

Abstract
Mosaicism is defined as the coexistence of cells with different genetic composition within an individual, caused by postzygotic somatic mutation. Although somatic mosaicism for chromosomal abnormalities is a well-established cause of developmental and somatic disorders and has also been detected in different tissues, its frequency and extent in the adult normal population are still unknown. We provide here a genome-wide survey of mosaic genomic variation obtained by analyzing Illumina 1M SNP array data from blood or buccal DNA samples of 1991 adult individuals from the Spanish Bladder Cancer/EPICURO genome-wide association study. We found mosaic abnormalities in autosomes in 1.7% of samples, including 23 segmental uniparental disomies, 8 complete trisomies, and 11 large (1.5–37 Mb) copy-number variants. Alterations were observed across the different autosomes with recurrent events in chromosomes 9 and 20. No case-control differences were found in the frequency of events or the percentage of cells affected, thus indicating that most rearrangements found are not central to the development of bladder cancer. However, five out of six events tested were detected in both blood and bladder tissue from the same individual, indicating an early developmental origin. The high cellular frequency of the anomalies detected and their presence in normal adult individuals suggest that this type of mosaicism is a widespread phenomenon in the human genome. Somatic mosaicism should be considered in the expanding repertoire of inter- and intraindividual genetic variation, some of which may cause somatic human diseases but also contribute to modifying inherited disorders and/or late-onset multifactorial traits.
Main Text
Genetic mosaicism results from a postzygotic mutation during development that is propagated to only a subset of adult cells. It can occur in either or both somatic and germline cells, the latter with the potential of passage to offspring.1 Among the somatic or germline mutations described in genetic mosaicism are point changes and small rearrangements, as well as structural and numerical chromosome aberrations.1,2 The most common form of mosaicism detected by karyotyping in pre- and perinatal diagnosis involves chromosomal aneuploidy, found in ∼50% of preimplantation embryos, 1% of chorionic villous samples, 0.2%–0.3% of amniotic fluids, and 0.1% of newborns.3,4 In single differentiated neurons, the average frequency of aneuploidy has been determined as 1.25%–1.45% per chromosome (30%–35% overall), with perhaps lower frequency in other cell types.5,6 Acquired monosomy of the X chromosome is a common type of mosaicism observed in normal individuals that is associated with aging.7 For large chromosomal structural variants, such as copy-number variations (CNVs), mosaicism has been more recently described on the basis of comparative analysis of differentiated human tissues from adult individuals8 as well as divergence between identical twins;8,9 the estimated frequency of postzygotic CNV could approach 5%. Molecular karyotyping with microarrays has also been used to detect mosaicism for chromosomal rearrangements and predict mutational mechanisms in clinical samples referred for routine diagnostic analysis.10–15 Although the frequency of uniparental disomy (UPD), the occurrence of two copies of a particular chromosome from the same parent, is unknown, it has been invoked as an important mechanism in carcinogenesis.16
The consequences of mosaicism nominally depend on the altered genetic architecture and specifically how it affects developmental and cell-specific pathways. So far, the majority of somatic mutations have been described in relation to clinical samples with a known phenotype, thus representing mosaic aberrations with strong effect, even though the mosaicism may result in either a milder or unusual disease phenotype.10–15,17,18 However, mosaic somatic changes can have no apparent phenotypic effect, and their occurrence can go undetected with most high-throughput methods of genome analysis applied to DNA obtained from samples containing large numbers of cells. Thus, the frequency and relevance of mosaicism are likely underestimated.
We provide here a survey of mosaic UPDs and segmental and complete aneuploidies of the human genome by molecular karyotyping with SNP arrays in 1991 adult individuals included in the Spanish Bladder Cancer/EPICURO study: 1034 patients and 957 hospital-based controls with a mean age of 63.7 years (range 20–82 years), 87% of whom were male.19,20 Cases were patients newly diagnosed with urothelial cell carcinoma of the bladder (MIM 109800). Controls were age-, sex-, and hospital-matched inpatients mainly recruited from the general surgery and traumatology departments with diagnoses not associated with bladder cancer risk factors. The study was approved by the institutional ethics committees of each participating hospital and the institutional review board of the National Cancer Institute (NCI, USA). Written informed consent was obtained from all individuals. DNA was extracted from peripheral blood with the Puregene DNA Isolation Kit (Gentra Systems) for most cases (n = 1107) and controls (n = 1032) included in the analysis. DNA from an additional 43 cases and 117 controls was extracted from mouthwash samples with phenol/chloroform. Formalin-fixed paraffin-embedded tissue blocks of tumors obtained at surgery were also available from several cases.
Native genomic DNA was screened and analyzed at the NCI according to the sample handling process of the Core Genotyping Facility prior to analysis with the HumanHap 1M BeadChip (Illumina, Inc.) via the Infinium Assay following manufacturer recommendations. Overall, 2.6% of controls were performed in duplicate, with SNP calling concordance greater than 99.94%. Good-quality data were obtained from 1991 samples, 1034 patients and 957 controls. Following a set of standard quality-control metrics that used a hidden Markov model-based method21 with stringent filtering criteria,22 we identified 26,198 presumably nonmosaic CNVs (see Figure S1 and Table S1 available online). Among the samples discarded by filtering, we observed a few with an unusually high number of putative CNVs concentrated across a single chromosome (n = 20). Inspection of the signal intensity log R ratio (LogR) and fraction of the total signal that was due to a specific allele (B allele frequency, BAF) value plots of the affected regions revealed single large aberrations with abnormal average BAF value for heterozygous SNPs (not centered at 0.5) and either (1) normal average LogR value around 0, indicating probable copy-number neutral change with allelic imbalance suggestive of a segmental UPD in mosaicism, or (2) altered LogR values not reaching the chosen threshold for heterozygous deletions or duplications (LogR > 0.2), suggesting mosaic CNVs (Figure 1). We validated the predicted mosaic rearrangements by multiplex ligation-dependent probe amplification (MLPA) and microsatellite analysis (see Table S5 for sequence details) on the same source of DNA used for the SNP array in all samples studied. We then performed a specific analysis to capture all BAF anomalies that might correspond to large mosaic rearrangements in the entire sample set in an unbiased manner (Figure S2). We used R software (version 2.8.1) and the zoo package by basically assessing B deviation values > 0.05 with LogR < 0.2. By using a sliding-windows system (250 SNPs), we analyzed the genome hybridization output of each sample with the established cutoff fixed parameters (≥75 SNPs with designated B deviation and LogR values) to identify trend changes along the chromosome analyzed (Figure S2). The analysis parameters were first set up with the 20 samples known to harbor mosaic abnormalities already confirmed by other techniques, and the tool was then applied to the whole data set. The performance of the method was tested by the reanalysis of samples with previously defined mosaic rearrangements, obtaining a 95% detection rate without false positives in the remaining chromosomal regions.
Figure 1.
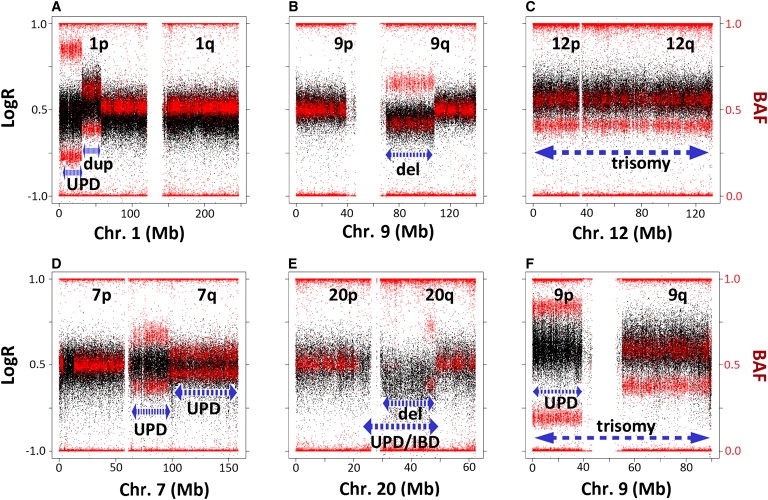
Examples of Different Types of Mosaic Rearrangements, Including Complex Rearrangements
The plots show the signal intensity log R ratio (LogR) (black dots, scale on the left side) and B allele frequency (BAF) (red dots, scale on the right side) values along the entire chromosome carrying the rearrangements in selected samples. The length of the aberration is designated by the dashed blue lines; the type of rearrangement is annotated below.
(A) Mosaic uniparental disomy (UPD) in distal 1p characterized by unchanged LogR and abnormal heterozygous BAF in the indicated interval; the interstitial mosaic duplication of the adjacent fragment shows elevated LogR (lower than the 0.2 cutoff for heterozygous duplication calling) and abnormal heterozygous BAF.
(B) Large mosaic deletion in chromosome arm 9q showing decreased LogR and abnormal heterozygous BAF without complete loss of heterozygosity.
(C) Mosaic trisomy 12 with a pattern similar to that of duplications along the entire chromosome.
(D) Mosaic UPD with different degrees of mosaicism for two adjacent regions of chromosome arm 7q.
(E) Large mosaic deletion in chromosome arm 20q (decreased LogR > −0.2 and abnormal heterozygous BAF). The complete loss of heterozygosity of a genomic region overlapping with the mosaic deletion suggests the presence of complete UPD or homozygosity resulting from identity by descent (IBD).
(F) Mosaic trisomy for the entire chromosome 9 (increased LogR). The different B deviation value between 9p and 9q with identical LogR ratios in this sample also suggests the presence of UPD for 9p in addition to the trisomy.
We detected 23 potential UPD regions in 13 different chromosomes from 20 individuals (13 patients and 7 controls). All UPDs involved segmental and terminal chromosome fragments ranging in size from 6 Mb on chromosome 2p to ∼96 Mb on 13q. Large CNVs with mosaicism were identified in 11 samples (5 cases and 6 controls), ranging in size from 1.1 to 37.7 Mb. Only one was a duplication-type mosaic CNV, 26.3 Mb in size, interstitial but adjacent to a terminal region of mosaic UPD on 1p. Eight entire chromosome gains suggestive of mosaic trisomies (or other polysomies) affecting six autosomes were identified in 7 samples (3 cases and 4 controls). Six individuals (4 cases and 2 controls) showed more than one large mosaic rearrangement. Some rearrangements were complex, with adjacent regions showing different degrees of mosaicism or combination of CNV and UPD (Table 1; Table S2; Figure 2).
Table 1.
Summary of Mosaic Rearrangements Detected
| Sample | Rearrangement | Source | Chr | Start | End | Size (Mb) | Validation | % of Cells | Bladder |
|---|---|---|---|---|---|---|---|---|---|
| Control 468a | UPD | blood | 1p | pter | 31,505,375 | 31.5 | Mi, MLPA | 55% | |
| duplication | blood | 1p | 31,508,099 | 58,012,249 | 26.5 | MLPA | 74% | ||
| UPD | blood | 7q | 69,769,236 | qter | 89 | MLPA | 48% | ||
| Case 197 | deletion | blood | 5q | 107,759,583 | 131,769,397 | 24 | MLPA | 39% | |
| Control 771 | deletion | blood | 9q | 70,096,379 | 107,838,079 | 37.7 | MLPA | 39% | |
| Case 1044 | deletion | blood | 9q | 82,074,397 | 104,355,425 | 22.2 | MLPA | 76% | |
| Control 1014a | deletion | blood | 11q | 93,126,656 | 116,093,059 | 22.9 | Mi | 39% | |
| trisomy/polysomy | blood | 12 | pter | qter | 132 | Mi, MLPA | 69%c | ||
| Control 1017 | deletion | blood | 16p | pter | 3,888,919 | 3.8 | MLPA | 58% | |
| Case 1079 | deletion | blood | 20q | 30,488,149 | 31,745,200 | 1.2 | MLPA | 60% | |
| Case 571 | deletion | blood | 20q | 30,491,175 | 48,812,965 | 18.3 | Mi, MLPA | 44% | |
| Control 837 | deletion | blood | 20q | 30,824,044 | 48,140,963 | 17.3 | Mi, MLPA | 40% | |
| Control 191b | deletion | blood | 20q | 30,489,196 | 48,819,540 | 18.3 | Mi, MLPA | 53% | |
| Case 426a | deletion | blood | 20q | 33,993,320 | 53,443,077 | 19.4 | Mi, MLPA | 89% | Mi, MLPA, FISH |
| trisomy/polysomy | blood | 9 | pter | qter | 140 | Mi, MLPA | 95%c | FISH | |
| Control 577 | trisomy/polysomy | blood | 8 | pter | qter | 146 | Mi, MLPA | 62%c | |
| Control 152 | trisomy/polysomy | blood | 9 | pter | qter | 140 | Mi, MLPA | 63%c | |
| Case 1185a | trisomy/polysomy | blood | 9 | pter | qter | 140 | Mi, MLPA | 72%c | Mi, MLPA, FISH |
| UPD | blood | 9p | pter | 38,987,691 | 38.9 | Mi, MLPA | d | ||
| trisomy/polysomy | blood | 22 | pter | qter | 49.7 | Mi, MLPA | 74%c | FISH negative | |
| Case 511 | trisomy/polysomy | blood | 15 | pter | qter | 100 | MLPA | 60%c | |
| Control 541 | trisomy/polysomy | blood | 19 | pter | qter | 63.8 | Mi, MLPA | 98%c | |
| Control 776 | UPD | blood | 2p | pter | 5,974,108 | 5.9 | MLPA | 28% | |
| Control 196 | UPD | blood | 2q | 210,673,136 | qter | 32.3 | Mi, MLPA | 51% | |
| Case 954 | UPD | blood | 2q | 218,476,667 | qter | 24.5 | MLPA | 22% | |
| Case 155 | UPD | blood | 3p | pter | 43,770,009 | 43.3 | MLPA | 18% | |
| Case 1105e | UPD | blood | 7q | 62,401,114 | 96,812,073 | 34.4 | MLPA | 29% | |
| UPD | blood | 7q | 96,934,617 | qter | 61.9 | MLPA | 18% | ||
| Control 586 | UPD | blood | 9p | pter | 39,102,964 | 39.1 | MLPA | 83% | |
| Control 843 | UPD | blood | 11q | 65,547,103 | qter | 69.2 | MLPA | 22% | |
| Case 125 | UPD | blood | 12q | 55,481,646 | qter | 82.3 | MLPA | 17% | |
| Case 234 | UPD | blood | 13q | 17,956,717 | qter | 96.2 | MLPA | 18% | |
| Case 962a | UPD | blood | 13q | 19,554,439 | qter | 94.5 | Mi | 28% | |
| UPD | blood | 17p | pter | 18,649,825 | 18.6 | Mi | 39% | Mi | |
| Case 787 | UPD | blood | 14q | 23,303,146 | qter | 83 | Mi | 34% | |
| Case 758 | UPD | blood | 14q | 74,454,224 | qter | 31.9 | MLPA | 18% | |
| Case 1205 | UPD | buccal | 16p | pter | 14,565,117 | 14.5 | Mi, MLPA | 25% | |
| Case 815 | UPD | blood | 17p | pter | 4,724,664 | 4.7 | Mi, MLPA | 34% | Mi, MLPA, FISH |
| Case 369 | UPD | blood | 17q | 37,339,650 | qter | 41.4 | MLPA | 28% | |
| Control 1007 | UPD | blood | 17q | 53,007,738 | qter | 25.8 | Mi, MLPA | 21% | |
| Control 670 | UPD | blood | 19q | 48,312,997 | qter | 15.7 | MLPA | 21% | |
| Case 138 | UPD | blood | 21q | 31,600,986 | qter | 14.9 | MLPA | 20% |
A total of 42 rearrangements were found in 34 individuals (19 cases and 15 controls). All observations (42 of 42) were confirmed on the original DNA used for SNP arrays. The start and end point of each rearrangement correspond to the coordinates of the first SNP or probe located within the rearrangement, based on B allele frequency (BAF) and signal intensity log R ratio (LogR) DNA segment values detected with described tools (see text and Figure S2). Additional studies on bladder tumor DNA and tissue were performed in a subset of samples (column “Bladder”). The following abbreviations are used: UPD, uniparental disomy; pter and qter, p-terminal and q-terminal ends of chromosomes; Mi, microsatellite markers; MLPA, multiplex ligation probe-dependent amplification; FISH, fluorescence in situ hybridization.
Samples with more than one rearrangement.
Complete loss of heterozygosity with identical LogR values in an overlapping region (Mb 27–44) in 20q, suggestive of complete UPD and/or identity by descent and mosaic (distal) deletions (Figure 1).
Mosaicism estimation assuming all the cells with the rearrangement are trisomic for the indicated chromosome. The proportion may differ from the estimation if there are cells with other polysomies (tetrasomic or other) in the samples.
Different BAF and identical LogR values between 9p and 9q, suggestive of 9p UPD in addition to whole chromosome 9 trisomy.
Different BAF value between the two regions of 7q with similar average LogR, suggestive of different degrees of mosaicism for UPD.
Figure 2.
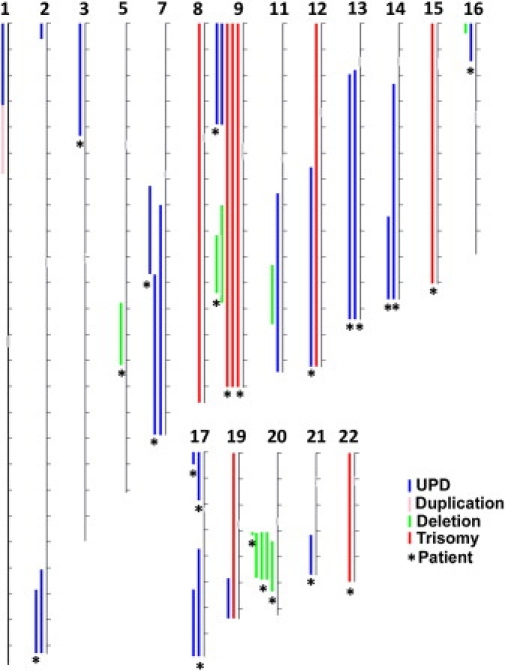
Genomic Distribution of Mosaic Events Detected
The illustration summarizes the chromosomal location and approximate size of all the mosaic events detected along the autosomal chromosomes: 23 UPDs (blue lines), 8 trisomies (red lines), one duplication (pink line), and 10 deletions (green lines). No alterations involving chromosomes 4, 6, 10, or 18 were found. Chromosomes are drawn to scale (tick marks indicate 10 Mb). Asterisks (∗) indicate events found in a bladder cancer patient.
In order to estimate the proportion of cells with mosaicism in every case, we used BAF values from central populations of data points according to Illumina technical notes. A sample with central populations of data points at 0.55 and 0.45 BAF values for heterozygous SNPs was considered to have 55% of chromosomes with a specific allele and 45% of chromosomes with the other allele (best estimates). We then used the B deviation (Bdev, deviation from the expected BAF value of 0.5 for heterozygous SNPs) to calculate the proportion of cells with the rearrangement depending on the type of mosaic rearrangement: loss (deletion/monosomy; genotypes A/− and A/B), gain (duplication/trisomy; genotypes AA/B and A/B), or copy-number neutral change (UPD; genotypes A/A and A/B). The simplified formulae used were as follows:
In order to avoid false positive results due to experimental data of poor quality, we discarded samples with an average standard deviation of the BAF value above 0.05. Given that the B deviation cutoff chosen for mosaic rearrangement calling was then >0.05, our method can be estimated to detect mosaicism only when the proportion of affected cells is above 10%, 18%, or 23% for UPD, deletions, and duplications and/or trisomies, respectively. In theory, for samples yielding high-quality data (i.e., with standard deviation of BAF < 0.025), it would be possible to detect much lower levels of mosaicism with SNP arrays (about half of the above figures). The percentage of cells carrying each rearrangement ranged from 17% to 82% in UPDs, 39% to 89% in deletions, 74% in the only mosaic duplication, and 62% to 98% in trisomies (Table 1). The high proportion of affected cells in most detected rearrangements suggests either the arousal of mutations early in development or a positive selection for the rearranged cells. Furthermore, a significant proportion of mosaic rearrangements might remain undetected, mainly those present in a lower proportion of cells, smaller than 1 Mb in size, or involving copy-number gains. Formal testing for case-control differences in the frequency of rearrangements, divided by broad category of event or percentage of cells affected, was clearly null in all cases, thus suggesting that most rearrangements found are probably not central to the development of bladder cancer (Table S3).
We validated by MLPA and microsatellite analysis on the same source of DNA all 42 predicted mosaic rearrangements (Table 1; Figure S3; see also Table S5 for sequence details). We further analyzed tumor DNA and tumor tissue sections from four cases of bladder cancer in which we had detected alterations in blood DNA (cases 962, 426, 815, and 1185). Using microsatellite analysis, we confirmed the allelic imbalances in tumor DNA indicative of the 17p UPDs, 20q deletion, and chromosome 9 trisomies (Figure 3); by contrast, the chromosome 22 gain was not detected in the tumor cells. The mosaic rearrangements were also confirmed by fluorescence in situ hybridization (FISH) with specific probes in tumor cells of three samples (Figure 3). Therefore, mosaicism was present in two tissues from the same individual with different embryonic origin, suggesting that the 17p UPDs, 20q deletion, and trisomies 9 (5 out of 6 tested rearrangements) must have arisen early in development. A previous report identified mosaic CNVs, ranging in size from 82 to 176 kb, in a diverse range of organs and tissues, some cell-type specific and some present in all tissues.9 Therefore, mutational events and chromosome instability leading to mosaicism can occur early in embryonic development but also in late mitosis affecting a single tissue.23–26
Figure 3.
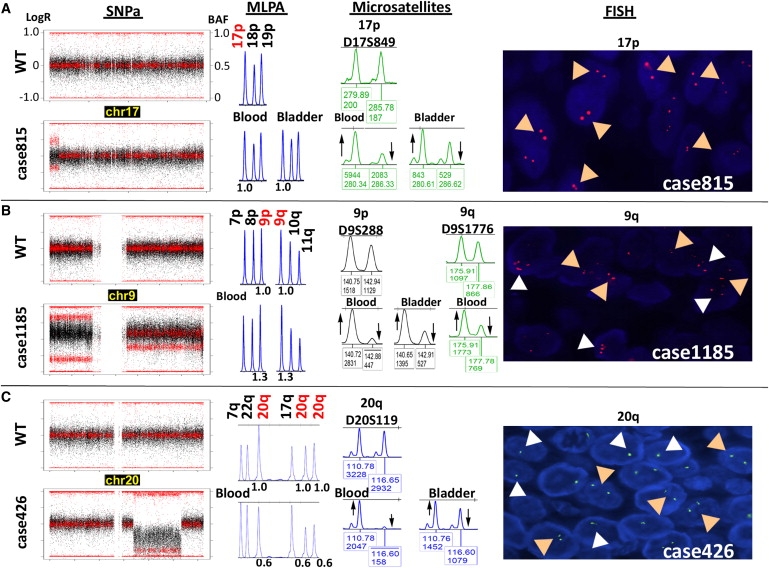
Representative Mosaic Rearrangements Validated by Independent Methods
Validation was performed on the same source of DNA used for the SNP array (multiplex ligation-dependent probe amplification [MLPA] and microsatellites) as well as on DNA (microsatellites) and tumor tissue sections (fluorescence in situ hybridization [FISH]) from bladder tumor tissue. The plots at left show the LogR ratio (black dots) and BAF (red dots) values along an entire chromosome in selected samples (bottom) compared to the wild-type pattern (WT, top).
(A) Segmental UPD of 17p terminal in case 815. MLPA (P060 panel) confirmed the disomic state at the UPD loci with no gain or loss of genetic material (1.0 relative peak height [RPH]), microsatellite analysis ratified the allelic imbalance both in blood and bladder tumor DNA reflected by an aberrant ratio between allelic peaks with respect to the control sample, and FISH showed two signals corresponding to disomy in all nuclei (tan arrowheads in panel at right).
(B) Full trisomy 9 with additional UPD of 9p. MLPA confirmed a gain of genetic material at the trisomic locus (1.3 RPH), and microsatellite analysis ratified the allelic imbalance without detecting third alleles in both blood and bladder tumor DNA (shown by upward and downward arrows in middle panel). Right panel: FISH on bladder tumor tissue revealed a mosaic pattern of interphase cell nuclei with two (disomic, tan arrowheads), three (trisomic, white arrowheads; ∼30%), or four or more (tetrasomic or polysomic; ∼10%) signals.
(C) Large (19.4 Mb) deletion-type copy-number variation (CNV) on chromosome 20q. MLPA confirmed a loss of genetic material at three loci within the interval, and microsatellite analysis ratified an allelic imbalance in both blood and bladder tumor DNA. Right panel: FISH on bladder tumor tissue revealed a mosaic pattern of interphase cell nuclei with either one (monosomic, white arrowheads) or two (disomic, tan arrowheads) signals. The percentage of cells carrying the rearrangement was higher in blood than in tumor cells in all three cases, as revealed by the greater degree of allelic imbalance.
Although none of the mosaic UPD regions detected in different individuals were identical in size and/or breakpoints, shared genomic intervals occurred at 2q, 7q, 13q, 17p, and 17q. Interestingly, mosaic UPD including almost the entire 13q arm was found in blood DNA from two unrelated bladder cancer patients who also had deletion-type heterozygous CNVs overlapping ∼833 kb at 13q14 (Figure 4). This deletion maps in the vicinity of the RB1 locus commonly deleted in bladder tumors, encompasses several candidate cancer genes (DLEU1, DLEU2, and DLEU7), and could alter expression of two cancer-related microRNAs (miR-15a and miR-16-1) located in an intron of DLEU2.27–29 The co-occurrence in two unrelated patients with bladder cancer of two rare chromosome abnormalities, germline 13q14 deletion and mosaic UPD 13q, suggests that these abnormalities could be mechanistically linked and/or related to disease. Heterozygous deletions can represent susceptibility factors for mitotic instability leading to UPD, as shown for some meiotic rearrangements.30 The potential pathogenic involvement of these rearrangements may depend on the proportion of UPD, susceptibility to nullizygosity, or tissue-specific gene effects.
Figure 4.
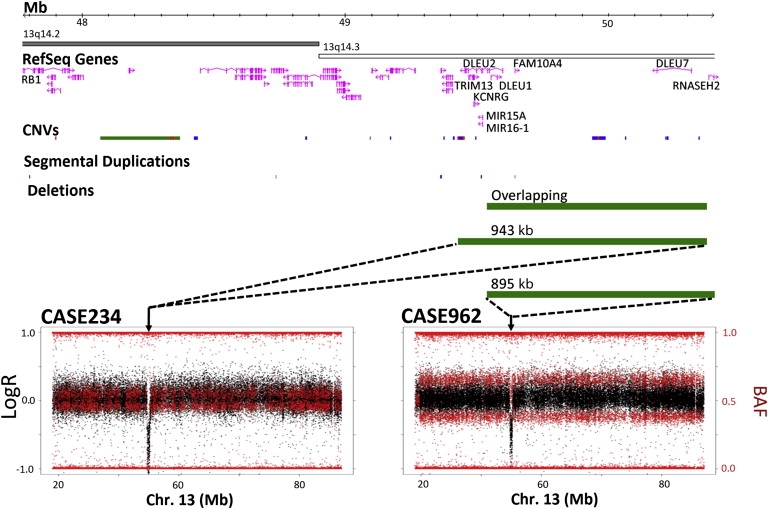
Overlapping Rearrangements in Leukocyte DNA Samples from Two Patients with Bladder Cancer
The plots on the bottom show the mosaic UPDs including almost the entire chromosome 13q found in blood DNA of two bladder cancer patients who also shared deletion-type heterozygous CNVs overlapping ∼833 kb at 13q14 (chr13 coordinates: 49539177–50372054). Analysis of the average LogR of the probes within the CNV yielded below-average LogR ratio for heterozygous deletion values in case 234 (left) and average values in case 962 (right); loss of heterozygosity was also found in case 234, whereas case 962 displayed values compatible with heterozygosity in 1/3 of SNPs within the interval, indicating that the disomic chromosome was the one carrying the CNV in case 234 whereas the other chromosome carried the CNV in case 962. The size of the deletions and overlapping fragment is represented by green bars illustrating their relative location with respect to the chromosome 13 ideogram and showing related genomic features: gene content (including distant RB1 gene), CNVs (Database of Genomic Variants, March 2010 version), and segmental duplications.
Whereas 10 of 11 mosaic CNVs were interstitial, all 23 segmental UPDs detected were terminal, likely resulting from a postfertilization error during early mitotic divisions mediated by single events of somatic homologous recombination. The mechanisms of somatic reshuffling leading to UPD are not well defined, and some argue that hot spots can play a role.31,32 We defined the breakpoint intervals for all segmental rearrangements as the regions between two informative SNP probes (within and outside the rearrangement) and analyzed whether they shared any genomic features, including their overlap with recombination hot spots of the human genome,33 segmental duplications, or structural variation.34,35 We calculated permutation p values by randomizing the positions of breakpoints across the genome 1000 times and measuring the number of times that the breakpoints overlapped with different genomic features; p values were calculated as the number of times that the observed value equaled or exceeded the expected value in the randomized set, divided by the total number of permutations plus one. Six UPD breakpoint intervals localized to segmental duplications (28.5%, ∼5× enrichment, p = 0.003), six overlapped with meiotic crossing-over hot spots (∼4.3× enrichment, p = 0.209), and ten mapped within copy-number-variable regions (∼2× enrichment, p = 0.035), suggesting that mitotic rearrangements are mediated by mechanisms similar to those of meiotic recombination (Table S4). No significant enrichment of sequence motifs was identified in the 19 breakpoints of the large CNVs. However, we found five overlapping deletion-type CNVs in chromosome 20q, some of which shared breakpoints (Figure 5; Table S4). Interestingly, similar deletions that may harbor tumor suppressor genes have been recurrently reported in myelodysplastic syndromes and in Philadelphia chromosome-negative myeloproliferative disorders.36,37 These findings further indicate the existence of hot spots for somatic recombination in chromosome 20q.
Figure 5.
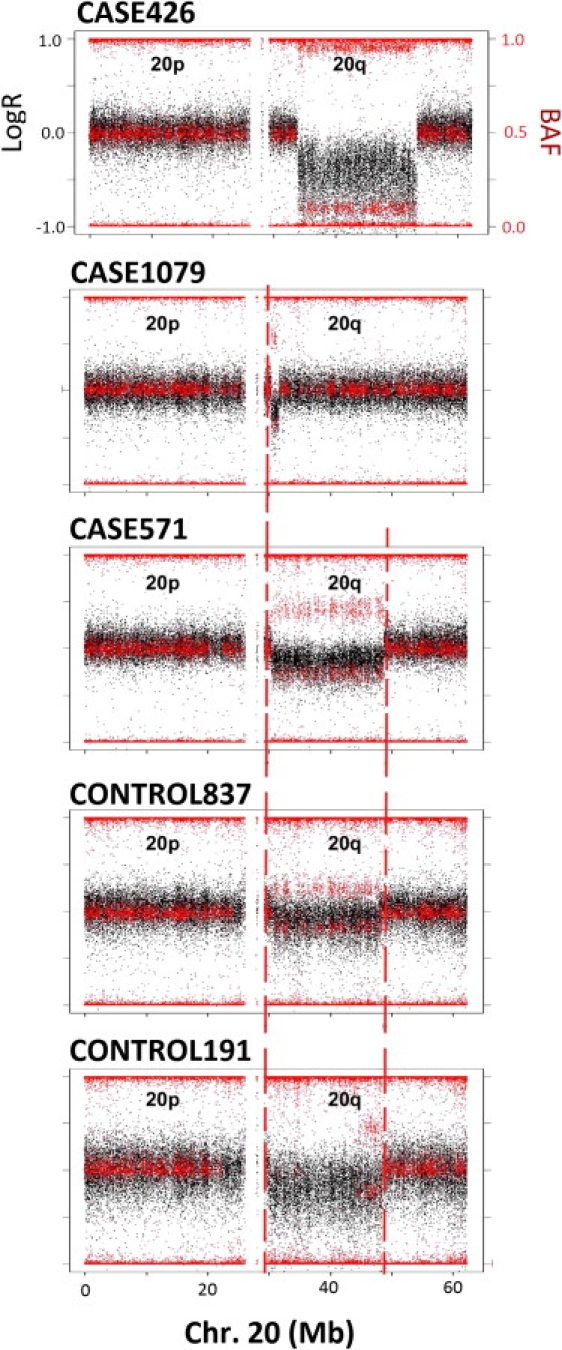
Overlapping Mitotic Interstitial Deletions of Chromosome 20q in Five Unrelated Individuals
Aligned plots of LogR ratio (black) and BAF (red) values along the entire chromosome 20 of the five individuals, showing the rearrangements and the similar breakpoints (joined by dashed vertical red lines).
In our survey, chromosome 9 had the highest rearrangement rate (two large 9q deletions, two 9p UPDs, and three complete trisomies), suggesting an increased somatic instability for this chromosome, but further studies are needed to evaluate this observation. It is plausible that chromosome 9 structural variants, such as the polymorphic pericentric inversion present in ∼0.85% of the population, may behave as susceptibility factors for somatic instability.38
Although large-scale mosaic events have been detected before, they were primarily observed in subjects from clinic-based studies, where screened individuals have high prior likelihood for one or more genetic causes associated with their condition. Our findings provide evidence for a greater complexity of the human genome with respect to structural events. Somatic mosaicism for large structural autosomal chromosome abnormalities, including CNVs, aneuploidies, and copy-number neutral changes due to segmental UPD, appears to be present in blood or buccal cells, both DNA sources with a spectrum of well-differentiated cell types, of 1.7% of the adult population. The high cellular frequency of most mosaic anomalies detected and their presence in normal adult individuals suggest that this type of mosaicism is a widespread phenomenon of the human genome with possible phenotypic consequences, though almost half of our observed events occurred in otherwise healthy elderly controls. Somatic mosaicism should be considered in the expanding repertoire of inter- and intraindividual genetic variation, some of which causes somatic human diseases but also modifies penetrance and/or expressivity of inherited disorders and late-onset multifactorial traits. When affecting the germline, these abnormalities could also contribute to infertility, recurrent miscarriages, or recurrent anomalies in offspring.39
It is highly possible that the mosaic occurrence of genomic variants, especially UPD, may have been missed previously with the standard analytical procedures applied to CNV detection in published studies with SNP arrays. Moreover, our quality-control metrics could also lead to an underestimation of the events, even within our study. It is also plausible that some of the CNVs registered in the database of genomic variants might correspond to mosaic rearrangements. Thus, optimization of the analysis of data obtained with SNP arrays, as well as the development of similar algorithms for the analysis of high-depth coverage data obtained with next-generation sequencing, is required to improve the accurate detection of these events in human populations. Capturing and classifying all relevant genomic variation features in cells from different tissues, and at different developmental stages, constitutional or acquired, should lead to a better understanding of the complex and evolving human genome and its relation to both diseases and traits.
Acknowledgments
We thank D. Pastor, A. Alfaro, M. Márquez, T. Lobato, F. Fernández, M. Torà, J. Lloreta, C. Guerrero, C. Ampurdanés, A. Itsara, K. Wang, and M. Salido for technical assistance, as well as D.G. Pisano, E. Andrés, R. Díaz-Uriarte, G. Gómez, A. González-Neira, G. Pita, and I. Cuscó for critical comments. This work was supported by the Intramural Research Program of the NCI Division of Cancer Epidemiology and Genetics (to N.R., D.S., and S.J.C.), the Asociación Española Contra el Cáncer (to F.X.R., N.M., and L.A.P.-J.), EU-6FP grants LSHG-CT-2006-037627 (to L.A.P.-J.) and HEALTH-STREP-2006-037739-DropTop (to N.M. and F.X.R.), Fondo de Investigación Sanitaria grants PI076832 (to L.A.P.-J.) and PI061614 (to F.X.R.), Consolider ONCOBIO (to F.X.R.), National Institutes of Health grant R01 CA089715-06A2 (to N.M. and F.X.R.), Red Temática de Investigación Cooperativa en Cáncer (to N.M. and A.C.), Fundació La Marató de TV3 (to N.M.), and EU-7FP grant agreements #201663-UROMOL (to N.M. and F.X.R.) and #201333-DECanBIO (to N.M.). B.R.-S. and G.M. were supported by a postdoctoral fellowship (FIS CD06/00019) and a predoctoral fellowship (FI09/00205) of the Fondo Investigación Sanitaria, respectively.
L.A. and L.A.P.-J. are the executive director and a member of the scientific advisory board of qGenomics, respectively.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Database of Genomic Variants, http://projects.tcag.ca/cgi-bin/variation/gbrowse/hg18/
Illumina Beadchip information, http://www.illumina.com/documents/products/appnotes/appnote_cytogenetics.pdf
NCI Core Genotyping Facility, http://cgf.nci.nih.gov
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
OXSTATS Recombination Map, http://mathgen.stats.ox.ac.uk/Recombination.html
R software, http://www.r-project.org
Segmental Duplication Database, http://humanparalogy.gs.washington.edu/build36/build36.htm
zoo package, http://cran.r-project.org/web/packages/zoo/index.html
References
- 1.Youssoufian H., Pyeritz R.E. Mechanisms and consequences of somatic mosaicism in humans. Nat. Rev. Genet. 2002;3:748–758. doi: 10.1038/nrg906. [DOI] [PubMed] [Google Scholar]
- 2.Notini A.J., Craig J.M., White S.J. Copy number variation and mosaicism. Cytogenet. Genome Res. 2008;123:270–277. doi: 10.1159/000184717. [DOI] [PubMed] [Google Scholar]
- 3.Hsu L.Y., Kaffe S., Jenkins E.C., Alonso L., Benn P.A., David K., Hirschhorn K., Lieber E., Shanske A., Shapiro L.R. Proposed guidelines for diagnosis of chromosome mosaicism in amniocytes based on data derived from chromosome mosaicism and pseudomosaicism studies. Prenat. Diagn. 1992;12:555–573. doi: 10.1002/pd.1970120702. [DOI] [PubMed] [Google Scholar]
- 4.Hsu L.Y., Yu M.T., Richkind K.E., Van Dyke D.L., Crandall B.F., Saxe D.F., Khodr G.S., Mennuti M., Stetten G., Miller W.A., Priest J.H. Incidence and significance of chromosome mosaicism involving an autosomal structural abnormality diagnosed prenatally through amniocentesis: A collaborative study. Prenat. Diagn. 1996;16:1–28. doi: 10.1002/(SICI)1097-0223(199601)16:1<1::AID-PD816>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 5.Rehen S.K., Yung Y.C., McCreight M.P., Kaushal D., Yang A.H., Almeida B.S., Kingsbury M.A., Cabral K.M., McConnell M.J., Anliker B. Constitutional aneuploidy in the normal human brain. J. Neurosci. 2005;25:2176–2180. doi: 10.1523/JNEUROSCI.4560-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yurov Y.B., Iourov I.Y., Vorsanova S.G., Liehr T., Kolotii A.D., Kutsev S.I., Pellestor F., Beresheva A.K., Demidova I.A., Kravets V.S. Aneuploidy and confined chromosomal mosaicism in the developing human brain. PLoS ONE. 2007;2:e558. doi: 10.1371/journal.pone.0000558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guttenbach M., Koschorz B., Bernthaler U., Grimm T., Schmid M. Sex chromosome loss and aging: In situ hybridization studies on human interphase nuclei. Am. J. Hum. Genet. 1995;57:1143–1150. [PMC free article] [PubMed] [Google Scholar]
- 8.Bruder C.E., Piotrowski A., Gijsbers A.A., Andersson R., Erickson S., Diaz de Ståhl T., Menzel U., Sandgren J., von Tell D., Poplawski A. Phenotypically concordant and discordant monozygotic twins display different DNA copy-number-variation profiles. Am. J. Hum. Genet. 2008;82:763–771. doi: 10.1016/j.ajhg.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piotrowski A., Bruder C.E., Andersson R., Diaz de Ståhl T., Menzel U., Sandgren J., Poplawski A., von Tell D., Crasto C., Bogdan A. Somatic mosaicism for copy number variation in differentiated human tissues. Hum. Mutat. 2008;29:1118–1124. doi: 10.1002/humu.20815. [DOI] [PubMed] [Google Scholar]
- 10.Ballif B.C., Rorem E.A., Sundin K., Lincicum M., Gaskin S., Coppinger J., Kashork C.D., Shaffer L.G., Bejjani B.A. Detection of low-level mosaicism by array CGH in routine diagnostic specimens. Am. J. Med. Genet. A. 2006;140:2757–2767. doi: 10.1002/ajmg.a.31539. [DOI] [PubMed] [Google Scholar]
- 11.Cheung S.W., Shaw C.A., Scott D.A., Patel A., Sahoo T., Bacino C.A., Pursley A., Li J., Erickson R., Gropman A.L. Microarray-based CGH detects chromosomal mosaicism not revealed by conventional cytogenetics. Am. J. Med. Genet. A. 2007;143A:1679–1686. doi: 10.1002/ajmg.a.31740. [DOI] [PubMed] [Google Scholar]
- 12.Conlin L.K., Thiel B.D., Bonnemann C.G., Medne L., Ernst L.M., Zackai E.H., Deardorff M.A., Krantz I.D., Hakonarson H., Spinner N.B. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum. Mol. Genet. 2010;19:1263–1275. doi: 10.1093/hmg/ddq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu X.Y., Phung M.T., Shaw C.A., Pham K., Neil S.E., Patel A., Sahoo T., Bacino C.A., Stankiewicz P., Kang S.H. Genomic imbalances in neonates with birth defects: High detection rates by using chromosomal microarray analysis. Pediatrics. 2008;122:1310–1318. doi: 10.1542/peds.2008-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Menten B., Maas N., Thienpont B., Buysse K., Vandesompele J., Melotte C., de Ravel T., Van Vooren S., Balikova I., Backx L. Emerging patterns of cryptic chromosomal imbalance in patients with idiopathic mental retardation and multiple congenital anomalies: A new series of 140 patients and review of published reports. J. Med. Genet. 2006;43:625–633. doi: 10.1136/jmg.2005.039453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scott S.A., Cohen N., Brandt T., Toruner G., Desnick R.J., Edelmann L. Detection of low-level mosaicism and placental mosaicism by oligonucleotide array comparative genomic hybridization. Genet. Med. 2010;12:85–92. doi: 10.1097/GIM.0b013e3181cc75d0. [DOI] [PubMed] [Google Scholar]
- 16.Tuna M., Knuutila S., Mills G.B. Uniparental disomy in cancer. Trends Mol. Med. 2009;15:120–128. doi: 10.1016/j.molmed.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Erickson R.P. Somatic gene mutation and human disease other than cancer. Mutat. Res. 2003;543:125–136. doi: 10.1016/s1383-5742(03)00010-3. [DOI] [PubMed] [Google Scholar]
- 18.Hall J.G. Review and hypotheses: Somatic mosaicism: Observations related to clinical genetics. Am. J. Hum. Genet. 1988;43:355–363. [PMC free article] [PubMed] [Google Scholar]
- 19.Guey L.T., García-Closas M., Murta-Nascimento C., Lloreta J., Palencia L., Kogevinas M., Rothman N., Vellalta G., Calle M.L., Marenne G., EPICURO/Spanish Bladder Cancer Study investigators Genetic susceptibility to distinct bladder cancer subphenotypes. Eur. Urol. 2010;57:283–292. doi: 10.1016/j.eururo.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samanic C., Kogevinas M., Dosemeci M., Malats N., Real F.X., Garcia-Closas M., Serra C., Carrato A., García-Closas R., Sala M. Smoking and bladder cancer in Spain: Effects of tobacco type, timing, environmental tobacco smoke, and gender. Cancer Epidemiol. Biomarkers Prev. 2006;15:1348–1354. doi: 10.1158/1055-9965.EPI-06-0021. [DOI] [PubMed] [Google Scholar]
- 21.Wang K., Li M., Hadley D., Liu R., Glessner J., Grant S.F., Hakonarson H., Bucan M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Itsara A., Cooper G.M., Baker C., Girirajan S., Li J., Absher D., Krauss R.M., Myers R.M., Ridker P.M., Chasman D.I. Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frank S.A. Evolution in health and medicine Sackler colloquium: Somatic evolutionary genomics: Mutations during development cause highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proc. Natl. Acad. Sci. USA. 2010;107(Suppl 1):1725–1730. doi: 10.1073/pnas.0909343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang Q., Conte N., Skarnes W.C., Bradley A. Extensive genomic copy number variation in embryonic stem cells. Proc. Natl. Acad. Sci. USA. 2008;105:17453–17456. doi: 10.1073/pnas.0805638105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mkrtchyan H., Gross M., Hinreiner S., Polytiko A., Manvelyan M., Mrasek K., Kosyakova N., Ewers E., Nelle H., Liehr T. Early embryonic chromosome instability results in stable mosaic pattern in human tissues. PLoS ONE. 2010;5:e9591. doi: 10.1371/journal.pone.0009591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vanneste E., Voet T., Le Caignec C., Ampe M., Konings P., Melotte C., Debrock S., Amyere M., Vikkula M., Schuit F. Chromosome instability is common in human cleavage-stage embryos. Nat. Med. 2009;15:577–583. doi: 10.1038/nm.1924. [DOI] [PubMed] [Google Scholar]
- 27.Lee S., Jeong J., Majewski T., Scherer S.E., Kim M.S., Tuziak T., Tang K.S., Baggerly K., Grossman H.B., Zhou J.H. Forerunner genes contiguous to RB1 contribute to the development of in situ neoplasia. Proc. Natl. Acad. Sci. USA. 2007;104:13732–13737. doi: 10.1073/pnas.0701771104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lerner M., Harada M., Lovén J., Castro J., Davis Z., Oscier D., Henriksson M., Sangfelt O., Grandér D., Corcoran M.M. DLEU2, frequently deleted in malignancy, functions as a critical host gene of the cell cycle inhibitory microRNAs miR-15a and miR-16-1. Exp. Cell Res. 2009;315:2941–2952. doi: 10.1016/j.yexcr.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 29.Migliazza A., Bosch F., Komatsu H., Cayanis E., Martinotti S., Toniato E., Guccione E., Qu X., Chien M., Murty V.V. Nucleotide sequence, transcription map, and mutation analysis of the 13q14 chromosomal region deleted in B-cell chronic lymphocytic leukemia. Blood. 2001;97:2098–2104. doi: 10.1182/blood.v97.7.2098. [DOI] [PubMed] [Google Scholar]
- 30.Cuscó I., Corominas R., Bayés M., Flores R., Rivera-Brugués N., Campuzano V., Pérez-Jurado L.A. Copy number variation at the 7q11.23 segmental duplications is a susceptibility factor for the Williams-Beuren syndrome deletion. Genome Res. 2008;18:683–694. doi: 10.1101/gr.073197.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Engel E. A fascination with chromosome rescue in uniparental disomy: Mendelian recessive outlaws and imprinting copyrights infringements. Eur. J. Hum. Genet. 2006;14:1158–1169. doi: 10.1038/sj.ejhg.5201619. [DOI] [PubMed] [Google Scholar]
- 32.Kotzot D. Complex and segmental uniparental disomy updated. J. Med. Genet. 2008;45:545–556. doi: 10.1136/jmg.2008.058016. [DOI] [PubMed] [Google Scholar]
- 33.Myers S., Bottolo L., Freeman C., McVean G., Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- 34.Cheung J., Estivill X., Khaja R., MacDonald J.R., Lau K., Tsui L.C., Scherer S.W. Genome-wide detection of segmental duplications and potential assembly errors in the human genome sequence. Genome Biol. 2003;4:R25. doi: 10.1186/gb-2003-4-4-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharp A.J., Locke D.P., McGrath S.D., Cheng Z., Bailey J.A., Vallente R.U., Pertz L.M., Clark R.A., Schwartz S., Segraves R. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bench A.J., Nacheva E.P., Hood T.L., Holden J.L., French L., Swanton S., Champion K.M., Li J., Whittaker P., Stavrides G., UK Cancer Cytogenetics Group (UKCCG) Chromosome 20 deletions in myeloid malignancies: Reduction of the common deleted region, generation of a PAC/BAC contig and identification of candidate genes. Oncogene. 2000;19:3902–3913. doi: 10.1038/sj.onc.1203728. [DOI] [PubMed] [Google Scholar]
- 37.White N.J., Nacheva E., Asimakopoulos F.A., Bloxham D., Paul B., Green A.R. Deletion of chromosome 20q in myelodysplasia can occur in a multipotent precursor of both myeloid cells and B cells. Blood. 1994;83:2809–2816. [PubMed] [Google Scholar]
- 38.Amiel A., Sardos-Albertini F., Fejgin M.D., Sharony R., Diukman R., Bartoov B. Interchromosomal effect leading to an increase in aneuploidy in sperm nuclei in a man heterozygous for pericentric inversion (inv 9) and C-heterochromatin. J. Hum. Genet. 2001;46:245–250. doi: 10.1007/s100380170073. [DOI] [PubMed] [Google Scholar]
- 39.Iourov I.Y., Vorsanova S.G., Yurov Y.B. Chromosomal mosaicism goes global. Mol. Cytogenet. 2008;1:26. doi: 10.1186/1755-8166-1-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.