PubMed] [Google Scholar]
- 15.Bonnefond L., Fender A., Rudinger-Thirion J., Giegé R., Florentz C., Sissler M. Toward the full set of human mitochondrial aminoacyl-tRNA synthetases: Characterization of AspRS and TyrRS. Biochemistry. 2005;44:4805–4816. doi: 10.1021/bi047527z. [DOI] [PubMed] [Google Scholar]
- 16.Antonellis A., Green E.D. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu. Rev. Genomics Hum. Genet. 2008;9:87–107. doi: 10.1146/annurev.genom.9.081307.164204. [DOI] [PubMed] [Google Scholar]
- 17.Bonnefond L., Frugier M., Touzé E., Lorber B., Florentz C., Giegé R., Sauter C., Rudinger-Thirion J. Crystal structure of human mitochondrial tyrosyl-tRNA synthetase reveals common and idiosyncratic features. Structure. 2007;15:1505–1516. doi: 10.1016/j.str.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 18.Barrett J.C., Fry B., Maller J., Daly M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 19.Fernández-Vizarra E., Tiranti V., Zeviani M. Assembly of the oxidative phosphorylation system in humans: What we have learned by studying its defects. Biochim. Biophys. Acta. 2009;1793:200–211. doi: 10.1016/j.bbamcr.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 20.Mootha V.K., Bunkenborg J., Olsen J.V., Hjerrild M., Wisniewski J.R., Stahl E., Bolouri M.S., Ray H.N., Sihag S., Kamal M. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115:629–640. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- 21.Antonicka H., Sasarman F., Kennaway N.G., Shoubridge E.A. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum. Mol. Genet. 2006;15:1835–1846. doi: 10.1093/hmg/ddl106. [DOI] [PubMed] [Google Scholar]
- 22.Remels A.H., Langen R.C., Schrauwen P., Schaart G., Schols A.M., Gosker H.R. Regulation of mitochondrial biogenesis during myogenesis. Mol. Cell. Endocrinol. 2010;315:113–120. doi: 10.1016/j.mce.2009.09.029. [DOI] [PubMed] [Google Scholar]
- 23.Koenig M.K. Presentation and diagnosis of mitochondrial disorders in children. Pediatr. Neurol. 2008;38:305–313. doi: 10.1016/j.pediatrneurol.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DiMauro S., Hirano M., Schon E. Parthenon Publishing; Lancaster, UK: 2006. Mitochondrial Medicine. [Google Scholar]
- 25.Park S.G., Schimmel P., Kim S. Aminoacyl tRNA synthetases and their connections to disease. Proc. Natl. Acad. Sci. USA. 2008;105:11043–11049. doi: 10.1073/pnas.0802862105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Casas K., Bykhovskaya Y., Mengesha E., Wang D., Yang H., Taylor K., Inbal A., Fischel-Ghodsian N. Gene responsible for mitochondrial myopathy and sideroblastic anemia (MSA) maps to chromosome 12q24.33. Am. J. Med. Genet. A. 2004;127A:44–49. doi: 10.1002/ajmg.a.20652. [DOI] [PubMed] [Google Scholar]
- 27.Jühling F., Mörl M., Hartmann R.K., Sprinzl M., Stadler P.F., Pütz J. tRNAdb 2009: Compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37(Database issue):D159–D162. doi: 10.1093/nar/gkn772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fechter P., Rudinger-Thirion J., Théobald-Dietrich A., Giegé R. Identity of tRNA for yeast tyrosyl-tRNA synthetase: Tyrosylation is more sensitive to identity nucleotides than to structural features. Biochemistry. 2000;39:1725–1733. doi: 10.1021/bi992276t. [DOI] [PubMed] [Google Scholar]
- 29.Giegé R., Lapointe J. Transfer RNA aminoacylation and modified nucleosides. In: Grosjean H., editor. DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution. Landes Bioscience; Georgetown, TX, USA: 2009. pp. 475–492. [Google Scholar]
- 30.Bykhovskaya Y., Mengesha E., Fischel-Ghodsian N. Pleiotropic effects and compensation mechanisms determine tissue specificity in mitochondrial myopathy and sideroblastic anemia (MLASA) Mol. Genet. Metab. 2007;91:148–156. doi: 10.1016/j.ymgme.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yaremchuk A., Kriklivyi I., Tukalo M., Cusack S. Class I tyrosyl-tRNA synthetase has a class II mode of cognate tRNA recognition. EMBO J. 2002;21:3829–3840. doi: 10.1093/emboj/cdf373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.