

Abstract
Age-related hearing loss is due to death over time, primarily by apoptosis, of hair cells in the inner ear. Studies of mutant genes responsible for inherited progressive hearing loss have suggested possible mechanisms for hair cell death, but critical connections between these mutations and the causes of progressive hearing loss have been elusive. In an Israeli kindred, dominant, adult-onset, progressive nonsyndromic hearing loss DFNA51 is due to a tandem inverted genomic duplication of 270 kb that includes the entire wild-type gene encoding the tight junction protein TJP2 (ZO-2). In the mammalian inner ear, TJP2 is expressed mainly in tight junctions, and also in the cytoplasm and nuclei. TJP2 expression normally decreases with age from embryonic development to adulthood. In cells of affected family members, TJP2 transcript and protein are overexpressed, leading to decreased phosphorylation of GSK-3β and to altered expression of genes that regulate apoptosis. These results suggest that TJP2- and GSK-3β-mediated increased susceptibility to apoptosis of cells of the inner ear is the mechanism for adult-onset hearing loss in this kindred and may serve as one model for age-related hearing loss in the general population.
Main Text
Age-related hearing loss is an extremely common problem worldwide. It is caused by the loss over time, primarily by apoptosis, of nonregenerative hair cells in the inner ear.1,2 In a large kindred from Israel, a gene responsible for progressive adult-onset hearing loss provides a clue to the mechanisms linking apoptosis to age-related hearing loss. Heretofore, genes responsible for hearing loss have been identified through point mutations, insertions, or deletions. Array comparative genomic hybridization (arrayCGH) enables genome-wide discovery of more complex mutations, such as microdeletions or microduplications, responsible for hearing loss. With arrayCGH, we identified DFNA51 (MIM 612642) as an inverted genomic duplication of the tight junction protein gene TJP2 (MIM 607709), leading to overexpression of the TJP2 protein and altered expression of genes that regulate apoptosis. Overexpression of TJP2 implicates the GSK-3β pathway in apoptosis leading to progressive hearing loss.
Family T, of Jewish ancestry, immigrated to Israel from Tunisia in 1951. In recent years, relatives in the kindred have sought medical advice for progressive hearing loss (Figure 1A). Pure tone audiometry revealed hearing loss with onset in the fourth decade, progressing first at high frequencies and ultimately becoming severe to profound at all frequencies (Figure 1B). There were no complaints of vertigo, dizziness, disequilibrium, or imbalance in affected individuals, as evaluated by the ocular motor and vestibular examination.3 Genome-wide linkage analysis, under a model postulating a dominant highly penetrant susceptibility allele, was undertaken for 58 relatives from family T aged 30 years and older. Of the 350 microsatellite markers tested, only one marker yielded a lod score > 3.0: at D9S175, Z = 6.56 (theta = 0). Genotypes of multiple additional microsatellite markers flanking D9S175 revealed perfect linkage of the phenotype to chromosome 9p13.3-q21.13 from 38.177 MB to 79.921 MB (hg19) (Figure 1C). This genomic region harbors more than 80 known genes, including the transmembrane channel gene TMC1 (MIM 606706), mutations in which are responsible for DFNA36 (MIM 606705) and DFNB7/11 (MIM 600974).4 We sequenced TMC1 from genomic DNA and from cDNA isolated from lymphoblasts of affected individuals of family T and found no rare variants coinherited with hearing loss. In addition, heterozygosity at rs2589615 in TMC1 exon 6 indicated that both TMC1 messages were present in lymphoblasts of affected individuals. By using genomic DNA from relatives of family T, we sequenced exons and flanking regulatory regions of 20 other genes (including TJP2) in the linked region. No deleterious mutations were detected in these 21 genes.
Figure 1.
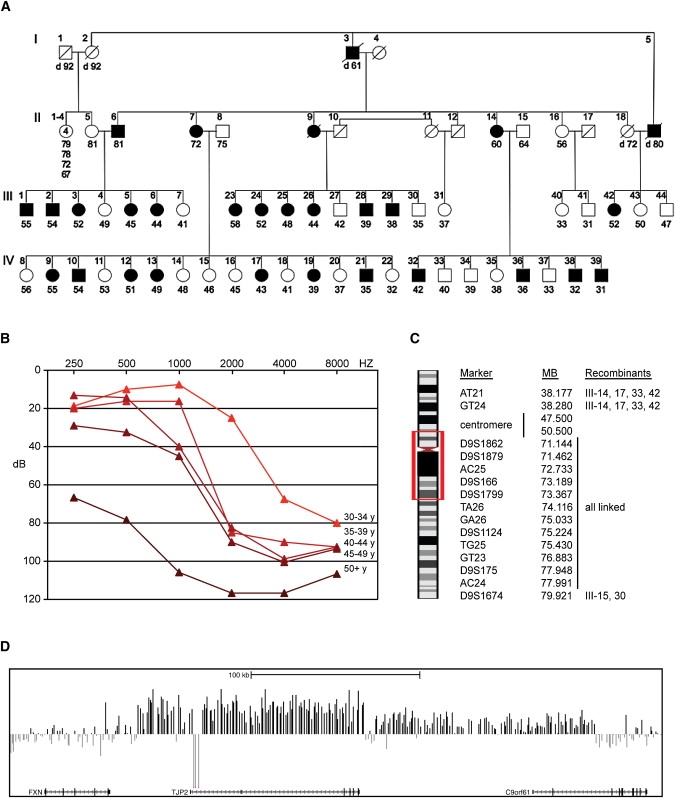
Linkage Analysis and Identification of the Inverted Genomic Duplication in Family T
(A) Family T with progressive hearing loss indicated by filled symbols.
(B) Audiogram of hearing loss by age.
(C) Chromosome 9 region of linkage to the hearing loss phenotype. Recombinant individuals are indicated in the notation of (A).
(D) arrayCGH scan indicating the family-specific duplication of ∼270 kb in genomic DNA of II-7. arrayCGH also detected homozygosity for a polymorphic deletion of 8760 bp within the larger novel duplicated region. The study was approved by the Helsinki Committees of Tel Aviv University and the Israel Ministry of Health and by the Human Subjects Division of the University of Washington.
In humans, the pericentromeric region of chromosome 9 is densely packed with segmental genomic duplications (segdups) and is prone to microdeletions and microduplications.5 In order to evaluate this region for microdeletions and microduplications in family T, we screened genomic DNA from affected individual II-7 by arrayCGH with the Nimblegen HD2 platform with the previously described CHP-SKN sample6 as the reference. Data were normalized and CNVs were called by identifying regions where Z-scores consistently deviated from the diploid mean. At 9q21.11, a genomic duplication of ∼270 kb was apparent in the genomic DNA of II-7 (Figure 1D). The duplication spanned 218 probes, with a median Z-score of 2.2. This duplication was not observed in the Database of Genomic Variants (March 25, 2010 update).
Genomic duplications may or may not be in tandem with their parent segment and may be either in the same or inverted orientation.7 We developed primers that would uniquely amplify genomic DNA with the duplication under each of these conditions. Forward (5′-CCCAGCAGAAGCAATGGTGGTAGCC-3′) and reverse (5′-GGTGGTGAATCCAAAAACACAAGAACAAAGTC-3′) primers diagnostic for a tandem inverted duplication (Figure 2A) yielded products of expected size in family T relatives with hearing loss, but yielded no product in unaffected family T relatives (Figure 2B). Genotypes of all 58 participating relatives in family T indicated that the tandem inverted duplication was coinherited with hearing loss. The duplication spans approximately positions 71,705,804 to 71,974,823 (hg19) on chromosome 9 for a size of ∼269,023 bp. The duplication includes the entire locus for the tight junction protein TJP2, which spans positions 71,788,971 to 71,870,124 (hg19).
Figure 2.
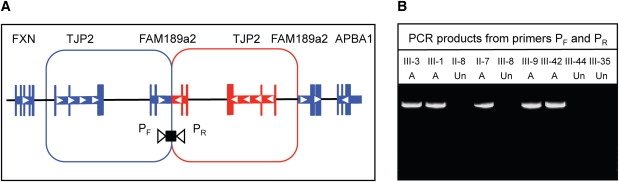
Characterization of the Inverted Duplication at TJP2
(A) Sketch of the inverted duplication.
(B) PCR products, ∼7 kb in size, amplified with primers specific to the inverted duplication, from genomic DNA of affected (A) and unaffected (Un) relatives of family T.
The distal breakpoint of the inverted duplication occurs in intron 2 of FAM189A2 (also named c9orf61). The annotated human sequence of FAM189A2 indicates exon 1 to be noncoding and exon 2 to include only four translated codons. However, at the orthologous mouse sequence Fam189a2, most of exon 1 is an open reading frame beginning with Met, and together with exon 2 is predicted to encode 150 amino acids. Genomic sequence of human and mouse are very similar in this region. We carried out RT-PCR from human fetal brain and human lymphoblast RNA and found that the human and mouse transcripts are conserved (data not shown). However, with 3′ rapid amplification of cDNA ends (3′ RACE), we found no evidence of a mutant FAM189A2 transcript in carriers of the genomic duplication, suggesting that no truncated FAM189A2 product was made. In addition, there was no correlation of genotype with expression of full-length FAM189A2 transcripts among family T relatives (Figure S1 available online). We thus focused on the duplication of TJP2 as the likely cause of the hearing loss in family T.
In order to characterize expression of Tjp2 during development, we assessed levels of Tjp2 transcript expression in the mouse ear by quantitative RT-PCR of RNA from wild-type C3H mice at ages E16.5, P0, 1 week, 1 month, 3 months, and 9 months (Figure 3A). Total RNA was harvested from mouse whole inner ears and reverse transcribed with random primers. Quantitative PCR (qPCR) was performed on an ABI 7900HT Real-Time PCR System, according to manufacturer's instructions and with the TaqMan Gene Expression Assays indicated in Table S1. Relative to levels of the internal control Hprt, Tjp2 expression decreased rapidly between E16.5 and 1 week to a level in adult mice approximately 50% the level at birth.
Figure 3.
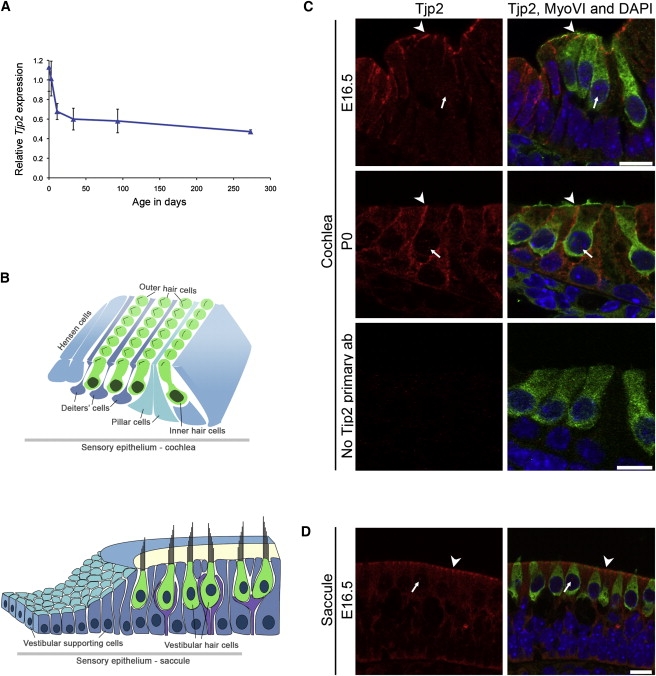
Expression of Tjp2 in the Mouse Inner Ear
(A) Expression of Tjp2 transcript at ages E16.5, P0, 1 week, 1 month, 3 months, and 9 months, determined by real-time qPCR, reveals decline with age until after hearing maturation at P30, then a stable level during adulthood. Values represent mean ± SD of samples run in triplicate. The experiment was repeated twice; a representative experiment is shown.
(B) Schematic representation of the P0 cochlea (top) and saccule (bottom). The inner and outer hair cells of the cochlea make up the sensory epithelium; nonsensory supporting cells include Deiters', pillar, and Hensen cells. The saccule contains sensory hair cells and nonsensory supporting cells.
(C and D) Immunofluorescence confocal images of the cochlea (C) at E16.5 and P0 and of the saccule (D) at E16.5. Tjp2 expression (red) is seen mainly at cell junctions (arrowheads) and also in the cytoplasm and nuclei (arrows) of hair cells and adjacent supporting cells. The vestibular pattern of expression was similar in the utricle and cristae at these ages (data not shown). Negative control staining of P0 cochlea without primary antibody against Tjp2 demonstrates the low background of the secondary antibody. Hair cell cytoplasm and nuclei are marked by myosin VI (MyoVI, green) and DAPI (blue), respectively. Paraffin-embedded sections were stained with the primary antibodies goat anti-myosin VI (Santa Cruz Biotechnology), rabbit anti-ZO-2 (Cell Signaling), and mouse anti-ZO-1 (Zymed) and the fluorescence-conjugated secondary antibodies donkey anti-goat 488, donkey anti-rabbit 594, and donkey anti-mouse rhodamine (Molecular Probes). Imaging was done with the LSM 510 confocal microscope (Zeiss). All procedures involving animals met NIH guidelines and were approved by the Animal Care and Use Committees of Tel Aviv University and the University of Washington. Scale bar represents 10 μm.
We evaluated localization of Tjp2 protein in the mouse inner ear at various ages by immunohistochemistry of paraffin-embedded sections (Figure 3). In the cochlea, Tjp2 is localized most prominently in membranes connecting hair cells and supporting cells (Figure 3C). Tjp2 is also localized in membranes connecting the cells of the vestibular system (Figure 3D). Localization is punctate and most concentrated at membrane boundaries, as expected for junctional staining.8 In the hair cell, Tjp2 is localized both at the apical edge, associated with tight junctions, and also along the basolateral side. In most epithelial cells, the basolateral side contains adherens junctions, but in the inner ear, apical and basal junctions are not distinct but form a combined structure, the tight-adherens junction, between outer hair cells and supporting Deiter cells.9 To a lesser extent, Tjp2 is also localized to both the cytoplasm and nucleus, presumably reflecting its role in signal transduction.10 The contrast between localization of Tjp2 and the closely related Tjp1 is illustrated in Figure S2.
By both genomic position and function, TJP2 is an excellent candidate for the gene responsible for hearing loss in family T. However, the sequence of TJP2 in family T is wild-type. In order to evaluate how genomic duplication of wild-type TJP2 might lead to progressive hearing loss, we compared expression of TJP2 in lymphoblasts of family T relatives carrying the duplication versus relatives not carrying the duplication. In cells of individuals with the duplication, endogenous levels of TJP2 message, relative to 18S rRNA, were elevated approximately 1.7-fold (Mann-Whitney U test, p = 0.002) (Figure 4A). Endogenous levels of TJP2 protein were elevated approximately 2-fold (Figures 4B and 4C).
Figure 4.
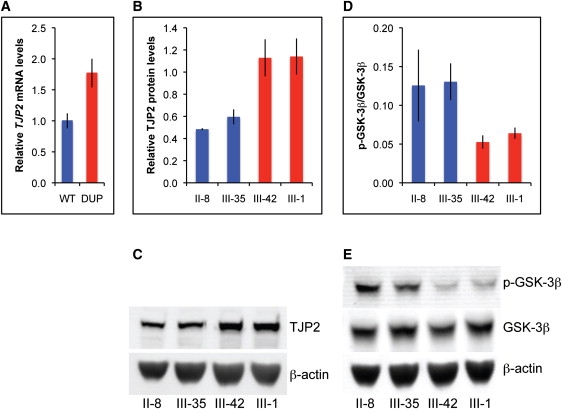
Overexpression of TJP2 and Reduced Phosphorylation of GSK-3β Associated with the Genomic Duplication
(A) Expression of TJP2 transcript in lymphoblasts of family T relatives with (DUP) and without (WT) the genomic duplication, determined by real-time qPCR. Values represent mean ± SEM of data from three independent experiments, which included cells from four individuals without the duplication and six with the duplication.
(B–E) Lymphoblast cell lysates of family T relatives were analyzed by immunoblotting with primary antibodies rabbit anti-TJP2 (Santa Cruz Biotechnology), mouse anti-GSK-3β (BD Transduction Laboratories), rabbit anti-phospho(Ser9)-GSK-3β (p-GSK-3β) (Cell Signaling Technology), and mouse anti-β-actin (Sigma) and IRDye-conjugated secondary antibodies (LI-COR Biosciences). Levels of TJP2 relative to β-actin (B) and levels of p-GSK-3β relative to GSK-3β (D) were quantified from western blots (C, E). Blue bars represent results from individuals without the duplication; red bars represent results for individuals with the duplication. Individuals are labeled as in Figure 1. Values in (B) and (D) represent mean ± SEM of data from three independent experiments; representative western blots are shown.
Experimental overexpression of TJP2 has been shown to decrease phosphorylation of the serine/threonine protein kinase GSK-3β at position serine 9.11 Phosphorylation at Ser9 is known to inhibit the kinase activity of GSK-3β.12,13 In order to determine the consequences of endogenous overexpression of TJP2, we evaluated phosphorylation of GSK-3β at Ser9 in cells of individuals with and without the genomic duplication (Figures 4D and 4E). The levels of total GSK-3β were similar, but the amount of phosphorylated GSK-3β was lower in cells of persons with the duplication. The decreased ratio of phosphorylated to unphosphorlylated GSK-3β suggests that GSK-3β activity was higher in cells with the duplication.
Apoptosis of cells in the cochlea, including transcriptional changes in genes involved in apoptosis, has been implicated in age-related hearing loss in mice,14–16 as well as in hearing loss induced by acoustic overstimulation17 and chemotherapeutic agents.18 Because GSK-3β has been shown to promote the mitochondrial intrinsic apoptosis pathway,13 and GSK-3β inhibitors can block cisplatin-induced ototoxicity in mice,18 we investigated whether genomic duplication of TJP2 led to changes in expression of apoptosis-related genes. We screened lymphoblast cells of four family T relatives, two with and two without the duplication, for differential expression of 92 apoptosis-related genes via human cellular apoptosis pathway plates (Applied Biosystems). Expression of the apoptosis-related genes was normalized to the expression of HPRT as an endogenous control. Genes were chosen for further analysis if expression by genotype differed at least 2-fold with t test p < 0.2. Four genes (BCL2L11, IL6, REL, and TSPO) were identified by these criteria (Table S2). On replication, all four genes showed significant differences in expression with p < 0.01 (Figure 5A). Expression of BCL2L11, REL, and TSPO was observed in mouse cochlea; IL6 was not detectably expressed (data not shown).
Figure 5.
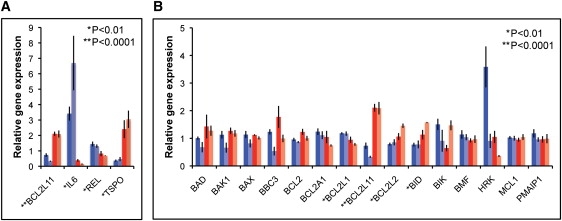
Effects of the TJP2 Duplication on Expression of Apoptosis-Related Genes
Real-time qPCR analyses of RNA from lymphoblasts of family T relatives (blue and red bars and individual samples as in Figure 4).
(A) Expression of genes identified as differentially expressed in the screen of apoptosis-related genes.
(B) Expression of BCL2 family genes. Gene expression was normalized to the geometric mean of the expression of the endogenous control genes HPRT, TBP, and UBC.41 Values represent mean ± SEM of data from three independent experiments. The result for BCL2L11 is indicated in both (A) and (B).
BCL2L11 (also known as BIM) and TSPO can both promote apoptosis via the intrinsic mitochondrial pathway19–21 and REL can suppress apoptosis as a subunit of the antiapoptotic transcription factor NF-κB.22 BCL2L11 (BIM) is a member of the BCL2 family of proteins, which includes both pro- and antiapoptotic members. The balance between pro- and antiapoptotic BCL2 family proteins influences the sensitivity of cells to apoptosis.20,21 Only a subset of BCL2 family genes were evaluated in our initial screen of 92 apoptosis-related genes. We were interested in whether any genes in this family in addition to BCL2L11 (BIM) were differentially expressed in cells with and without the genomic duplication. When the remaining BCL2 family members20,21 were evaluated, we found that three additional genes were differentially expressed with p < 0.01. Expression of the proapoptotic gene BID was elevated 1.7-fold and expression of the antiapoptotic isoform of BCL2L1 (BCL-xL) was decreased 1.4-fold. In addition, expression of the antiapoptotic BCL2L2 (BCL-w) was elevated 1.5-fold (Figure 5B). Based on the GEPIS and UniGene databases, all of these genes are expressed in mouse inner ear. Taken together, these data suggest that in cells carrying the genomic duplication, there is an overall shift in expression of BCL2 family genes that would favor apoptosis.
Tight junction proteins play multiple roles in epithelial cells.23 Claudins, tricellulins, and zona occludins (TJP) family members are critical to the formation of diffusion barriers that regulate cellular permeability.24 In the inner ear, tight junctions linking the cells of the sensory epithelia are crucial for the maintenance of separation between endolymphatic and perilymphatic fluids, which differ in their ionic composition.25 This separation and the controlled transfer of ions, namely K+, under specific sound and vibration conditions, allow the formation of the endocochlear potential and the conversion of fluid changes into mechanical stimuli. Tight junctions ensure the strict compartmentalization of these regions. Mutations in tight junction proteins claudin-11, claudin-14, and tricellulin lead to hearing loss.26–29
Tight junctions also participate in signal transduction mechanisms that regulate cell proliferation and gene expression. TJP2 and related proteins are localized both at tight junctions and in the nucleus. In the nucleus, TJP2 most probably functions in the regulation of gene expression by regulating nuclear import and export of transcription factors and other proteins.30 TJP2 has been shown to regulate GSK-3β,11 a serine/threonine kinase involved in signal transduction pathways that regulate a wide range of cellular processes. In response to various upstream signals, GSK-3β is phosphorylated at Ser9, inhibiting its kinase activity.12,13 Overexpression of TJP2 leads to a decrease in GSK-3β Ser9 phosphorylation,11 thereby increasing GSK-3β activity. Results for family T are consistent with this pattern, in that levels of TJP2 transcript and protein are elevated and GSK-3β-Ser9 phosphorylation is decreased in lymphoblast cells of family T members carrying the TJP2 duplication. Decreased GSK-3β-Ser9 phosphorylation is an indicator of increased GSK-3β activity in these cells.
In various cell types, including neurons, increased GSK-3β activity makes cells more susceptible to apoptosis.13 GSK-3β directly or indirectly regulates several key components of the intrinsic apoptotic pathway, including members of the BCL2 family. GSK-3β is required for the induction of BCL2L11 (BIM) protein during stress- and dexamethasone-induced apoptosis.31,32 Phosphorylation by GSK-3β activates the proapoptotic BAX,33 which was previously linked to age-related hearing loss,34 and destabilizes the antiapoptotic MCL1.35 GSK-3β activity also regulates permeability of the mitochondrial outer membrane through phosphorylation of VDAC.36 In addition, phosphorylation of tau or kinesin by GSK-3β can disrupt microtubules or intracellular protein transport, respectively, which can also contribute to apoptosis.13 The gene expression changes in lymphoblast cells of TJP2 duplication carriers indicate a shift toward increased expression of proapoptotic and decreased expression of antiapoptotic genes, consistent with the known role of GSK-3β in promoting apoptosis. Although the antiapoptotic BCL2L2 (BCL-w) gene is slightly elevated, expression changes seen for other BCL2 family members, as well as for TSPO and REL, probably shift the overall balance toward an increased sensitivity to apoptosis. In addition to these transcriptional changes, it is likely that elevated GSK-3β activity also results in posttranslational cellular changes favoring apoptosis.
We hypothesize that in the inner ear, as in lymphoblasts, TJP2 overexpression results in changes in GSK-3β phosphorylation and apoptosis-related gene expression and that these changes increase the susceptibility of inner ear cells to apoptosis. Consistent with the hypothesis that GSK-3β can regulate hair cell survival, inhibition of PI3 kinase signaling, which can negatively regulate GSK-3β, blocked the ability of dexamethasone to protect cells from ototoxicity in a rat model of trauma-induced hearing loss.37 TJP2 is widely expressed, yet TJP2 overexpression via genomic duplication leads only to hearing loss. We speculate that because the inner ear is a very sensitive organ, a subtle difference in expression of genes in apoptotic pathways is manifested in a hearing loss phenotype. Even a subtle increase in apoptotic susceptibility of inner ear hair cells could account for the progressive hearing loss of carriers of the TJP2 duplication. Our experiments suggest that overexpressed TJP2 in family T modulates intracellular signaling. This does not preclude the possibility that excess TJP2 may also perturb the stability of intercellular junctions and that this could also contribute to the hearing loss in family T.
Hearing loss associated with overexpression of TJP2 may resemble age-related hearing loss generally in that both involve apoptosis. The dramatic decrease in Tjp2 expression in the mouse ear by early adulthood suggests that maintaining relatively low levels of Tjp2 from this stage on may be important for maintaining normal inner ear function throughout adulthood. In principle, other gain-of-expression mutations in TJP2 could have a similar effect. An amino acid substitution in TJP2 that segregates with dominant progressive hearing loss has been reported in a Guatemalan family, although levels of TJP2 transcript and protein were not analyzed.38
A missense mutation in TJP2 has also been reported in Amish families with oligogenic inheritance of hypercholanemia (MIM 607748) involving mutations in both TJP2 and BAAT (MIM 602938). The TJP2 mutation, V48A, is in the N-terminal PDZ domain and affects protein folding, changing relative binding affinities of TJP2 for different claudins and leading to changes in ratios of claudins in tight junctions.39 The relatives of family T do not have any signs of familial hypercholanemia. In family T, TJP2 is overexpressed via genomic duplication, leading to modulation of intracellular signaling, and, we suggest, increasing the susceptibility of inner ear cells to apoptosis, leading to progressive hearing loss.
The general consensus is that age-related progressive hearing loss is due to loss of hair cells over time by apoptosis. Although this is a common theme, the routes to apoptosis are varied. Studies of mutant proteins responsible for progressive hearing loss have suggested possible mechanisms for hair cell death, including mechanical stress and aberrations of DNA repair, cell cycle progression, and cell signaling.40 In most of these cases, the critical connection between the mutation and the cause of the progressive hearing impairment remains elusive. The biology underlying progressive hearing loss in family T may offer one such connection. Overexpression of TJP2 in carriers of the genomic duplication suggests a mechanism for events leading to apoptosis, and thus eventually to death of hair cells and hearing loss in an extended family.
Acknowledgments
The authors would like to thank the members of family T for suggesting this project and for their continued commitment to it. We also thank Amiel Dror for schematic figures of the inner ear. This work was supported by National Institutes of Health grant R01DC005641 from the National Institute of Deafness and Communication Disorders, by the European Commission FP6 Integrated Project EUROHEAR LSHG-CT-2004-512063, and by NIH training grants 5T32ES007032 (to W.R.) and 1T32ES015459 (to A.S.N.) from the National Institute of Environmental Health Sciences.
Contributor Information
Mary-Claire King, Email: mcking@uw.edu.
Karen B. Avraham, Email: karena@post.tau.ac.il.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Database of Genomic Variants, http://projects.tcag.ca/variation/
Gene Hub–GEPIS, http://www.cgl.ucsf.edu/Research/genentech/genehub-gepis/index.html
Hereditary Hearing Loss Homepage, http://webhost.ua.ac.be/hhh/
NCBI UniGene, http://www.ncbi.nlm.nih.gov/unigene
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
R project for statistical computing, http://www.r-project.org
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Kwan T., White P.M., Segil N. Development and regeneration in the inner ear. Ann. N Y Acad. Sci. 2009;1170:28–33. doi: 10.1111/j.1749-6632.2009.04484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng A.G., Cunningham L.L., Rubel E.W. Mechanisms of hair cell death and protection. Curr. Opin. Otolaryngol. Head Neck Surg. 2005;13:343–348. doi: 10.1097/01.moo.0000186799.45377.63. [DOI] [PubMed] [Google Scholar]
- 3.Zee D.S., Fletcher W.A. Bedside examination. In: Baloh R.W., Halmagyi G.M., editors. Disorders of the Vestibular System. Oxford University Press; New York: 1996. pp. 178–190. [Google Scholar]
- 4.Kurima K., Peters L.M., Yang Y., Riazuddin S., Ahmed Z.M., Naz S., Arnaud D., Drury S., Mo J., Makishima T. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat. Genet. 2002;30:277–284. doi: 10.1038/ng842. [DOI] [PubMed] [Google Scholar]
- 5.Rocchi M., Archidiacono N., Ward D.C., Baldini A. A human chromosome 9-specific alphoid DNA repeat spatially resolvable from satellite 3 DNA by fluorescent in situ hybridization. Genomics. 1991;9:517–523. doi: 10.1016/0888-7543(91)90419-f. [DOI] [PubMed] [Google Scholar]
- 6.Walsh T., McClellan J.M., McCarthy S.E., Addington A.M., Pierce S.B., Cooper G.M., Nord A.S., Kusenda M., Malhotra D., Bhandari A. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- 7.Bailey J.A., Gu Z., Clark R.A., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki T., Oyamada M., Takamatsu T. Different regulation of connexin26 and ZO-1 in cochleas of developing rats and of guinea pigs with endolymphatic hydrops. J. Histochem. Cytochem. 2001;49:573–586. doi: 10.1177/002215540104900504. [DOI] [PubMed] [Google Scholar]
- 9.Nunes F.D., Lopez L.N., Lin H.W., Davies C., Azevedo R.B., Gow A., Kachar B. Distinct subdomain organization and molecular composition of a tight junction with adherens junction features. J. Cell Sci. 2006;119:4819–4827. doi: 10.1242/jcs.03233. [DOI] [PubMed] [Google Scholar]
- 10.Traweger A., Fuchs R., Krizbai I.A., Weiger T.M., Bauer H.C., Bauer H. The tight junction protein ZO-2 localizes to the nucleus and interacts with the heterogeneous nuclear ribonucleoprotein scaffold attachment factor-B. J. Biol. Chem. 2003;278:2692–2700. doi: 10.1074/jbc.M206821200. [DOI] [PubMed] [Google Scholar]
- 11.Tapia R., Huerta M., Islas S., Avila-Flores A., Lopez-Bayghen E., Weiske J., Huber O., González-Mariscal L. Zona occludens-2 inhibits cyclin D1 expression and cell proliferation and exhibits changes in localization along the cell cycle. Mol. Biol. Cell. 2009;20:1102–1117. doi: 10.1091/mbc.E08-03-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugden P.H., Fuller S.J., Weiss S.C., Clerk A. Glycogen synthase kinase 3 (GSK3) in the heart: A point of integration in hypertrophic signaling and a therapeutic target? A critical analysis. Br. J. Pharmacol. 2008;153:S137–S153. doi: 10.1038/sj.bjp.0707659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beurel E., Jope R.S. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 2006;79:173–189. doi: 10.1016/j.pneurobio.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sha S.H., Chen F.Q., Schacht J. Activation of cell death pathways in the inner ear of the aging CBA/J mouse. Hear. Res. 2009;254:92–99. doi: 10.1016/j.heares.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Someya S., Xu J., Kondo K., Ding D., Salvi R.J., Yamasoba T., Rabinovitch P.S., Weindruch R., Leeuwenburgh C., Tanokura M., Prolla T.A. Age-related hearing loss in C57BL/6J mice is mediated by Bak-dependent mitochondrial apoptosis. Proc. Natl. Acad. Sci. USA. 2009;106:19432–19437. doi: 10.1073/pnas.0908786106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tadros S.F., D'Souza M., Zhu X., Frisina R.D. Apoptosis-related genes change their expression with age and hearing loss in the mouse cochlea. Apoptosis. 2008;13:1303–1321. doi: 10.1007/s10495-008-0266-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu B.H., Cai Q., Manohar S., Jiang H., Ding D., Coling D.E., Zheng G., Salvi R. Differential expression of apoptosis-related genes in the cochlea of noise-exposed rats. Neuroscience. 2009;161:915–925. doi: 10.1016/j.neuroscience.2009.03.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park H.J., Kim H.J., Bae G.S., Seo S.W., Kim D.Y., Jung W.S., Kim M.S., Song M.Y., Kim E.K., Kwon K.B. Selective GSK-3b inhibitors attenuate the cisplatin-induced cytotoxicity of auditory cells. Hear. Res. 2009;257:53–62. doi: 10.1016/j.heares.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 19.Veenman L., Papadopoulos V., Gavish M. Channel-like functions of the 18-kDa translocator protein (TSPO): Regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr. Pharm. Des. 2007;13:2385–2405. doi: 10.2174/138161207781368710. [DOI] [PubMed] [Google Scholar]
- 20.Brenner D., Mak T.W. Mitochondrial cell death effectors. Curr. Opin. Cell Biol. 2009;21:871–877. doi: 10.1016/j.ceb.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 21.Chipuk J.E., Moldoveanu T., Llambi F., Parsons M.J., Green D.R. The BCL-2 family reunion. Mol. Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kucharczak J., Simmons M.J., Fan Y., Gélinas C. To be, or not to be: NF-kB is the answer–role of Rel/NF-kappaB in the regulation of apoptosis. Oncogene. 2003;8:8961–8982. doi: 10.1038/sj.onc.1207230. [DOI] [PubMed] [Google Scholar]
- 23.Matter K., Balda M.S. Epithelial tight junctions, gene expression and nucleo-junctional interplay. J. Cell Sci. 2007;120:1505–1511. doi: 10.1242/jcs.005975. [DOI] [PubMed] [Google Scholar]
- 24.Schulzke J.D., Fromm M. Tight junctions: Molecular structure meets function. Ann. N Y Acad. Sci. 2009;1165:1–6. doi: 10.1111/j.1749-6632.2009.04925.x. [DOI] [PubMed] [Google Scholar]
- 25.Wangemann P. K+ cycling and the endocochlear potential. Hear. Res. 2002;165:1–9. doi: 10.1016/s0378-5955(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 26.Wilcox E.R., Burton Q.L., Naz S., Riazuddin S., Smith T.N., Ploplis B., Belyantseva I., Ben-Yosef T., Liburd N.A., Morell R.J. Mutations in the gene encoding tight junction claudin-14 cause autosomal recessive deafness DFNB29. Cell. 2001;104:165–172. doi: 10.1016/s0092-8674(01)00200-8. [DOI] [PubMed] [Google Scholar]
- 27.Ben-Yosef T., Belyantseva I.A., Saunders T.L., Hughes E.D., Kawamoto K., Van Itallie C.M., Beyer L.A., Halsey K., Gardner D.J., Wilcox E.R. Claudin-14 knockout mice, a model for autosomal recessive deafness DFNB29, are deaf due to cochlear hair cell degeneration. Hum. Mol. Genet. 2003;12:2049–2061. doi: 10.1093/hmg/ddg210. [DOI] [PubMed] [Google Scholar]
- 28.Gow A., Davies C., Southwood C.M., Frolenkov G., Chrustowski M., Ng L., Yamauchi D., Marcus D.C., Kachar B. Deafness in Claudin-11 null mice reveals the critical contribution of basal cell tight junctions to stria vascularis function. J. Neurosci. 2004;24:7051–7062. doi: 10.1523/JNEUROSCI.1640-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Riazuddin S., Ahmed Z.M., Fanning A.S., Lagziel A., Kitajiri S., Ramzan K., Khan S.N., Chattaraj P., Friedman P.L., Anderson J.M. Tricellulin is a tight-junction protein necessary for hearing. Am. J. Hum. Genet. 2006;79:1040–1051. doi: 10.1086/510022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balda M.S., Matter K. Tight junctions and the regulation of gene expression. Biochim. Biophys. Acta. 2009;1788:761–767. doi: 10.1016/j.bbamem.2008.11.024. [DOI] [PubMed] [Google Scholar]
- 31.Nuutinen U., Ropponen A., Suoranta S., Eeva J., Eray M., Pellinen R., Wahlfors J., Pelkonen J. Dexamethasone-induced apoptosis and up-regulation of Bim is dependent on glycogen synthase kinase-3. Leuk. Res. 2009;33:1714–1717. doi: 10.1016/j.leukres.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 32.Hongisto V., Smeds N., Brecht S., Herdegen T., Courtney M.J., Coffey E.T. Lithium blocks the c-Jun stress response and protects neurons via its action on glycogen synthase kinase 3. Mol. Cell. Biol. 2003;23:6027–6036. doi: 10.1128/MCB.23.17.6027-6036.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linseman D.A., Butts B.D., Precht T.A., Phelps R.A., Le S.S., Laessig T.A., Bouchard R.J., Florez-McClure M.L., Heidenreich K.A. Glycogen synthase kinase-3b phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J. Neurosci. 2004;24:9993–10002. doi: 10.1523/JNEUROSCI.2057-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Riva C., Donadieu E., Magnan J., Lavieille J.P. Age-related hearing loss in CD/1 mice is associated to ROS formation and HIF target proteins up-regulation in the cochlea. Exp. Gerontol. 2007;42:327–336. doi: 10.1016/j.exger.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 35.Maurer U., Charvet C., Wagman A.S., Dejardin E., Green D.R. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell. 2006;21:749–760. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 36.Pastorino J.G., Hoek J.B., Shulga N. Activation of glycogen synthase kinase 3b disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005;65:10545–10554. doi: 10.1158/0008-5472.CAN-05-1925. [DOI] [PubMed] [Google Scholar]
- 37.Haake S.M., Dinh C.T., Chen S., Eshraghi A.A., VanDeWater T.R. Dexamethasone protects auditory hair cells against TNFalpha-initiated apoptosis via activation of PI3K/Akt and NFkB signaling. Hear. Res. 2009;255:22–32. doi: 10.1016/j.heares.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 38.Hilgert N., Alasti F., Dieltjens N., Pawlik B., Wollnik B., Uyguner O., Delmaghani S., Weil D., Petit C., Danis E. Mutation analysis of TMC1 identifies four new mutations and suggests an additional deafness gene at loci DFNA36 and DFNB7/11. Clin. Genet. 2008;74:223–232. doi: 10.1111/j.1399-0004.2008.01053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carlton V.E.H., Harris B.Z., Puffenberger E.G., Bata A.K., Knisely A.S., Robinson D.L., Strauss K.A., Shneider B.L., Lim W.A., Salen G. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat. Genet. 2003;34:91–96. doi: 10.1038/ng1147. [DOI] [PubMed] [Google Scholar]
- 40.Dror A.A., Avraham K.B. Hearing loss: Mechanisms revealed by genetics and cell biology. Annu. Rev. Genet. 2009;43:411–437. doi: 10.1146/annurev-genet-102108-134135. [DOI] [PubMed] [Google Scholar]
- 41.Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:1–12. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.